Viromes of Ten Alfalfa Plants in Australia Reveal Diverse Known Viruses and a Novel RNA Virus

 , , and
, , and 
Abstract
:1. Introduction
2. Results
2.1. Identification and Taxonomic Profiling of Viruses in Australian Alfalfa
2.2. Genome Assembly and Sequence Comparisons of AMV and BLRV Isolates
2.3. A putative New Emaravirus Associated with Alfalfa
2.4. Persistent dsRNA Viruses Associated with Alfalfa
2.4.1. Medicago sativa Alphapartitivirus 1
2.4.2. Medicago sativa Amalgavirus 1
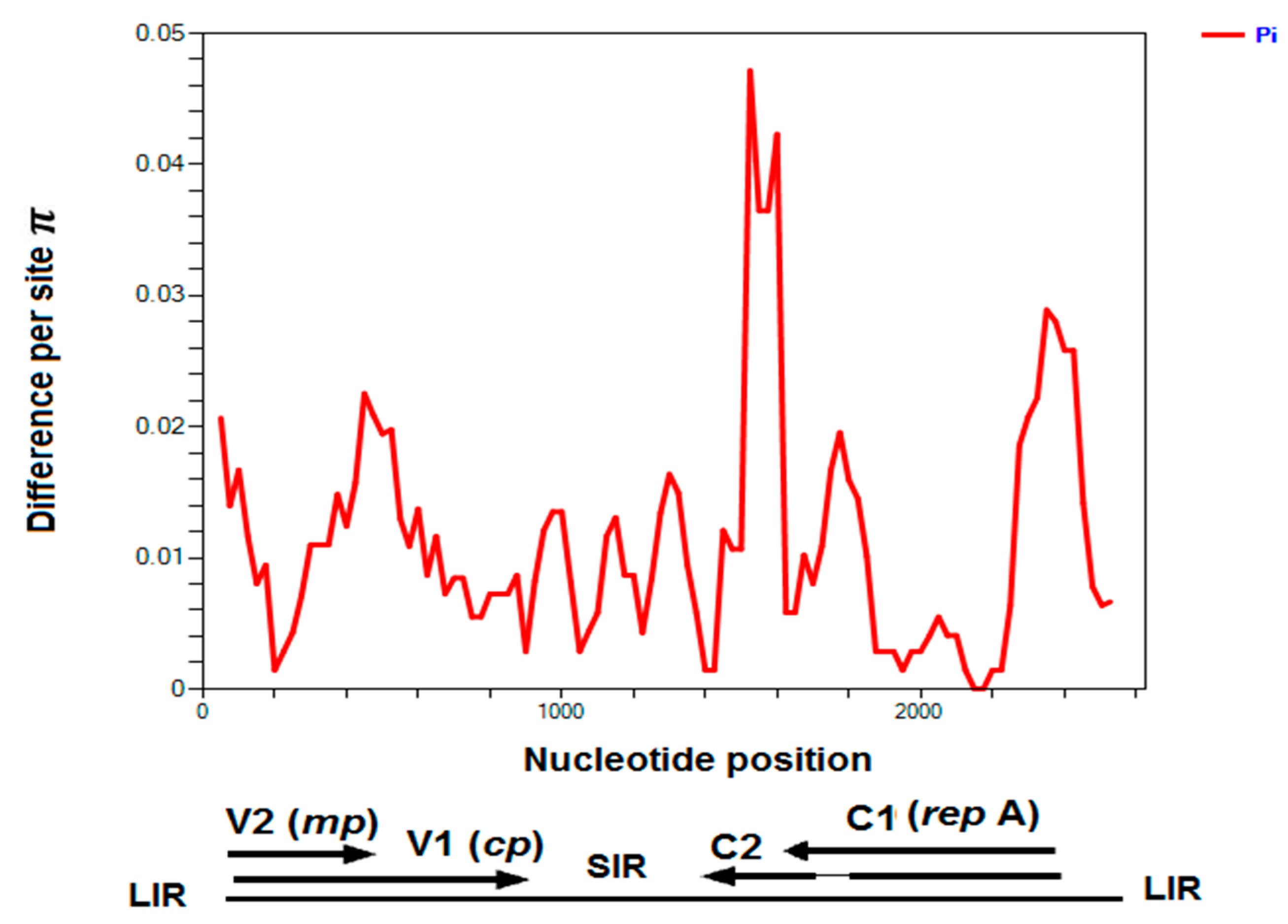
2.5. Chickpea Chlorosis Australia Virus
3. Discussion
4. Materials and Methods
4.1. Plant Material for Analysis
4.2. dsRNA Capture
4.3. High Throughput Sequencing of dsRNA
4.4. Identification of DNA Viruses
4.5. Bioinformatics Analyses
4.6. Verification of HTS-Based Virus Detection by RT-PCR and Sanger Sequencing
4.7. Phylogenetic and Sequence Diversity Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nichols, P.; Revell, C.; Humphries, A.; Howie, J.; Hall, E.; Sandral, G.; Ghamkhar, K.; Harris, C. Temperate pasture legumes in Australia—Their history, current use, and future prospects. Crop Pasture Sci. 2013, 63, 691–725. [Google Scholar] [CrossRef]
- Bouton, J. Breeding lucerne for persistence. Crop Pasture Sci. 2012, 63, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Van Leur, J.A.G.; Kumari, S.G. A survey of lucerne in northern New South Wales for viruses of importance to the winter legume industry. Australas. Plant Pathol. 2011, 40, 180–186. [Google Scholar] [CrossRef]
- Hajimorad, M.R.; Francki, R.I.B. Alfalfa mosaic virus isolates from lucerne in South Australia: Biological variability and antigenic similarity. Ann. Appl. Biol. 1988, 113, 45–54. [Google Scholar] [CrossRef]
- Jones, R.A.C. Occurrence of virus infection in seed stocks and 3-year-old pastures of lucerne (Medicago sativa). Aust. J. Agric. Res. 2004, 55, 757–764. [Google Scholar] [CrossRef]
- Freeman, A.J.; Aftab, M. Effective management of viruses in pulse crops in south eastern Australia should include management of weeds. Australas. Plant Pathol. 2011, 40, 430–441. [Google Scholar] [CrossRef]
- Bejerman, N.; Nome, C.; Giolitti, F.; Kitajima, E.; De Breuil, S.; Pérez Fernández, J.; Basigalup, D.; Cornacchione, M.; Lenardon, S. First report of a rhabdovirus infecting alfalfa in Argentina. Plant Dis. 2011, 95, 771. [Google Scholar] [CrossRef]
- Trucco, V.; de Breuil, S.; Bejerman, N.; Lenardon, S.; Giolitti, F. Complete nucleotide sequence of alfalfa mosaic virus isolated from alfalfa (Medicago sativa L.) in Argentina. Virus Genes 2014, 48, 562–565. [Google Scholar] [CrossRef]
- Trucco, V.; De Breuil, S.; Bejerman, N.; Lenardon, S.; Giolitti, F. Bean leafroll virus (BLRV) in Argentina: Molecular characterization and detection in alfalfa fields. Eur. J. Plant Pathol. 2016, 146, 207–212. [Google Scholar] [CrossRef]
- Bejerman, N.; Giolitti, F.; de Breuil, S.; Trucco, V.; Nome, C.; Lenardon, S.; Dietzgen, R.G. Complete genome sequence and integrated protein localization and interaction map for alfalfa dwarf virus, which combines properties of both cytoplasmic and nuclear plant rhabdoviruses. Virology 2015, 483, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Bejerman, N.; Giolitti, F.; Trucco, V.; De Breuil, S.; Dietzgen, R.G.; Lenardon, S. Complete genome sequence of a new enamovirus from Argentina infecting alfalfa plants showing dwarfism symptoms. Arch. Virol. 2016, 161, 2029–2032. [Google Scholar] [CrossRef] [PubMed]
- Bejerman, N.; Trucco, V.; De Breuil, S.; Pardina, P.R.; Lenardon, S.; Giolitti, F. Genome characterization of an Argentinean isolate of alfalfa leaf curl virus. Arch. Virol. 2018, 163, 799–803. [Google Scholar] [CrossRef]
- Gaafar, Y.Z.; Richert-Pöggeler, K.R.; Maaß, C.; Vetten, H.J.; Ziebell, H. Characterisation of a novel nucleorhabdovirus infecting alfalfa (Medicago sativa). Virol. J. 2019, 16, 55. [Google Scholar] [CrossRef] [Green Version]
- Nemchinov, L.G.; François, S.; Roumagnac, P.; Ogliastro, M.; Hammond, R.W.; Mollov, D.S.; Filloux, D. Characterization of alfalfa virus F, a new member of the genus Marafivirus. PLoS ONE 2018, 13, e0203477. [Google Scholar] [CrossRef] [PubMed]
- Nemchinov, L.G.; Grinstead, S.C.; Mollov, D.S. Alfalfa virus S, a new species in the family Alphaflexiviridae. PLoS ONE 2017, 12, e0178222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemchinov, L.G.; Lee, M.N.; Shao, J. First Report of alphapartitiviruses infecting alfalfa (Medicago sativa L.) in the United States. Microbiol. Resour. Announc. 2018, 7, e01266-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.; Park, D.; Hahn, Y. Identification of novel RNA viruses in alfalfa (Medicago sativa): An Alphapartitivirus, a Deltapartitivirus, and a Marafivirus. Gene 2018, 638, 7–12. [Google Scholar] [CrossRef]
- Bejerman, N.; Humberto, D.; Nome, C.; Cabrera-Mederos, D.; Trucco, V.; de Breuil, S.; Lenardon, S.; Giolitti, F. Redefining the medicago sativa alphapartitiviruses genome sequences. Virus Res. 2019, 265, 156–161. [Google Scholar] [CrossRef]
- Al Rwahnih, M.; Daubert, S.; Golino, D.; Islas, C.; Rowhani, A. Comparison of next-generation sequencing versus biological indexing for the optimal detection of viral pathogens in grapevine. Phytopathology 2015, 105, 758–763. [Google Scholar] [CrossRef] [Green Version]
- Villamor, D.E.; Ho, T.; Al Rwahnih, M.; Martin, R.R.; Tzanetakis, I. High throughput sequencing in plant virus detection and discovery. Phytopathology 2019. [Google Scholar] [CrossRef]
- Massart, S.; Chiumenti, M.; De Jonghe, K.; Glover, R.; Haegeman, A.; Koloniuk, I.; Komínek, P.; Kreuze, J.; Kutnjak, D.; Lotos, L.; et al. Virus detection by high-throughput sequencing of small RNAs: Large scale performance testing of sequence analysis strategies. Phytopathology 2019, 109, 488–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, I.P.; Fox, A.; Boonham, N.; Massart, S.; De Jonghe, K. The impact of high throughput sequencing on plant health diagnostics. Eur. J. Plant Pathol. 2018, 152, 909–919. [Google Scholar] [CrossRef]
- Li, Y.; Jia, A.; Qiao, Y.; Xiang, J.; Zhang, Y.; Wang, W. Virome analysis of lily plants reveals a new potyvirus. Arch. Virol. 2018, 163, 1079–1082. [Google Scholar] [CrossRef] [PubMed]
- Verbeek, M.; Dullemans, A.M.; van Raaij, H.M.; Verhoeven, J.T.J.; van der Vlugt, R.A. Lettuce necrotic leaf curl virus, a new plant virus infecting lettuce and a proposed member of the genus Torradovirus. Arch. Virol. 2014, 159, 801–805. [Google Scholar] [CrossRef]
- Alicai, T.; Ndunguru, J.; Sseruwagi, P.; Tairo, F.; Okao-Okuja, G.; Nanvubya, R.; Kiiza, L.; Kubatko, L.; Kehoe, M.A.; Boykin, L.M. Cassava brown streak virus has a rapidly evolving genome: Implications for virus speciation, variability, diagnosis and host resistance. Sci. Rep. 2016, 6, 36164. [Google Scholar] [CrossRef] [Green Version]
- Ndunguru, J.; Sseruwagi, P.; Tairo, F.; Stomeo, F.; Maina, S.; Djinkeng, A.; Kehoe, M.; Boykin, L.M. Analyses of twelve new whole genome sequences of cassava brown streak viruses and ugandan cassava brown streak viruses from East Africa: Diversity, supercomputing and evidence for further speciation. PLoS ONE 2015, 10, e0139321. [Google Scholar]
- Roossinck, M.J.; Martin, D.P.; Roumagnac, P. Plant virus metagenomics: Advances in virus discovery. Phytopathology 2015, 105, 716–727. [Google Scholar] [CrossRef] [Green Version]
- Adams, I.P.; Glover, R.H.; Monger, W.A.; Mumford, R.; Jackeviciene, E.; Navalinskiene, M.; Samuitiene, M.; Boonham, N. Next-generation sequencing and metagenomic analysis: A universal diagnostic tool in plant virology. Mol. Plant Pathol. 2009, 10, 537–545. [Google Scholar] [CrossRef]
- Candresse, T.; Filloux, D.; Muhire, B.; Julian, C.; Galzi, S.; Fort, G.; Bernardo, P.; Daugrois, J.H.; Fernandez, E.; Martin, D.P.; et al. Appearances can be deceptive: Revealing a hidden viral infection with deep sequencing in a plant quarantine context. PLoS ONE 2014, 9, e102945. [Google Scholar] [CrossRef] [Green Version]
- Kreuze, J.F.; Perez, A.; Untiveros, M.; Quispe, D.; Fuentes, S.; Barker, I.; Simon, R. Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: A generic method for diagnosis, discovery and sequencing of viruses. Virology 2009, 388, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Roossinck, M.J.; Saha, P.; Wiley, G.B.; Quan, J.; White, J.D.; Lai, H.; Chavarria, F.; Shen, G.; Roe, B.A. Ecogenomics: Using massively parallel pyrosequencing to understand virus ecology. Mol. Ecol. 2010, 19, 81–88. [Google Scholar] [CrossRef]
- Filloux, D.; Dallot, S.; Delaunay, A.; Galzi, S.; Jacquot, E.; Roumagnac, P. Metagenomics approaches based on virion-associated nucleic acids (VANA): An innovative tool for assessing without a priori viral diversity of plants. In Plant Pathology; Humana Press: New York, NY, USA, 2015; pp. 249–257. [Google Scholar]
- Al Rwahnih, M.; Daubert, S.; Golino, D.; Rowhani, A. Deep sequencing analysis of RNAs from a grapevine showing Syrah decline symptoms reveals a multiple virus infection that includes a novel virus. Virology 2009, 387, 395–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, R.J.; Wang, J.; Todd, A.K.; Bissielo, A.B.; Yen, S.; Strydom, H.; Moore, N.E.; Ren, X.; Huang, Q.S.; Carter, P.E.; et al. Evaluation of rapid and simple techniques for the enrichment of viruses prior to metagenomic virus discovery. J. Virol. Meth. 2014, 195, 194–204. [Google Scholar] [CrossRef] [Green Version]
- Blouin, A.G.; Ross, H.A.; Hobson-Peters, J.; O’Brien, C.A.; Warren, B.; MacDiarmid, R. A new virus discovered by immunocapture of double-stranded RNA, a rapid method for virus enrichment in metagenomic studies. Mol. Ecol. Res. 2016, 16, 1255–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Q.; Ding, S.W.; Zhang, Y.; Zhu, S. Identification of viruses and viroids by next-generation sequencing and homology-dependent and homology-independent algorithms. Annu. Rev. Phytopathol. 2015, 53, 425–444. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.W.; Lu, R. Virus-derived siRNAs and piRNAs in immunity and pathogenesis. Curr. Opin. Virol. 2011, 1, 533–544. [Google Scholar] [CrossRef] [Green Version]
- Pooggin, M.M. Small RNA-omics for plant virus identification, virome reconstruction and antiviral defense characterization. Front. Microbiol. 2018, 9, 2779. [Google Scholar] [CrossRef]
- Aliyari, R.; Wu, Q.; Li, H.W.; Wang, X.H.; Li, F.; Green, L.D.; Han, C.S.; Li, W.X.; Ding, S.W. Mechanism of induction and suppression of antiviral immunity directed by virus-derived small RNAs in Drosophila. Cell. Host Microbe. 2008, 4, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Cao, M.; Du, P.; Wang, X.; Yu, Y.Q.; Qiu, Y.H.; Li, W.; Gal-On, A.; Zhou, C.; Li, Y.; Ding, S.W. Virus infection triggers widespread silencing of host genes by a distinct class of endogenous siRNAs in Arabidopsis. Proc. Natl. Acad. Sci. USA 2014, 11, 14613–14618. [Google Scholar] [CrossRef] [Green Version]
- Adams, I.P.; Miano, D.W.; Kinyua, Z.M.; Wangai, A.; Kimani, E.; Phiri, N.; Reeder, R.; Harju, V.; Glover, R.; Hany, U.; et al. Use of next-generation sequencing for the identification and characterization of Maize chlorotic mottle virus and Sugarcane mosaic virus causing maize lethal necrosis in Kenya. Plant. Pathol. 2013, 62, 741–749. [Google Scholar] [CrossRef]
- Al Rwahnih, M.; Daubert, S.; Urbez-Torres, J.R.; Cordero, F.; Rowhani, A. Deep sequencing evidence from single grapevine plants reveals a virome dominated by mycoviruses. Arch. Virol. 2011, 156, 397–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coetzee, B.; Freeborough, M.J.; Maree, H.J.; Celton, J.M.; Rees, D.J.G.; Burger, J.T. Deep sequencing analysis of viruses infecting grapevines: Virome of a vineyard. Virology 2010, 400, 157–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quito-Avila, D.F.; Jelkmann, W.; Tzanetakis, I.E.; Keller, K.; Martin, R.R. Complete sequence and genetic characterization of raspberry latent virus, a novel member of the family Reoviridae. Virus Res. 2011, 155, 397–405. [Google Scholar] [CrossRef]
- Samarfard, S.; Bejerman, N.E.; Sharman, M.; Trucco, V.; Giolitti, F.; Dietzgen, R.G. Development and validation of PCR assays for detection of alfalfa dwarf disease-associated viruses in Australian lucerne pastures. Australas. Plant Pathol. 2018, 47, 215–225. [Google Scholar] [CrossRef]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef] [Green Version]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [Green Version]
- Maina, S.; Zheng, L.; Kinoti, W.M.; Aftab, M.; Nancarrow, N.; Trębicki, P.; King, S.; Constable, F.; Rodoni, B. Metagenomic analysis reveals a nearly complete genome sequence of alfalfa mosaic virus from a field pea in Australia. Microbiol. Resour. Announc. 2019, 8, e00766-19. [Google Scholar] [CrossRef] [Green Version]
- Von Bargen, S.; Dieckmann, H.L.; Candresse, T.; Mühlbach, H.P.; Roßbach, J.; Büttner, C. Determination of the complete genome sequence of European mountain ash ringspot-associated emaravirus from Sorbus intermedia reveals two additional genome segments. Arch. Virol. 2019, 164, 1937–1941. [Google Scholar] [CrossRef]
- Darzentas, N. Circoletto: Visualizing sequence similarity with Circos. Bioinformatics 2010, 26, 2620–2621. [Google Scholar] [CrossRef]
- Elbeaino, T.; Digiaro, M.; Mielke-Ehret, N.; Muehlbach, H.-P.; Martelli, G.P.; ICTV Report Consortium. ICTV virus taxonomy profile: Fimoviridae. J. Gen. Virol. 2018, 99, 1478–1479. [Google Scholar] [CrossRef]
- Zhan, B.; Cao, M.; Wang, K.; Wang, X.; Zhou, X. Detection and characterization of Cucumis melo cryptic virus, Cucumis melo amalgavirus 1, and melon necrotic spot virus in Cucumis melo. Viruses 2019, 11, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abudurexiti, A.; Adkins, S.; Alioto, D.; Alkhovsky, S.V.; Avšič-Županc, T.; Ballinger, M.J.; Bente, D.A.; Beer, M.; Bergeron, É.; Blair, C.D.; et al. Taxonomy of the order Bunyavirales: Update 2019. Arch. Virol. 2019, 164, 1949–1965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Bello, P.L.; Ho, T.; Tzanetakis, I.E. The evolution of emaraviruses is becoming more complex: Seven segments identified in the causal agent of rose rosette disease. Virus Res. 2015, 210, 241–244. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; McGavin, W.; Cock, P.J.; Schnettler, E.; Yan, F.; Chen, J.; MacFarlane, S. Newly identified RNAs of raspberry leaf blotch virus encoding a related group of proteins. J. Gen. Virol. 2015, 96, 3432–3439. [Google Scholar] [CrossRef]
- Ishikawa, K.; Maejima, K.; Komatsu, K.; Kitazawa, Y.; Hashimoto, M.; Takata, D.; Yamaji, Y.; Namba, S. Identification and characterization of two novel genomic RNA segments of fig mosaic virus, RNA5 and RNA6. J. Gen. Virol. 2012, 93, 1612–1619. [Google Scholar] [CrossRef]
- Buzkan, N.; Chiumenti, M.; Massart, S.; Sarpkaya, K.; Karadag, S. A new emaravirus discovered in Pistacia from Turkey. Virus Res. 2019, 263, 159–163. [Google Scholar] [CrossRef]
- Wang, X.; Zhai, Z.; Wen, S.; Yang, Z.; Wang, G.; Hong, N. Molecular characterization of a novel emaravirus infecting Actinidia spp. in China. Virus Res. 2020, 275, 197736. [Google Scholar] [CrossRef]
- Weber, F.; Wagner, V.; Rasmussen, S.B.; Hartmann, R.; Paludan, S.R. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 2006, 80, 5059–5064. [Google Scholar] [CrossRef] [Green Version]
- Alves-Freitas, D.M.; Pinheiro-Lima, B.; Faria, J.C.; Lacorte, C.; Ribeiro, S.G.; Melo, F.L. Double-stranded RNA high-throughput sequencing reveals a new cytorhabdovirus in a bean golden mosaic virus-resistant common bean transgenic line. Viruses 2019, 11, 90. [Google Scholar] [CrossRef] [Green Version]
- Teycheney, P.Y.; Geering, A.D. Endogenous viral sequences in plant genomes. In Recent Advances in Plant Virology; Caranta, C., Aranda, M.A., Tepfer, M., Lopez-Moya, J.J., Eds.; Caister Academic Press: Norfolk, UK, 2011; pp. 343–362. [Google Scholar]
- Al Rwahnih, M.A.; Dave, A.; Anderson, M.M.; Rowhani, A.; Uyemoto, J.K.; Sudarshana, M.R. Association of a DNA virus with grapevines affected by red blotch disease in California. Phytopathology 2013, 103, 1069–1076. [Google Scholar] [CrossRef] [Green Version]
- Hadfield, J.; Thomas, J.E.; Schwinghamer, M.W.; Kraberger, S.; Stainton, D.; Dayaram, A.; Parry, J.N.; Pande, D.; Martin, D.P.; Varsani, A. Molecular characterisation of dicot-infecting mastreviruses from Australia. Virus Res. 2012, 166, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Kraberger, S.; Harkins, G.W.; Kumari, S.G.; Thomas, J.E.; Schwinghamer, M.W.; Sharman, M.; Collings, D.A.; Briddon, R.W.; Martin, D.P.; Varsani, A. Evidence that dicot-infecting mastreviruses are particularly prone to inter-species recombination and have likely been circulating in Australia for longer than in Africa and the Middle East. Virology 2013, 444, 282–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roossinck, M.J. Lifestyles of plant viruses. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 1899–1905. [Google Scholar] [CrossRef] [Green Version]
- Roossinck, M.J. Persistent plant viruses: Molecular hitchhikers or epigenetic elements? In Viruses: Essential Agents of Life; Springer: Dordrecht, The Netherlands, 2012; pp. 177–186. [Google Scholar]
- Roossinck, M.J. Plants, viruses and the environment: Ecology and mutualism. Virology 2015, 479, 271–277. [Google Scholar] [CrossRef] [Green Version]
- Peyambari, M.; Warner, S.; Stoler, N.; Rainer, D.; Roossinck, M.J. A 1,000-year-old RNA virus. J. Virol. 2019, 93, e01188-18. [Google Scholar] [CrossRef] [Green Version]
- Safari, M.; Roossinck, M.J. How does the genome structure and lifestyle of a virus affect its population variation? Curr. Opin. Virol. 2014, 9, 39–44. [Google Scholar] [CrossRef]
- Svanella-Dumas, L.; Theil, S.; Barret, M.; Candresse, T. Complete genomic sequence of Raphanus sativus cryptic virus 4 (RsCV4), a novel alphapartitivirus from radish. Arch. Virol. 2018, 163, 1097–1100. [Google Scholar] [CrossRef]
- Lee, J.S.; Goh, C.J.; Park, D.; Hahn, Y. Identification of a novel plant RNA virus species of the genus Amalgavirus in the family Amalgaviridae from chia (Salvia hispanica). Genes Genom. 2019, 41, 507–514. [Google Scholar] [CrossRef]
- Nibert, M.L.; Pyle, J.D.; Firth, A.E. A +1 ribosomal frameshifting motif prevalent among plant amalgaviruses. Virology 2016, 498, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.P.; Zhang, J.X.; Wang, M.L.; Guan, Y.Z.; Qu, G.; Liu, J.Y.; Guo, Y.X.; Yan, X.B. First report of alfalfa leaf curl virus infecting alfalfa (Medicago sativa L.) in China. Plant Dis. Notes 2019. [Google Scholar] [CrossRef]
- Jiang, P.; Shao, J.; Nemchinov, L.G. Identification of emerging viral genomes in transcriptomic datasets of alfalfa (Medicago sativa L.). Virol. J. 2019, 16, 153. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Hobson-Peters, J.; Yam, A.W.Y.; Colmant, A.M.; McLean, B.J.; Prow, N.A.; Watterson, D.; Hall-Mendelin, S.; Warrilow, D.; Ng, M.L.; et al. Viral RNA intermediates as targets for detection and discovery of novel and emerging mosquito-borne viruses. PLoS Negl. Trop. Dis. 2015, 9, e0003629. [Google Scholar] [CrossRef] [Green Version]
- Doyle, J.; Doyle, J.L. Genomic plant DNA preparation from fresh tissue-CTAB method. Phytochem Bull. 1987, 19, 11–15. [Google Scholar]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Reuter, J.S.; Mathews, D.H. RNAstructure: Software for RNA secondary structure prediction and analysis. BMC Bioinform. 2010, 11, 129. [Google Scholar] [CrossRef] [Green Version]
- Garnier, J.; Osguthorpe, D.J.; Robson, B. Analysis of the accuracy and implications of simple methods for predicting the secondary structure of globular proteins. J. Mol. Biol. 1978, 120, 97–120. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample No. | No. of Contigs >200 bp | Total Reads After QC | AMV | BLRV * | ARaV | dsRNA Viruses | Total Viral Reads | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. | % | |||||||||||||
| RNA1 * | RNA2 * | RNA3 * | RNA1 * | RNA3 * | RNA4 * | MsAV1 * | MsAPV RdRP * | MsAPV CP * | ||||||
| 1 | 273 | 85,486 | 12,631 | 2292 | 3693 | 0 | 0 | 0 | 0 | 182 | 203 | 384 | 19,001 | 22.2 |
| 2 | 245 | 92,960 | 10,264 | 1027 | 1874 | 0 | 45 | 132 | 156 | 81 | 5209 | 6318 | 19,076 | 20.5 |
| 3 | 378 | 91,818 | 405 | 74 | 108 | 0 | 0 | 29 | 0 | 138 | 186 | 524 | 2223 | 2.4 |
| 4 | 551 | 74,794 | 296 | 32 | 81 | 0 | 0 | 0 | 0 | 78 | 86 | 287 | 1619 | 2.1 |
| 5 | 158 | 77,372 | 4627 | 359 | 697 | 0 | 0 | 9 | 0 | 730 | 497 | 1095 | 8129 | 10.5 |
| 6 | 306 | 97,815 | 824 | 56 | 93 | 0 | 0 | 0 | 0 | 6073 | 144 | 369 | 7458 | 7.6 |
| 7 | 146 | 76,427 | 1617 | 84 | 165 | 0 | 0 | 0 | 0 | 299 | 253 | 445 | 2959 | 3.8 |
| 8 | 140 | 83,257 | 481 | 65 | 118 | 0 | 0 | 0 | 0 | 108 | 210 | 372 | 1252 | 1.5 |
| 9 | 156 | 91,252 | 2439 | 149 | 488 | 0 | 34 | 33 | 0 | 1495 | 2504 | 2408 | 7983 | 8.7 |
| 10 | 244 | 94,339 | 8705 | 810 | 515 | 146 | 0 | 0 | 0 | 204 | 12,419 | 5495 | 23,866 | 25.2 |
| Sample No | Location | Symptom | Detected RNA Viruses and Detection Methods | ||||
|---|---|---|---|---|---|---|---|
| AMV | BLRV | MsAV1 * | MsAPV1 * | ARaV * | |||
| 1 | Victoria | Yellow mosaic | A, B, C | - | B | B | - |
| 2 | Victoria | Ringspot, yellow mosaic | A, B, C | - | B, C | B, C | B, C |
| 3 | South Australia | Ringspot | B, C | - | B | B | B, C |
| 4 | Victoria | Yellow patches, mild ringspot | B, C | - | B | B | - |
| 5 | Victoria | Ringspot | A, B, C | - | B | B, C | B |
| 6 | South Australia | Ringspot | B, C | - | B, C | B | - |
| 7 | Victoria | Reddening | A, B, C | - | B | B, C | - |
| 8 | Victoria | Leaf rolling, enation | B, C | - | B | B | - |
| 9 | South Australia | Ringspot, yellow patches | B, C | - | B, C | B, C | B, C |
| 10 | South Australia | Leaf curling | A, B, C | A, B, C | B | B, C | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Samarfard, S.; McTaggart, A.R.; Sharman, M.; Bejerman, N.E.; Dietzgen, R.G. Viromes of Ten Alfalfa Plants in Australia Reveal Diverse Known Viruses and a Novel RNA Virus. Pathogens 2020, 9, 214. https://doi.org/10.3390/pathogens9030214
Samarfard S, McTaggart AR, Sharman M, Bejerman NE, Dietzgen RG. Viromes of Ten Alfalfa Plants in Australia Reveal Diverse Known Viruses and a Novel RNA Virus. Pathogens. 2020; 9(3):214. https://doi.org/10.3390/pathogens9030214
Chicago/Turabian StyleSamarfard, Samira, Alistair R. McTaggart, Murray Sharman, Nicolás E. Bejerman, and Ralf G. Dietzgen. 2020. "Viromes of Ten Alfalfa Plants in Australia Reveal Diverse Known Viruses and a Novel RNA Virus" Pathogens 9, no. 3: 214. https://doi.org/10.3390/pathogens9030214