
Genetic Variability of Bovine Leukemia Virus: Evidence of Dual Infection, Recombination and Quasi-Species
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
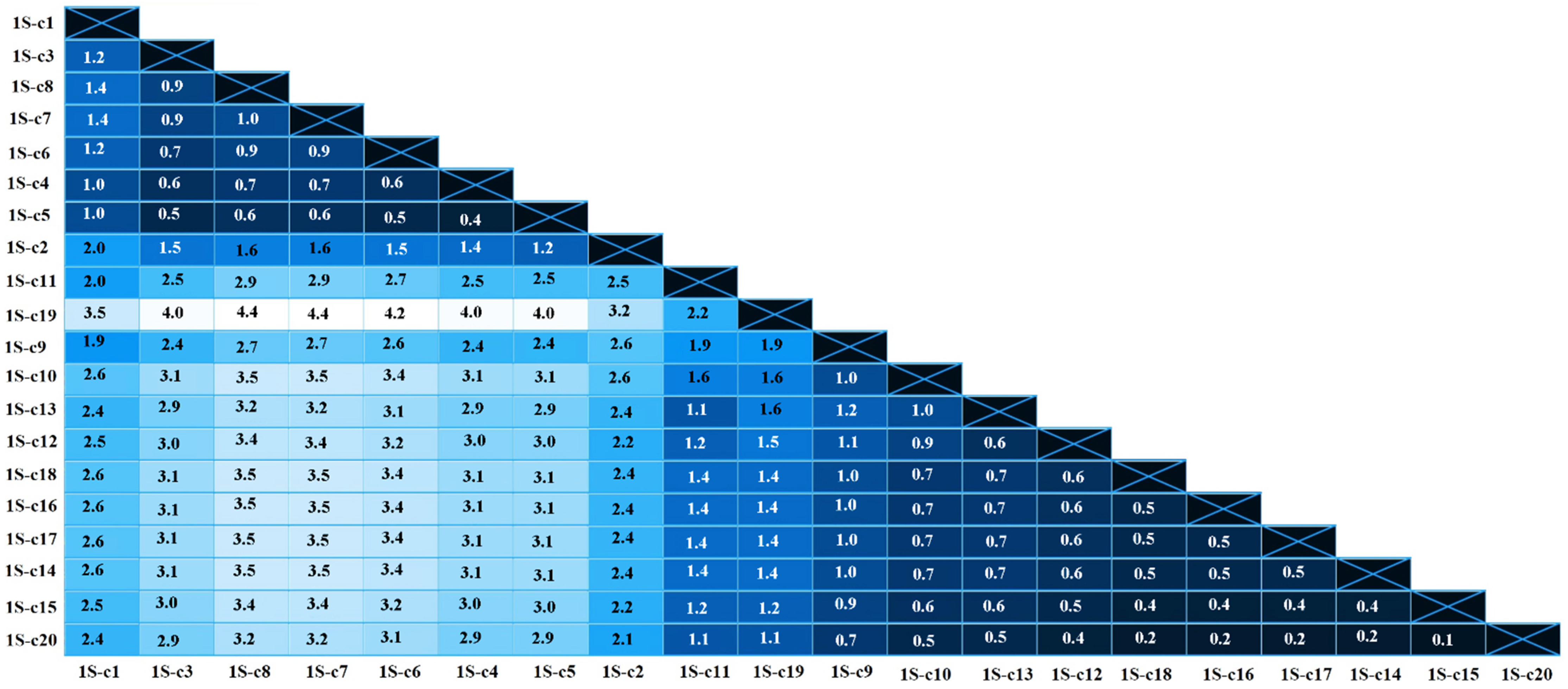
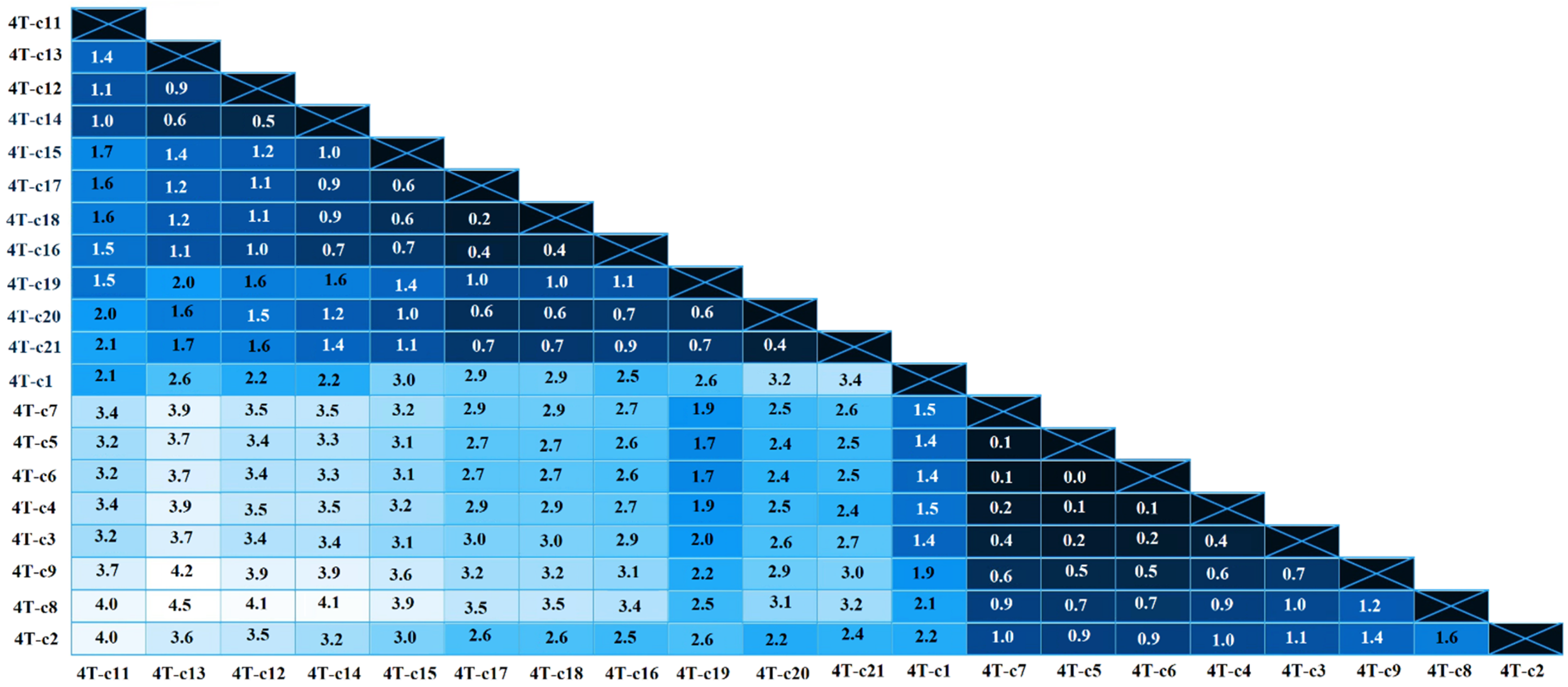
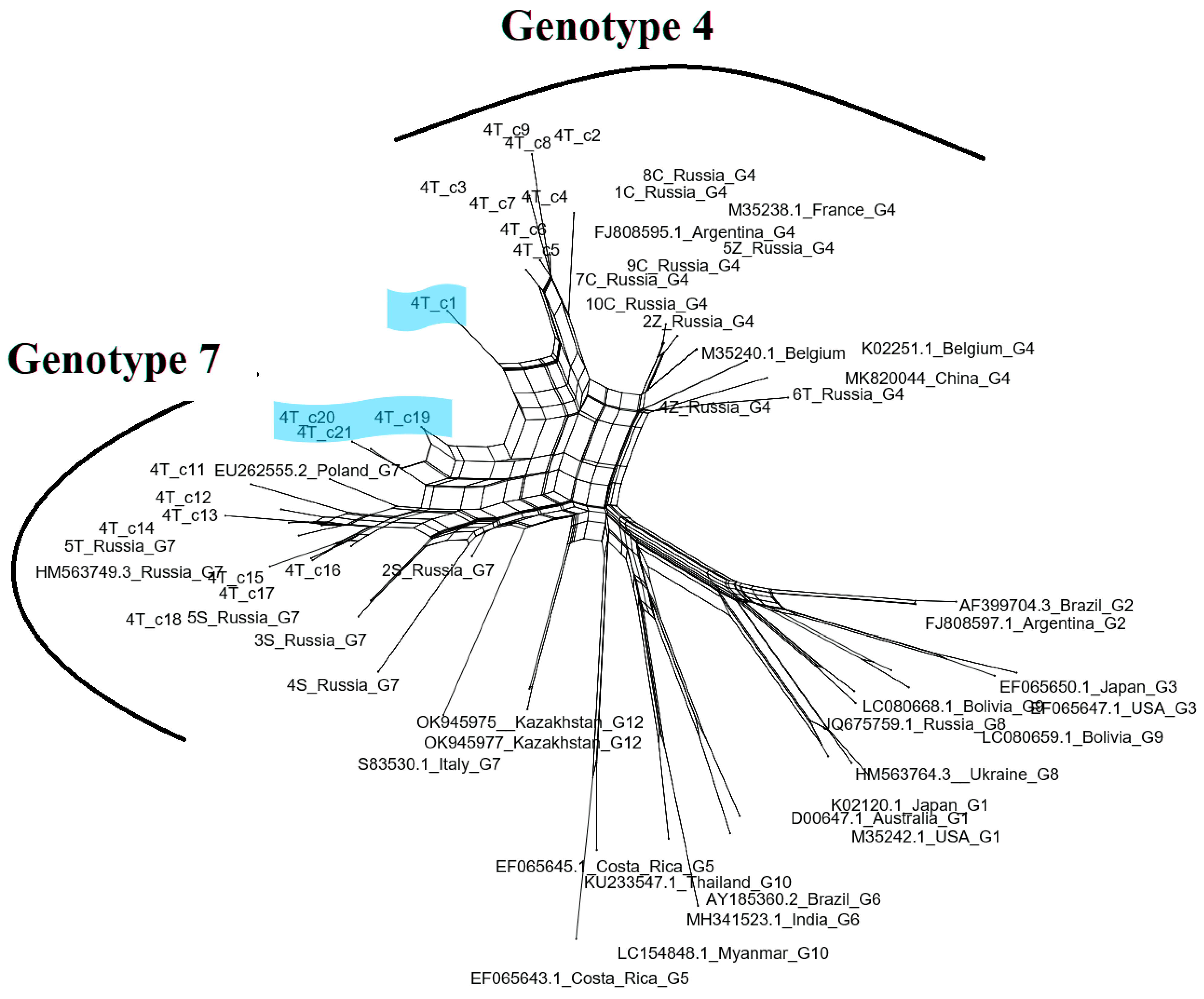
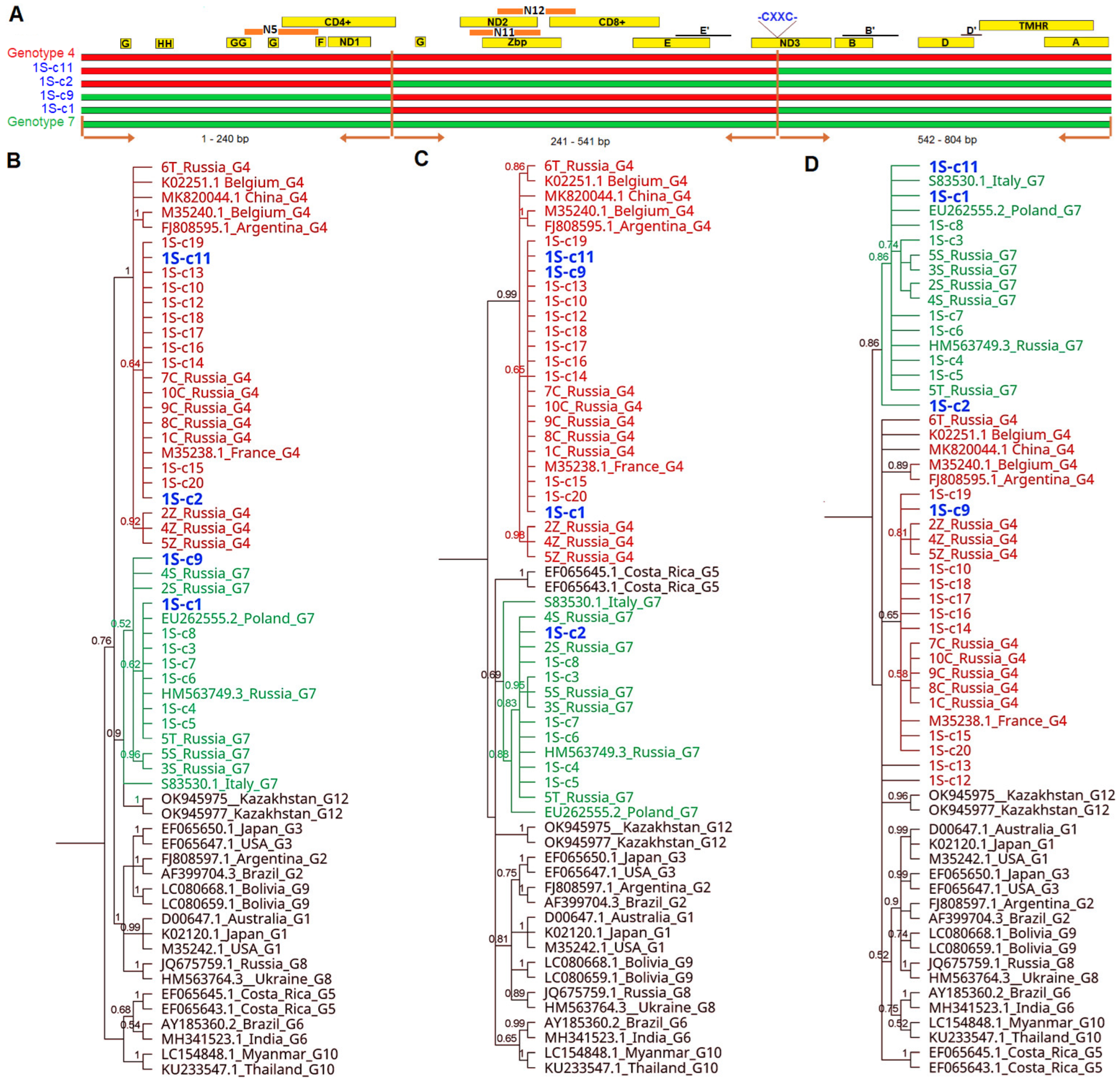
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Domingo, E.; Martin, V.; Perales, C.; Grande-Pérez, A.; García-Arriaza, J.; Arias, A. Viruses as quasispecies: Biological implications. Curr. Top. Microbiol. Immunol. 2006, 299, 51–82. [Google Scholar]
- Mukhopadhyay, S.; Ringe, R.; Patil, A.; Paranjape, R.; Bhattacharya, J. Characterization of circulating HIV type 1 env genes in plasma of two antiretroviral-naive slow progressing patients with brfoad neutralizing antibody response with evidence of recombination. AIDS Res. Hum. Retroviruses 2012, 28, 739–745. [Google Scholar] [CrossRef]
- Charpentier, C.; Nora, T.; Tenaillon, O.; Clavel, F.; Hance, A.J. Extensive recombination among human immunodeficiency virus type 1 quasispecies makes an important contribution to viral diversity in individual patients. J. Virol. 2006, 80, 2472–2482. [Google Scholar] [CrossRef] [PubMed]
- Kim, F.J.; Lavanya, M.; Gessain, A.; Gallego, S.; Battini, J.L.; Sitbon, M.; Courgnaud, V. Intrahost variations in the envelope receptor-binding domain (RBD) of HTLV-1 and STLV-1 primary isolates. Retrovirology 2006, 3, 29. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Gojobori, T. The origin and evolution of human T-cell lymphotropic virus types I and II. Virus Genes 1998, 16, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Costa, J.M.; Segurado, A.C. Molecular evidence of human T-cell lymphotropic virus types 1 and 2 (HTLV-1 and HTLV-2) infections in HTLV seroindeterminate individuals from São Paulo, Brazil. J. Clin. Virol. 2009, 44, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Courgnaud, V.; Van Dooren, S.; Liegeois, F.; Pourrut, X.; Abela, B.; Loul, S.; Mpoudi-Ngole, E.; Vandamme, A.; Delaporte, E.; Peeters, M. Simian T-cell leukemia virus (STLV) infection in wild primate populations in Cameroon: Evidence for dual STLV type 1 and type 3 infection in agile mangabeys (Cercocebus agilis). J. Virol. 2004, 78, 4700–4709. [Google Scholar] [CrossRef]
- Desrames, A.; Cassar, O.; Gout, O.; Hermine, O.; Taylor, G.P.; Afonso, P.V.; Gessain, A. Northern African strains of human T-lymphotropic virus type 1 arose from a recombination event. J. Virol. 2014, 88, 9782–9788. [Google Scholar] [CrossRef]
- Ghysdael, J.; Bruck, C.; Kettmann, R.; Burny, A. Bovine leukemia virus. Curr. Top. Microbiol. Immunol. 1984, 112, 1–19. [Google Scholar]
- Rodríguez, S.M.; Florins, A.; Gillet, N.; de Brogniez, A.; Sánchez-Alcaraz, M.T.; Boxus, M.; Boulanger, F.; Gutiérrez, G.; Trono, K.; Alvarez, I.; et al. Preventive and therapeutic strategies for bovine leukemia virus: Lessons for HTLV. Viruses 2011, 3, 1210–1248. [Google Scholar] [CrossRef]
- Gutiérrez, G.; Alvarez, I.; Politzki, R.; Lomónaco, M.; Dus Santos, M.J.; Rondelli, F.; Fondevila, N.; Trono, K. Natural progression of Bovine Leukemia Virus infection in Argentinean dairy cattle. Vet. Microbiol. 2011, 151, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Ott, S.L.; Johnson, R.; Wells, S.J. Association between bovine-leukosis virus seroprevalence and herd-level productivity on US dairy farms. Prev. Vet. Med. 2003, 61, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Kobayashi, S.; Konishi, M.; Kameyama, K.; Yamamoto, T.; Tsutsui, T. The recent prevalence of bovine leukemia virus (BLV) infection among Japanese cattle. Vet. Microbiol. 2011, 148, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Balić, D.; Lojkić, I.; Periškić, M.; Bedeković, T.; Jungić, A.; Lemo, N.; Roić, B.; Cač, Z.; Barbić, L.; Madić, J. Identification of a new genotype of bovine leukemia virus. Arch. Virol. 2012, 157, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Rola-Luszczak, M.; Pluta, A.; Olech, M.; Donnik, I.; Petropavlovskiy, M.; Gerilovych, A.; Vinogradova, I.; Choudhury, B.; Kuzmak, J. The molecular characterization of bovine leukaemia virus isolates from Eastern Europe and Siberia and its impact on phylogeny. PLoS ONE 2013, 8, e58705. [Google Scholar] [CrossRef] [PubMed]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M.; Kumar, S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. USA 2004, 101, 11030–11035. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Sagata, N.; Yasunaga, T.; Tsuzuku-Kawamura, J.; Ohishi, K.; Ogawa, Y.; Ikawa, Y. Complete nucleotide sequence of the genome of bovine leukemia virus: Its evolutionary relationship to other retroviruses. Proc. Natl. Acad. Sci. USA 1985, 82, 677–681. [Google Scholar] [CrossRef]
- Coulston, J.; Naif, H.; Brandon, R.; Kumar, S.; Khan, S.; Daniel, R.C.; Lavin, M.F. Molecular cloning and sequencing of an Australian isolate of proviral bovine leukaemia virus DNA: Comparison with other isolates. J. Gen. Virol. 1990, 71, 1737–1746. [Google Scholar] [CrossRef]
- Mamoun, R.Z.; Morisson, M.; Rebeyrotte, N.; Busetta, B.; Couez, D.; Kettmann, R.; Hospital, M.; Guillemain, B. Sequence variability of bovine leukemia virus env gene and its relevance to the structure and antigenicity of the glycoproteins. J. Virol. 1990, 64, 4180–4188. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, S.M.; Golemba, M.D.; Campos, R.H.; Trono, K.; Jones, L.R. Bovine leukemia virus can be classified into seven genotypes: Evidence for the existence of two novel clades. J. Gen. Virol. 2009, 90, 2788–2797. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Buehring, G.C. Natural genetic variations in bovine leukemia virus envelope gene: Possible effects of selection and escape. Virology 2007, 366, 150–165. [Google Scholar] [CrossRef] [PubMed]
- Rice, N.R.; Stephens, R.M.; Couez, D.; Deschamps, J.; Kettmann, R.; Burny, A.; Gilden, R.V. The nucleotide sequence of the env gene and post-env region of bovine leukemia virus. Virology 1984, 138, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chen, L.; Dong, M.; Huang, W.; Hao, X.; Peng, Y.; Gong, Z.; Qin, A.; Shang, S.; Yang, Z. Molecular characterization of bovine leukemia virus reveals existence of genotype 4 in Chinese dairy cattle. Virol. J. 2019, 16, 108. [Google Scholar] [CrossRef] [PubMed]
- Gautam, S.; Mishra, N.; Kalaiyarasu, S.; Jhade, S.K.; Sood, R. Molecular Characterization of Bovine Leukaemia Virus (BLV) Strains Reveals Existence of Genotype 6 in Cattle in India with evidence of a new subgenotype. Transbound. Emerg. Dis. 2018, 65, 1968–1978. [Google Scholar] [CrossRef] [PubMed]
- Molteni, E.; Agresti, A.; Meneveri, R.; Marozzi, A.; Malcovati, M.; Bonizzi, L.; Poli, G.; Ginelli, E. Molecular characterization of a variant of proviral bovine leukaemia virus (BLV). Zentralbl Vet. B 1996, 43, 201–211. [Google Scholar] [CrossRef]
- Polat, M.; Takeshima, S.N.; Hosomichi, K.; Kim, J.; Miyasaka, T.; Yamada, K.; Arainga, M.; Murakami, T.; Matsumoto, Y.; de la Barra Diaz, V.; et al. A new genotype of bovine leukemia virus in South America identified by NGS-based whole genome sequencing and molecular evolutionary genetic analysis. Retrovirology 2016, 13, 4. [Google Scholar] [CrossRef]
- Lee, E.; Kim, E.J.; Ratthanophart, J.; Vitoonpong, R.; Kim, B.H.; Cho, I.S.; Song, J.Y.; Lee, K.K.; Shin, Y.K. Molecular epidemiological and serological studies of bovine leukemia virus (BLV) infection in Thailand cattle. Infect. Genet. Evol. 2016, 41, 245–254. [Google Scholar] [CrossRef]
- Moe, K.K.; Polat, M.; Borjigin, L.; Matsuura, R.; Hein, S.T.; Moe, H.H.; Aida, Y. New evidence of bovine leukemia virus circulating in Myanmar cattle through epidemiological and molecular characterization. PLoS ONE 2020, 15, e0229126. [Google Scholar] [CrossRef]
- Sultanov, A.; Rola-Luszczak, M.; Mamanova, S.; Rylo, A.; Osinski, Z.; Saduakassova, M.A.; Bashenova, E.; Kuzmak, J. Molecular Characterization of Bovine Leukemia Virus with the Evidence of a New Genotype Circulating in Cattle from Kazakhstan. Pathogens 2022, 11, 180. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Bryant, D. Application of Phylogenetic Networks in Evolutionary Studies. Mol. Biol. Evol. 2005, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Boussau, B.; Guéguen, L.; Gouy, M. A mixture model and a hidden markov model to simultaneously detect recombination breakpoints and reconstruct phylogenies. Evol. Bioinform. Online 2009, 5, 67–79. [Google Scholar] [CrossRef] [PubMed]
- van der Kuyl, A.C.; Cornelissen, M. Identifying HIV-1 dual infections. Retrovirology 2007, 4, 67. [Google Scholar] [CrossRef] [PubMed]
- Asfaw, Y.; Tsuduku, S.; Konishi, M.; Murakami, K.; Tsuboi, T.; Wu, D.; Sentsui, H. Distribution and superinfection of bovine leukemia virus genotypes in Japan. Arch. Virol. 2005, 150, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Camargos, M.F.; Rajão, D.S.; Leite, R.C.; Stancek, D.; Heinemann, M.B.; Reis, J.K. Genetic variation of bovine leukemia virus (BLV) after replication in cell culture and experimental animals. Genet. Mol. Res. 2014, 13, 1717–1723. [Google Scholar] [CrossRef] [PubMed]
- Kuczewski, A.; Orsel, K.; Barkema, H.W.; Mason, S.; Erskine, R.; van der Meer, F. Invited review: Bovine leukemia virus—Transmission, control, and eradication. J. Dairy Sci. 2021, 104, 6358–6375. [Google Scholar] [CrossRef]
- Suárez Archilla, G.; Gutiérrez, G.; Camussone, C.; Calvinho, L.; Abdala, A.; Alvarez, I.; Petersen, M.; Franco, L.; Destefano, G.; Monti, G.; et al. A safe and effective vaccine against bovine leukemia virus. Front. Immunol. 2022, 13, 980514. [Google Scholar] [CrossRef]
- Krutko, K.S.; Kinareikina, A.G.; Serkova, M.I.; Silivanova, E.A.; Fedorova, O.A. Detection of genetic material of causative agents of animal viral diseases in blood-sucking dipterans from the Tyumen Region. Russ. J. Parasitol. 2023, 16, 389–402. [Google Scholar] [CrossRef]
- Sprygin, A.V.; Fedorova, O.A.; Babin, Y.Y.; Kononov, A.V.; Karaulov, A.K. Blood-sucking midges from the genus Culicoides (Diptera: Ceratopogonidae) act as field vectors of human and animal diseases. Agric. Biol. 2015, 50, 183–197. [Google Scholar] [CrossRef]
- Dogan, F.; Bilge Dagalp, S.; Dik, B.; Farzani, T.A.; Alkan, F. Detection of genotype 1 bovine leukemia virus from a C. schultzei pool: Do Culicoides spp. have a role on the transmission of bovine leukemia virus? Infect. Genet. Evol. 2020, 85, 104469. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, G.; Rodríguez, S.M.; de Brogniez, A.; Gillet, N.; Golime, R.; Burny, A.; Jaworski, J.P.; Alvarez, I.; Vagnoni, L.; Trono, K.; et al. Vaccination against δ-retroviruses: The bovine leukemia virus paradigm. Viruses 2014, 6, 2416–2427. [Google Scholar] [CrossRef] [PubMed]
- Pomier, C.; Alcaraz, M.T.; Debacq, C.; Lancon, A.; Kerkhofs, P.; Willems, L.; Wattel, E.; Mortreux, F. Early and transient reverse transcription during primary deltaretroviral infection of sheep. Retrovirology 2008, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Inoue, E.; Mori, H.; Osawa, Y.; Okazaki, K. Delayed-onset enzootic bovine leukosis possibly caused by superinfection with bovine leukemia virus mutated in the pol gene. Arch. Virol. 2015, 160, 2087–2091. [Google Scholar] [CrossRef] [PubMed]
- Gillet, N.A.; Gutiérrez, G.; Rodriguez, S.M.; de Brogniez, A.; Renotte, N.; Alvarez, I.; Trono, K.; Willems, L. Massive Depletion of Bovine Leukemia Virus Proviral Clones Located in Genomic Transcriptionally Active Sites during Primary Infection. PLOS Pathog. 2013, 9, e1003687. [Google Scholar] [CrossRef] [PubMed]
- Gillet, N.; Florins, A.; Boxus, M.; Burteau, C.; Nigro, A.; Vandermeers, F.; Balon, H.; Bouzar, A.-B.; Defoiche, J.; Burny, A.; et al. Mechanisms of leukemogenesis induced by bovine leukemia virus: Prospects for novel anti-retroviral therapies in human. Retrovirology 2007, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Pluta, A.; Albritton, L.M.; Rola-Łuszczak, M.; Kuźmak, J. Computational analysis of envelope glycoproteins from diverse geographical isolates of bovine leukemia virus identifies highly conserved peptide motifs. Retrovirology 2018, 15, 2. [Google Scholar] [CrossRef]
- Fechner, H.; Blankenstein, P.; Looman, A.C.; Elwert, J.; Geue, L.; Albrecht, C.; Kurg, A.; Beier, D.; Marquardt, O.; Ebner, D. Provirus variants of the bovine leukemia virus and their relation to the serological status of naturally infected cattle. Virology 1997, 237, 261–269. [Google Scholar] [CrossRef]
- Bai, L.; Takeshima, S.-n.; Isogai, E.; Kohara, J.; Aida, Y. Novel CD8+ cytotoxic T cell epitopes in bovine leukemia virus with cattle. Vaccine 2015, 33, 7194–7202. [Google Scholar] [CrossRef]
- Elemans, M.; Florins, A.; Willems, L.; Asquith, B. Rates of CTL killing in persistent viral infection in vivo. PLoS Comput. Biol. 2014, 10, e1003534. [Google Scholar] [CrossRef]
- Gutiérrez, G.; Alvarez, I.; Merlini, R.; Rondelli, F.; Trono, K. Dynamics of perinatal bovine leukemia virus infection. BMC Vet. Res. 2014, 10, 82. [Google Scholar] [CrossRef]
- Yu, C.; Wang, X.; Zhou, Y.; Wang, Y.; Zhang, X.; Zheng, Y. Genotyping bovine leukemia virus in dairy cattle of Heilongjiang, northeastern China. BMC Vet. Res. 2019, 15, 179. [Google Scholar] [CrossRef] [PubMed]
- Ochirkhuu, N.; Konnai, S.; Odbileg, R.; Nishimori, A.; Okagawa, T.; Murata, S.; Ohashi, K. Detection of bovine leukemia virus and identification of its genotype in Mongolian cattle. Arch. Virol. 2016, 161, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Kim, E.-J.; Joung, H.-K.; Kim, B.-H.; Song, J.-Y.; Cho, I.-S.; Lee, K.-K.; Shin, Y.-K. Sequencing and phylogenetic analysis of the gp51 gene from Korean bovine leukemia virus isolates. Virol. J. 2015, 12, 64. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, K.; Inoue, E.; Osawa, Y.; Okazaki, K. Molecular epidemiology of bovine leukemia virus associated with enzootic bovine leukosis in Japan. Virus Res. 2011, 155, 343–348. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No | GenBank Accession No | Geographic Region | Genotype | Identity Code & Source |
|---|---|---|---|---|
| 1 | K02120 | Japan | 1 | Sagata et al. (1985) [19] |
| 2 | D00647 | Australia | 1 | Coulston et al. (1990) [20] |
| 3 | M35242 | USA | 1 | Mamoun et al. (1990) [21] |
| 4 | FJ808597.1 | Argentina | 2 | Rodriguez et al. (2009) [22] |
| 5 | AF399704.3 | Brazil | 2 | Camargos et al. (2004) ‡ |
| 6 | EF065650.1 | Japan | 3 | Zhao et al. (2007) [23] |
| 7 | EF065647.1 | USA | 3 | Zhao et al. (2007) [23] |
| 8 | K02251.1 | Belgium | 4 | Rice et al. (1984) [24] |
| 9 | M35238 | France | 4 | Mamoun et al. (1990) [21] |
| 10 | MK820044 | China | 4 | Yang et al. (2019) [25] |
| 11 | M35240.1 | Belgium | 4 | Mamoun et al. (1990) [21] |
| 12 | FJ808595 | Argentina | 4 | Rodriguez et al. (2009) [22] |
| 13 | EF065645.1 | Costa Rica | 5 | Zhao et al. (2007) [23] |
| 14 | EF065643.1 | Costa Rica | 5 | Zhao et al. (2007) [23] |
| 15 | AY185360.2 | Brazil | 6 | Camargos et al. (2004) ‡ |
| 16 | MH341523 | India | 6 | Gautam et al. (2018) [26] |
| 17 | S83530.1 | Italy | 7 | Molteni et al. (1996) [27] |
| 18 | HM563749.3 | Russia | 7 | Rola-Łuszczak (2013) [15] |
| 19 | EU262555 | Poland | 7 | Rola-Łuszczak (2013) [15] |
| 20 | JQ675759.1 | Russia | 8 | Lomakina et al. (2013) § |
| 21 | HM563764 | Ukraine | 8 | Rola-Łuszczak (2013) [15] |
| 22 | LC080668 | Bolivia | 9 | Polat et al. (2016) [28] |
| 23 | LC080659 | Bolivia | 9 | Polat et al. (2016) [28] |
| 24 | KU233547 | Thailand | 10 | Lee et al. (2016) [29] |
| 25 | LC154848 | Myanmar | 10 | Moe et al. (2020) [30] |
| 26 | OK945975 | Kazakhstan | 12 | Sultanov et al. (2022) [31] |
| 27 | OK945977 | Kazakhstan | 12 | Sultanov et al. (2022) [31] |
| No | GenBank Accession No | Genotype | Identity Code of Samples |
|---|---|---|---|
| 1 | JQ353633 | 7 | 1S-c6 |
| 2 | JQ353634 | 7 | 1S-c7 |
| 3 | JQ353635 | 7 | 1S-c5 |
| 4 | JQ353636 | 7 | 1S-c8 |
| 5 | JQ353637 | 4 | 1S-c12 |
| 6 | JQ353638 | recombinant 4/7 | 1S-c2 |
| 7 | JQ353639 | 4 | 1S-c19 |
| 8 | JQ353640 | recombinant 4/7 | 1S-c9 |
| 9 | JQ353641 | 4 | 1S-c15 |
| 10 | JQ353642 | 4 | 1S-c14 |
| 11 | JQ353643 | 4 | 1S-c13 |
| 12 | JQ353644 | 4 | 1S-c17 |
| 13 | JQ353645 | 7 | 1S-c3 |
| 14 | JQ353646 | recombinant 4/7 | 1S-c11 |
| 15 | JQ353647 | 4 | 1S-c20 |
| 16 | JQ353648 | 4 | 1S-c18 |
| 17 | JQ353649 | recombinant 4/7 | 1S-c1 |
| 18 | JQ353650 | 4 | 1S-c10 |
| 19 | JQ353651 | 7 | 1S-c4 |
| 20 | JQ353652 | 4 | 1S-c16 |
| 21 | JQ353653 | 7 | 4T-c15 |
| 22 | JQ353654 | 7 | 4T-c13 |
| 23 | JQ353655 | recombinant 4/7 | 4T-c19 |
| 24 | JQ353656 | 7 | 4T-c11 |
| 25 | JQ353657 | 4 | 4T-c7 |
| 26 | JQ353658 | recombinant 4/7 | 4T-c1 |
| 27 | JQ353659 | 4 | 4T-c6 |
| 28 | JQ353660 | 4 | 4T-c3 |
| 29 | JQ353661 | recombinant 4/7 | 4T-c20 |
| 30 | JQ353662 | 7 | 4T-c12 |
| 31 | JQ353663 | recombinant 4/7 | 4T-c21 |
| 32 | JQ353664 | 7 | 4T-c17 |
| 33 | JQ353665 | 7 | 4T-c16 |
| 34 | JQ353666 | 4 | 4T-c2 |
| 35 | JQ353667 | 7 | 4T-c18 |
| 36 | JQ353668 | 4 | 4T-c5 |
| 37 | JQ353669 | 4 | 4T-c4 |
| 38 | JQ353670 | 4 | 4T-c8 |
| 39 | JQ353671 | 7 | 4T-c14 |
| 40 | JQ353672 | 4 | 4T-c9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pluta, A.; Rola-Łuszczak, M.; Hoffmann, F.G.; Donnik, I.; Petropavlovskiy, M.; Kuźmak, J. Genetic Variability of Bovine Leukemia Virus: Evidence of Dual Infection, Recombination and Quasi-Species. Pathogens 2024, 13, 178. https://doi.org/10.3390/pathogens13020178
Pluta A, Rola-Łuszczak M, Hoffmann FG, Donnik I, Petropavlovskiy M, Kuźmak J. Genetic Variability of Bovine Leukemia Virus: Evidence of Dual Infection, Recombination and Quasi-Species. Pathogens. 2024; 13(2):178. https://doi.org/10.3390/pathogens13020178
Chicago/Turabian StylePluta, Aneta, Marzena Rola-Łuszczak, Federico G. Hoffmann, Irina Donnik, Maxim Petropavlovskiy, and Jacek Kuźmak. 2024. "Genetic Variability of Bovine Leukemia Virus: Evidence of Dual Infection, Recombination and Quasi-Species" Pathogens 13, no. 2: 178. https://doi.org/10.3390/pathogens13020178