



Rift Valley Fever Virus Primes Immune Responses in Aedes aegypti Cells

 ,
,
Abstract
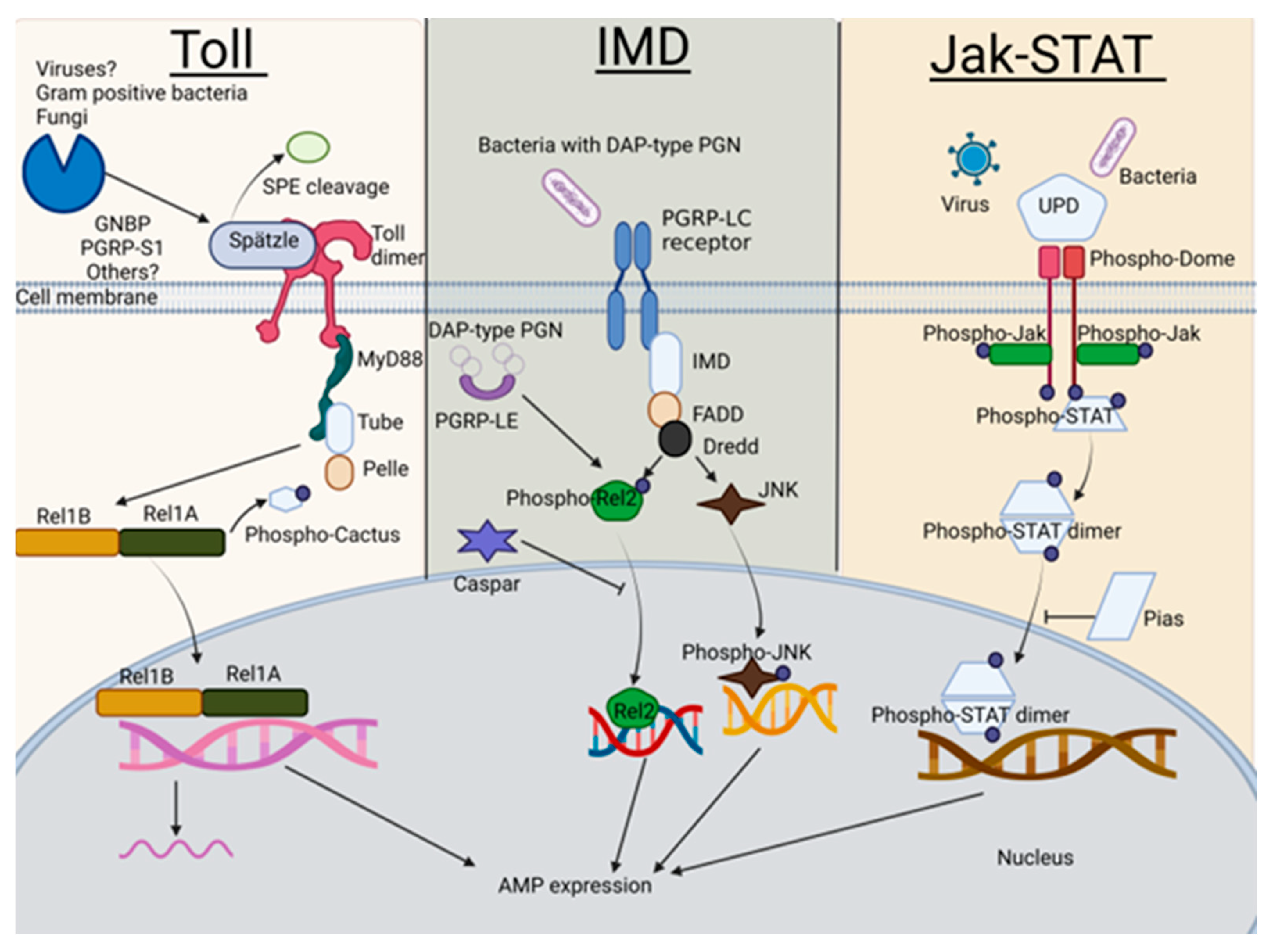
:1. Introduction

2. Materials and Methods
2.1. Cells and Viruses
2.2. Bacterial Immune Stimulation and Virus Infections
- (i)
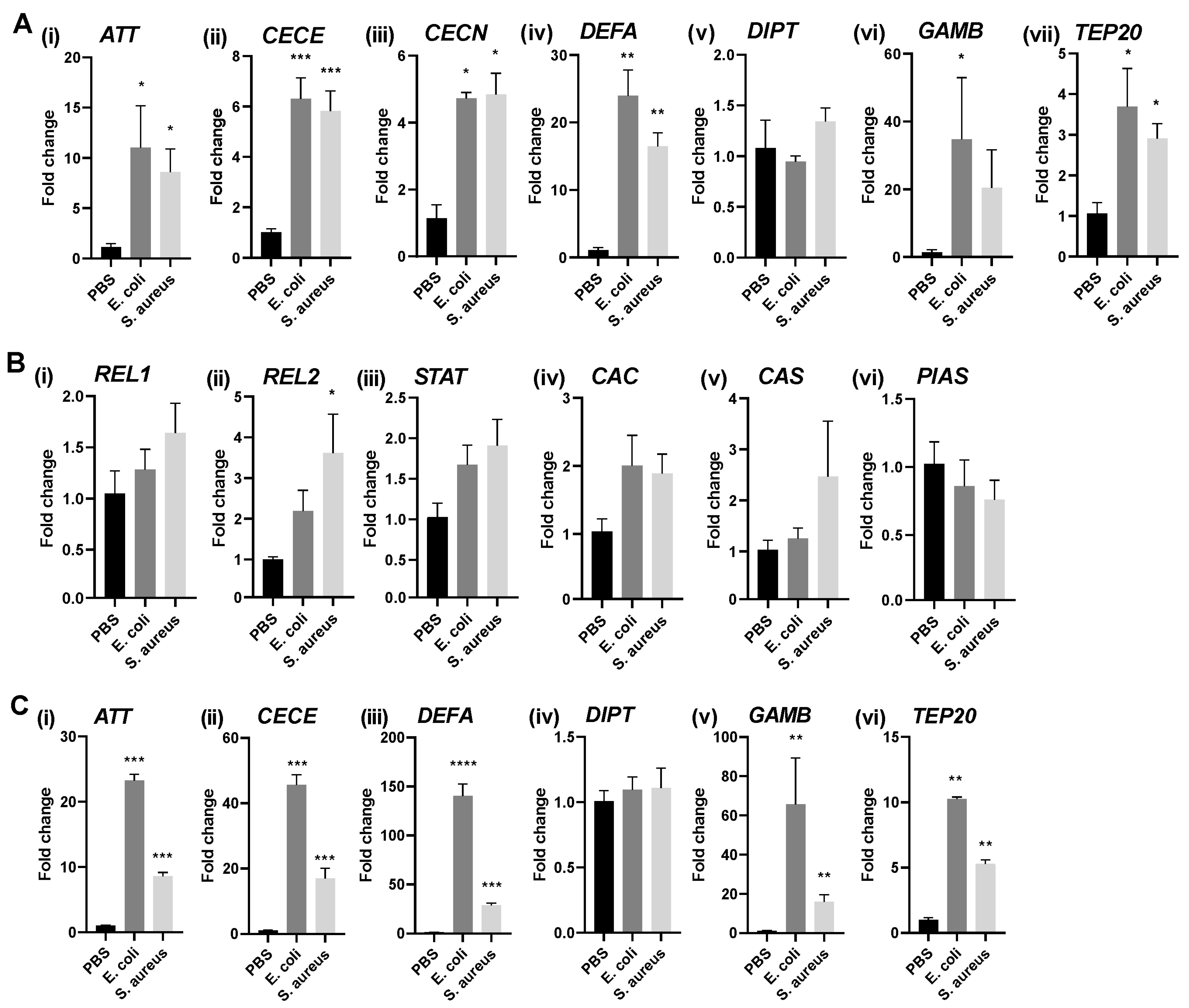
- To induce immune signaling using heat-inactivated bacteria, Aag2 cells were seeded at a density of 3 × 105 cells per well in 24-well plates and left to adhere overnight. Cultures of Escherichia coli (strain JM109; Promega, Chilworth, UK) and Staphylococcus aureus (ATCC) were grown in 5 mL LB broth without antibiotics and incubated at 37 °C for 16 h. Serial dilutions of the cultures were prepared, and cell forming units per mL were determined on LB agar plates following an overnight incubation at 37 °C. The remainder of the cultures were centrifuged at 1174× g for 20 min at 4 °C. The bacterial pellets were washed twice in phosphate-buffered saline (PBS), resuspended in 500 µL PBS, and heat-inactivated for 10 min at 80 °C. Aag2 cells were stimulated with heat-inactivated E. coli and S. aureus [multiplicity of infection (MOI) of 300 CFU/cell] for 16 h at 28 °C. PBS was used as control. Cells were washed with PBS and lysed in TRIzol Reagent (Life Technologies, Paisley, UK) according to the manufacturer’s instructions;
- (ii)
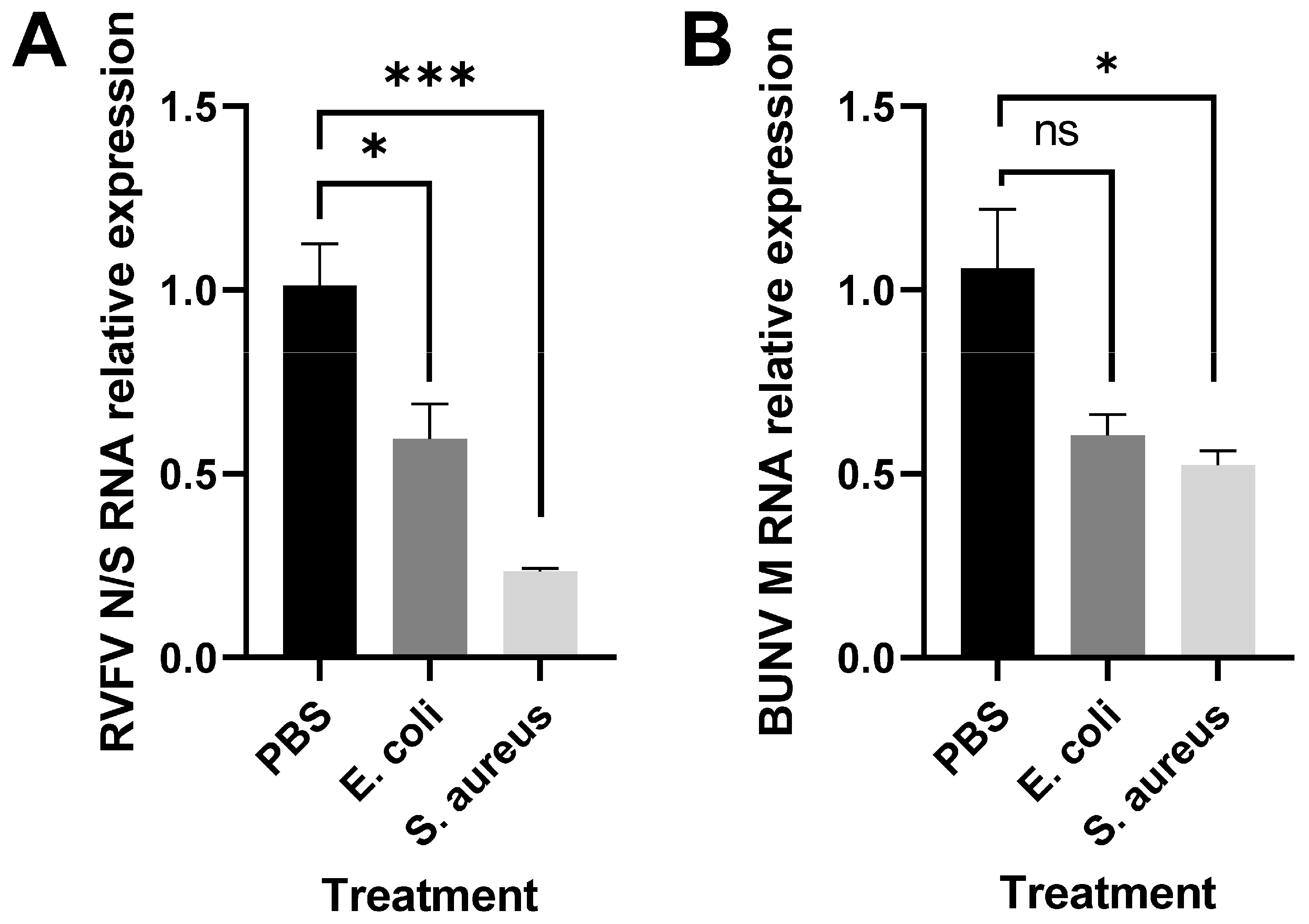
- To assess the impact of bacterial immune stimulation on viral replication, Aag2 cells were treated with heat-killed bacteria for 16 h. Cells were then infected with RVFV rMP-12 or rBUNV at MOI 0.1 or left uninfected for 24 h, and cells lysed in TRIzol Reagent;
- (iii)
- To quantify the effect of viral infection on immune gene expression and RNA levels of insect-specific viruses, cells were mock-infected or infected with RVFV rMP-12, RVFV rMP-12:delNSm, RVFV rMP-12:delNSs or rBUNV at MOI 1 for 24 h. Cells were lysed in TRIzol Reagent;
- (iv)
- Lastly, to determine if virus infection alters immune gene expression in response to bacterial stimulation of Aag2 cells, cells were mock-infected or infected with RVFV rMP-12 or rBUNV at MOI 1 for 24 h, followed by bacterial or PBS stimulation for 16 h. Cells were lysed in TRIzol Reagent.
2.3. Quantification of Gene Expression
2.4. Plaque Assays
2.5. Pathway Sensors and Luciferase Assays
2.6. Caspase Assays
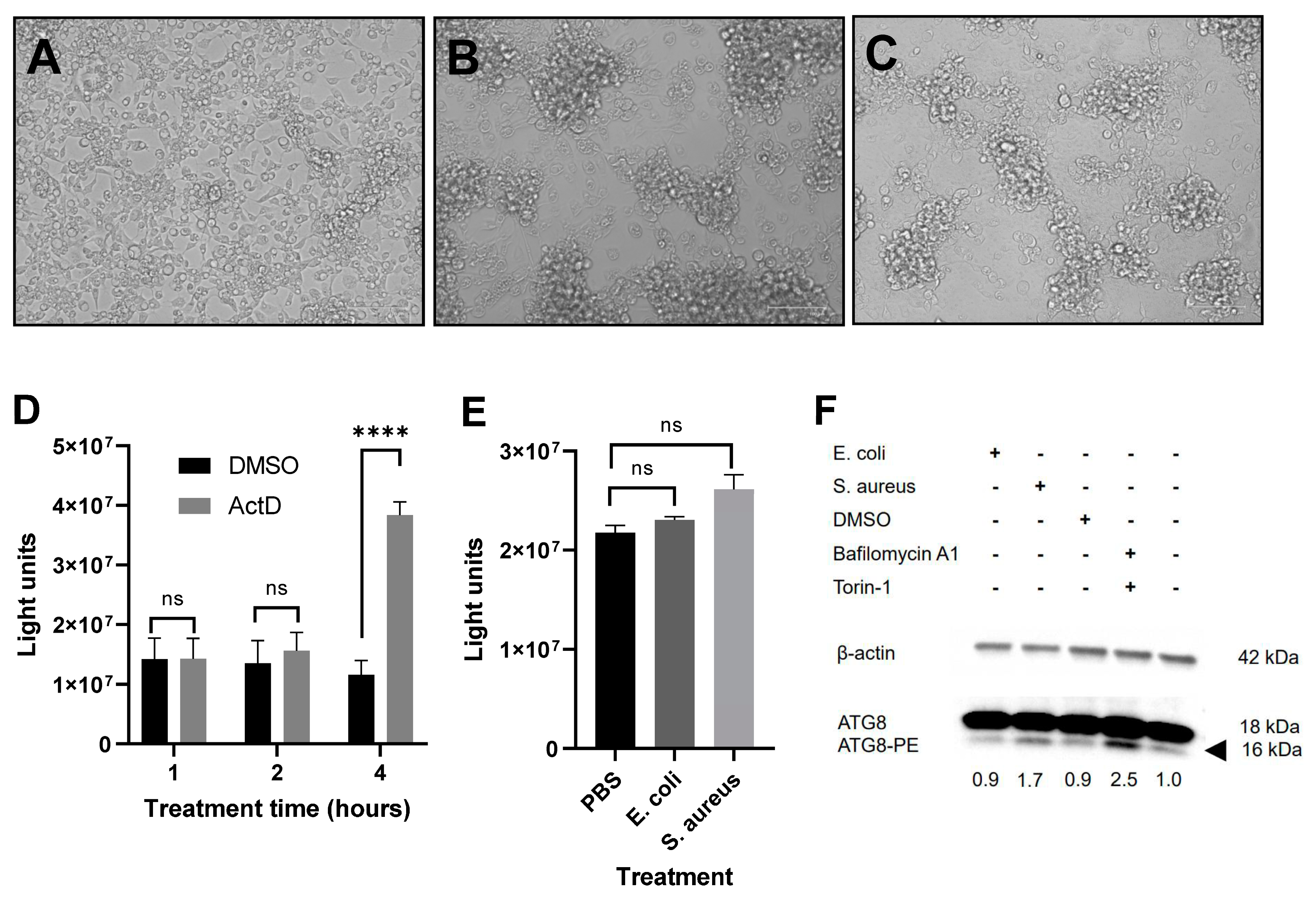
2.7. Induction of Autophagy and Immunoblotting
2.8. Statistical Analyses
3. Results
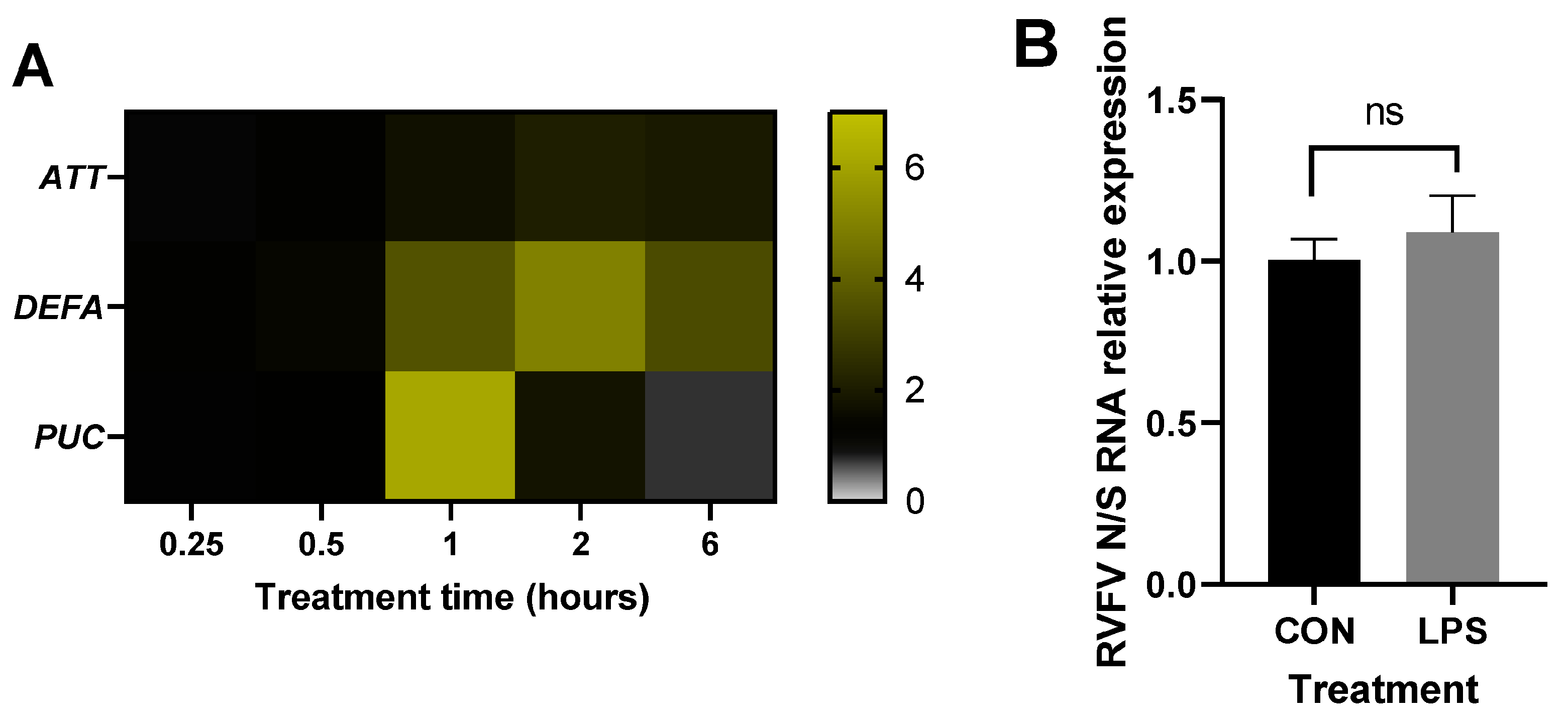
3.1. Immune Stimulation of Different Aag2 Cell Isolates Induces AMP Transcription to Varying Degrees
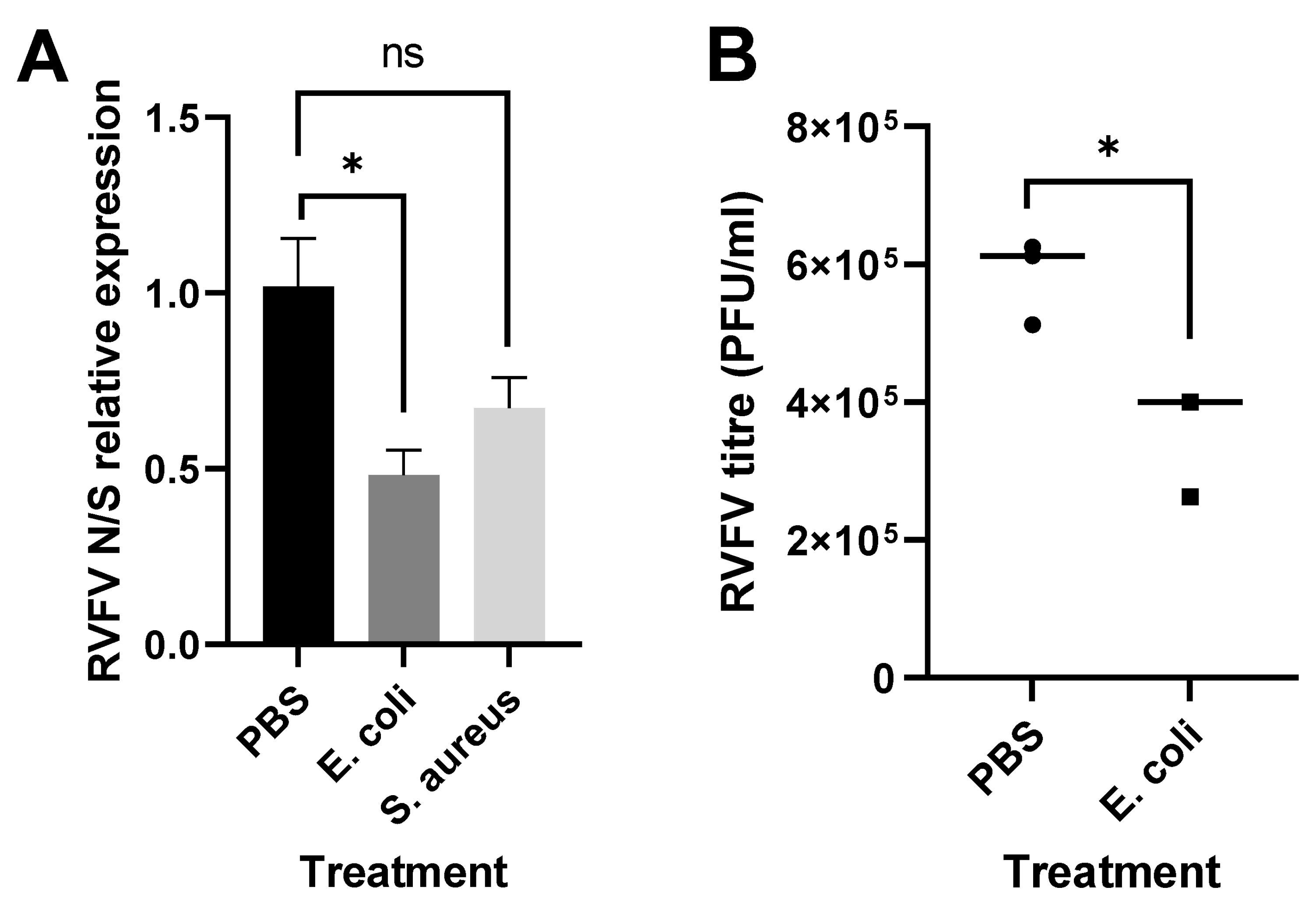
3.2. Immune Stimulation Reduces RVFV Replication
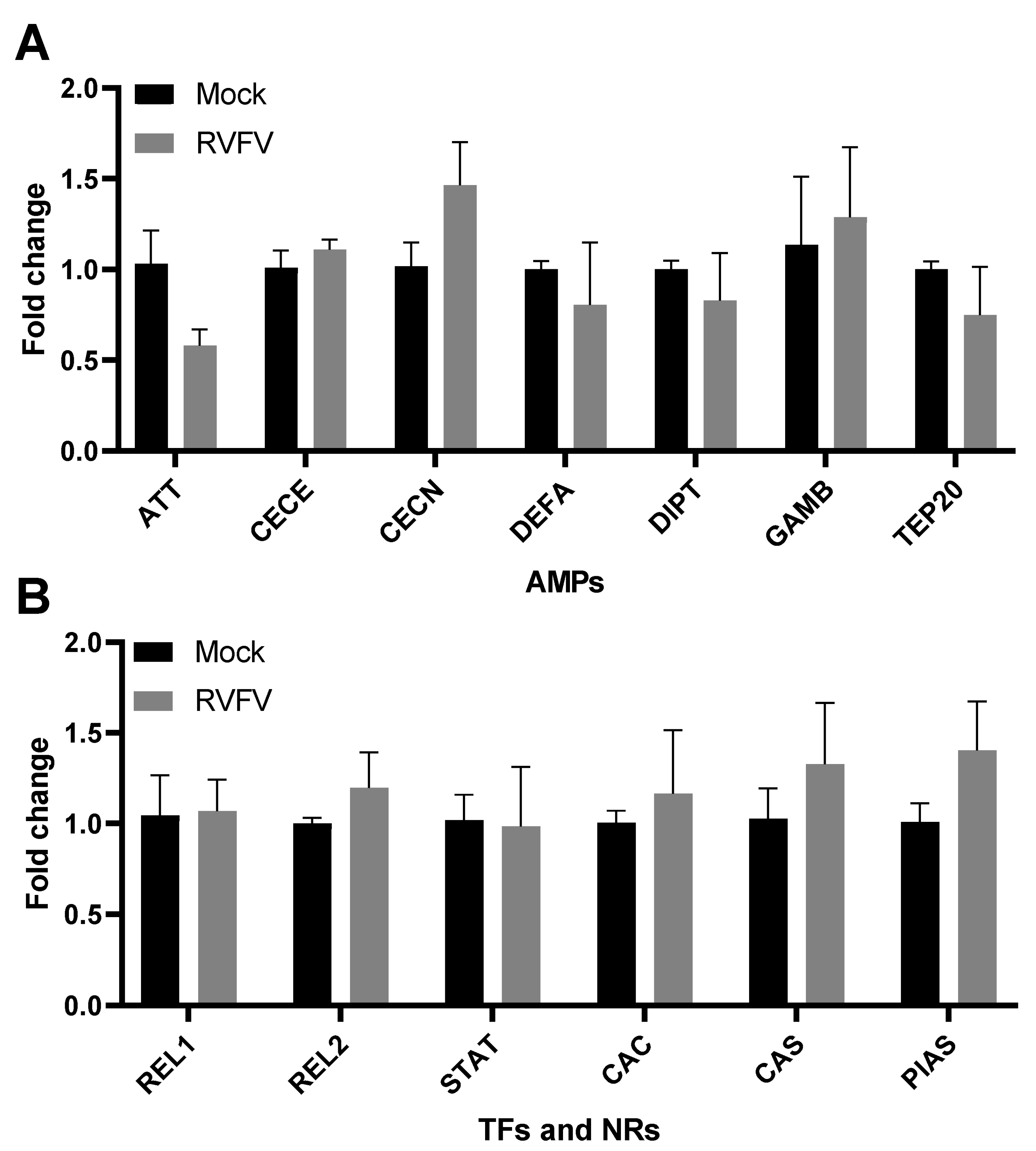
3.3. RVFV Infection Does Not Elicit an Immune Response in Infected Aag2 Cells
3.4. RVFV Infection of Aag2 Cells Primes Immune Responses to Bacteria
3.5. RVFV Infection Regulates Expression of Selected Pattern Recognition Receptors
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kingsolver, M.B.; Hardy, R.W. Making connections in insect innate immunity. Proc. Natl. Acad. Sci. USA 2012, 109, 18639–18640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merkling, S.H.; van Rij, R.P. Beyond RNAi: Antiviral defense strategies in Drosophila and mosquito. J. Insect Physiol. 2013, 59, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Tikhe, C.V.; Dimopoulos, G. Mosquito antiviral immune pathways. Dev. Comp. Immunol. 2021, 116, 103964. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, I.; Jansen, S.; Fall, G.; Lorenzen, S.; Rudolf, M.; Huber, K.; Heitmann, A.; Schicht, S.; Ndiaye, E.H.; Watson, M.; et al. RNA Interference Restricts Rift Valley Fever Virus in Multiple Insect Systems. mSphere 2017, 2, e00090-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, I.; Shi, X.; McFarlane, M.; Watson, M.; Blomstrom, A.L.; Skelton, J.K.; Kohl, A.; Elliott, R.M.; Schnettler, E. The Antiviral RNAi Response in Vector and Non-vector Cells against Orthobunyaviruses. PLoS. Negl. Trop. Dis. 2017, 11, e0005272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, S.W.; Bian, G.; Raikhel, A.S. A toll receptor and a cytokine, Toll5A and Spz1C, are involved in toll antifungal immune signaling in the mosquito Aedes aegypti. J. Biol. Chem. 2006, 281, 39388–39395. [Google Scholar] [CrossRef] [Green Version]
- Sim, S.; Dimopoulos, G. Dengue virus inhibits immune responses in Aedes aegypti cells. PLoS ONE 2010, 5, e10678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, Z.; Ramirez, J.L.; Dimopoulos, G. The Aedes aegypti toll pathway controls dengue virus infection. PLoS Pathog. 2008, 4, e1000098. [Google Scholar] [CrossRef]
- Russell, T.A.; Ayaz, A.; Davidson, A.D.; Fernandez-Sesma, A.; Maringer, K. Imd pathway-specific immune assays reveal NF-kappaB stimulation by viral RNA PAMPs in Aedes aegypti Aag2 cells. PLoS Negl. Trop. Dis. 2021, 15, e0008524. [Google Scholar] [CrossRef]
- Barletta, A.B.; Nascimento-Silva, M.C.; Talyuli, O.A.; Oliveira, J.H.; Pereira, L.O.; Oliveira, P.L.; Sorgine, M.H. Microbiota activates IMD pathway and limits Sindbis infection in Aedes aegypti. Parasites Vectors 2017, 10, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carissimo, G.; Pondeville, E.; McFarlane, M.; Dietrich, I.; Mitri, C.; Bischoff, E.; Antoniewski, C.; Bourgouin, C.; Failloux, A.B.; Kohl, A.; et al. Antiviral immunity of Anopheles gambiae is highly compartmentalized, with distinct roles for RNA interference and gut microbiota. Proc. Natl. Acad. Sci. USA 2015, 112, E176–E185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behura, S.K.; Gomez-Machorro, C.; Harker, B.W.; deBruyn, B.; Lovin, D.D.; Hemme, R.R.; Mori, A.; Romero-Severson, J.; Severson, D.W. Global cross-talk of genes of the mosquito Aedes aegypti in response to dengue virus infection. PLoS Negl. Trop. Dis. 2011, 5, e1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colpitts, T.M.; Cox, J.; Vanlandingham, D.L.; Feitosa, F.M.; Cheng, G.; Kurscheid, S.; Wang, P.; Krishnan, M.N.; Higgs, S.; Fikrig, E. Alterations in the Aedes aegypti transcriptome during infection with West Nile, dengue and yellow fever viruses. PLoS Pathog. 2011, 7, e1002189. [Google Scholar] [CrossRef] [Green Version]
- Souza-Neto, J.A.; Sim, S.; Dimopoulos, G. An evolutionary conserved function of the JAK-STAT pathway in anti-dengue defense. Proc. Natl. Acad. Sci. USA 2009, 106, 17841–17846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barillas-Mury, C.; Han, Y.S.; Seeley, D.; Kafatos, F.C. Anopheles gambiae Ag-STAT, a new insect member of the STAT family, is activated in response to bacterial infection. EMBO J. 1999, 18, 959–967. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.C.; Chou, C.M.; Hsu, Y.L.; Lien, J.C.; Wang, Y.M.; Chen, S.T.; Tsai, S.C.; Hsiao, P.W.; Huang, C.J. Characterization of two mosquito STATs, AaSTAT and CtSTAT. Differential regulation of tyrosine phosphorylation and DNA binding activity by lipopolysaccharide treatment and by Japanese encephalitis virus infection. J. Biol. Chem. 2004, 279, 3308–3317. [Google Scholar] [CrossRef] [Green Version]
- Bahia, A.C.; Kubota, M.S.; Tempone, A.J.; Araujo, H.R.; Guedes, B.A.; Orfano, A.S.; Tadei, W.P.; Rios-Velasquez, C.M.; Han, Y.S.; Secundino, N.F.; et al. The JAK-STAT pathway controls Plasmodium vivax load in early stages of Anopheles aquasalis infection. PLoS Negl. Trop. Dis. 2011, 5, e1317. [Google Scholar] [CrossRef] [Green Version]
- Rolin, A.I.; Berrang-Ford, L.; Kulkarni, M.A. The risk of Rift Valley fever virus introduction and establishment in the United States and European Union. Emerg. Microbes. Infect. 2013, 2, e81. [Google Scholar] [CrossRef]
- Jang, I.H.; Chosa, N.; Kim, S.H.; Nam, H.J.; Lemaitre, B.; Ochiai, M.; Kambris, Z.; Brun, S.; Hashimoto, C.; Ashida, M.; et al. A Spatzle-processing enzyme required for toll signaling activation in Drosophila innate immunity. Dev. Cell 2006, 10, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Yagi, Y.; Tanji, T.; Zhou, S.; Ip, Y.T. Multimerization and interaction of Toll and Spatzle in Drosophila. Proc. Natl. Acad. Sci. USA 2004, 101, 9369–9374. [Google Scholar] [CrossRef] [Green Version]
- Weber, A.N.; Tauszig-Delamasure, S.; Hoffmann, J.A.; Lelievre, E.; Gascan, H.; Ray, K.P.; Morse, M.A.; Imler, J.L.; Gay, N.J. Binding of the Drosophila cytokine Spatzle to Toll is direct and establishes signaling. Nat. Immunol. 2003, 4, 794–800. [Google Scholar] [CrossRef] [PubMed]
- Horng, T.; Medzhitov, R. Drosophila MyD88 is an adapter in the Toll signaling pathway. Proc. Natl. Acad. Sci. USA 2001, 98, 12654–12658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tauszig-Delamasure, S.; Bilak, H.; Capovilla, M.; Hoffmann, J.A.; Imler, J.L. Drosophila MyD88 is required for the response to fungal and Gram-positive bacterial infections. Nat. Immunol. 2002, 3, 91–97. [Google Scholar] [CrossRef]
- Whalen, A.M.; Steward, R. Dissociation of the dorsal-cactus complex and phosphorylation of the dorsal protein correlate with the nuclear localization of dorsal. J. Cell. Biol. 1993, 123, 523–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemaitre, B.; Meister, M.; Govind, S.; Georgel, P.; Steward, R.; Reichhart, J.M.; Hoffmann, J.A. Functional analysis and regulation of nuclear import of dorsal during the immune response in Drosophila. EMBO J. 1995, 14, 536–545. [Google Scholar] [CrossRef] [Green Version]
- Choe, K.M.; Werner, T.; Stoven, S.; Hultmark, D.; Anderson, K.V. Requirement for a peptidoglycan recognition protein (PGRP) in Relish activation and antibacterial immune responses in Drosophila. Science 2002, 296, 359–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottar, M.; Gobert, V.; Michel, T.; Belvin, M.; Duyk, G.; Hoffmann, J.A.; Ferrandon, D.; Royet, J. The Drosophila immune response against Gram-negative bacteria is mediated by a peptidoglycan recognition protein. Nature 2002, 416, 640–644. [Google Scholar] [CrossRef]
- Ramet, M.; Manfruelli, P.; Pearson, A.; Mathey-Prevot, B.; Ezekowitz, R.A. Functional genomic analysis of phagocytosis and identification of a Drosophila receptor for E. coli. Nature 2002, 416, 644–648. [Google Scholar] [CrossRef]
- Kaneko, T.; Silverman, N. Bacterial recognition and signalling by the Drosophila IMD pathway. Cell Microbiol. 2005, 7, 461–469. [Google Scholar] [CrossRef]
- Takehana, A.; Yano, T.; Mita, S.; Kotani, A.; Oshima, Y.; Kurata, S. Peptidoglycan recognition protein (PGRP)-LE and PGRP-LC act synergistically in Drosophila immunity. EMBO J. 2004, 23, 4690–4700. [Google Scholar] [CrossRef] [Green Version]
- Park, J.M.; Brady, H.; Ruocco, M.G.; Sun, H.; Williams, D.; Lee, S.J.; Kato, T., Jr.; Richards, N.; Chan, K.; Mercurio, F.; et al. Targeting of TAK1 by the NF-kappa B protein Relish regulates the JNK-mediated immune response in Drosophila. Genes Dev. 2004, 18, 584–594. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, T.; Yano, T.; Aggarwal, K.; Lim, J.H.; Ueda, K.; Oshima, Y.; Peach, C.; Erturk-Hasdemir, D.; Goldman, W.E.; Oh, B.H.; et al. PGRP-LC and PGRP-LE have essential yet distinct functions in the drosophila immune response to monomeric DAP-type peptidoglycan. Nat. Immunol. 2006, 7, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Lee, J.H.; Lee, S.Y.; Kim, E.; Chung, J. Caspar, a suppressor of antibacterial immunity in Drosophila. Proc. Natl. Acad. Sci. USA 2006, 103, 16358–16363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, D.A.; McCoon, P.E.; Binari, R.; Gilman, M.; Perrimon, N. Drosophila unpaired encodes a secreted protein that activates the JAK signaling pathway. Genes Dev. 1998, 12, 3252–3263. [Google Scholar] [CrossRef] [Green Version]
- Agaisse, H.; Petersen, U.M.; Boutros, M.; Mathey-Prevot, B.; Perrimon, N. Signaling role of hemocytes in Drosophila JAK/STAT-dependent response to septic injury. Dev. Cell 2003, 5, 441–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, S.; Hu, N.; Hombria, J.C. Novel level of signalling control in the JAK/STAT pathway revealed by in situ visualisation of protein-protein interaction during Drosophila development. Development 2003, 130, 3077–3084. [Google Scholar] [CrossRef] [Green Version]
- Hou, X.S.; Melnick, M.B.; Perrimon, N. Marelle acts downstream of the Drosophila HOP/JAK kinase and encodes a protein similar to the mammalian STATs. Cell 1996, 84, 411–419. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.W.; Chen, X.; Oh, S.W.; Marinissen, M.J.; Gutkind, J.S.; Hou, S.X. mom identifies a receptor for the Drosophila JAK/STAT signal transduction pathway and encodes a protein distantly related to the mammalian cytokine receptor family. Genes. Dev. 2002, 16, 388–398. [Google Scholar] [CrossRef] [Green Version]
- Arbouzova, N.I.; Zeidler, M.P. JAK/STAT signalling in Drosophila: Insights into conserved regulatory and cellular functions. Development 2006, 133, 2605–2616. [Google Scholar] [CrossRef] [Green Version]
- Betz, A.; Lampen, N.; Martinek, S.; Young, M.W.; Darnell, J.E., Jr. A Drosophila PIAS homologue negatively regulates stat92E. Proc. Natl. Acad. Sci. USA 2001, 98, 9563–9568. [Google Scholar] [CrossRef] [Green Version]
- Billecocq, A.; Gauliard, N.; Le May, N.; Elliott, R.M.; Flick, R.; Bouloy, M. RNA polymerase I-mediated expression of viral RNA for the rescue of infectious virulent and avirulent Rift Valley fever viruses. Virology 2008, 378, 377–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowen, A.C.; Noonan, C.; McLees, A.; Elliott, R.M. Efficient bunyavirus rescue from cloned cDNA. Virology 2004, 330, 493–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredericks, A.C.; Russell, T.A.; Wallace, L.E.; Davidson, A.D.; Fernandez-Sesma, A.; Maringer, K. Aedes aegypti (Aag2)-derived clonal mosquito cell lines reveal the effects of pre-existing persistent infection with the insect-specific bunyavirus Phasi Charoen-like virus on arbovirus replication. PLoS. Negl. Trop. Dis. 2019, 13, e0007346. [Google Scholar] [CrossRef] [Green Version]
- McFarlane, M.; Arias-Goeta, C.; Martin, E.; O’Hara, Z.; Lulla, A.; Mousson, L.; Rainey, S.M.; Misbah, S.; Schnettler, E.; Donald, C.L.; et al. Characterization of Aedes aegypti innate-immune pathways that limit Chikungunya virus replication. PLoS. Negl. Trop. Dis. 2014, 8, e2994. [Google Scholar] [CrossRef]
- Pan, X.; Zhou, G.; Wu, J.; Bian, G.; Lu, P.; Raikhel, A.S.; Xi, Z. Wolbachia induces reactive oxygen species (ROS)-dependent activation of the Toll pathway to control dengue virus in the mosquito Aedes aegypti. Proc. Natl. Acad. Sci. USA 2012, 109, E23–E31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Zhu, Y.; Pang, X.; Xiao, X.; Zhang, R.; Cheng, G. Regulation of Antimicrobial Peptides in Aedes aegypti Aag2 Cells. Front. Cell Infect. Microbiol. 2017, 7, 22. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.; Souza-Neto, J.; Xi, Z.; Kokoza, V.; Shin, S.W.; Dimopoulos, G.; Raikhel, A. Transcriptome analysis of Aedes aegypti transgenic mosquitoes with altered immunity. PLoS Pathog. 2011, 7, e1002394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chowdhury, A.; Modahl, C.M.; Tan, S.T.; Wong Wei Xiang, B.; Misse, D.; Vial, T.; Kini, R.M.; Pompon, J.F. JNK pathway restricts DENV2, ZIKV and CHIKV infection by activating complement and apoptosis in mosquito salivary glands. PLoS Pathog. 2020, 16, e1008754. [Google Scholar] [CrossRef]
- Barletta, A.B.; Silva, M.C.; Sorgine, M.H. Validation of Aedes aegypti Aag-2 cells as a model for insect immune studies. Parasit. Vectors. 2012, 5, 148. [Google Scholar] [CrossRef] [Green Version]
- Karsten, P.; Plischke, I.; Perrimon, N.; Zeidler, M.P. Mutational analysis reveals separable DNA binding and trans-activation of Drosophila STAT92E. Cell Signal 2006, 18, 819–829. [Google Scholar] [CrossRef]
- Tauszig, S.; Jouanguy, E.; Hoffmann, J.A.; Imler, J.L. Toll-related receptors and the control of antimicrobial peptide expression in Drosophila. Proc. Natl. Acad. Sci. USA 2000, 97, 10520–10525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, P.; Kuttenkeuler, D.; Gesellchen, V.; Zeidler, M.P.; Boutros, M. Identification of JAK/STAT signalling components by genome-wide RNA interference. Nature 2005, 436, 871–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saucereau, Y.; Wilson, T.H.; Tang, M.C.K.; Moncrieffe, M.C.; Hardwick, S.W.; Chirgadze, D.Y.; Soares, S.G.; Marcaida, M.J.; Gay, N.J.; Gangloff, M. Structure and dynamics of Toll immunoreceptor activation in the mosquito Aedes aegypti. Nat. Commun. 2022, 13, 5110. [Google Scholar] [CrossRef] [PubMed]
- Franzke, K.; Leggewie, M.; Sreenu, V.B.; Jansen, S.; Heitmann, A.; Welch, S.R.; Brennan, B.; Elliott, R.M.; Tannich, E.; Becker, S.C.; et al. Detection, infection dynamics and small RNA response against Culex Y virus in mosquito-derived cells. J. Gen. Virol. 2018, 99, 1739–1745. [Google Scholar] [CrossRef]
- Gao, Y.; Hernandez, V.P.; Fallon, A.M. Immunity proteins from mosquito cell lines include three defensin A isoforms from Aedes aegypti and a defensin D from Aedes albopictus. Insect. Mol. Biol. 1999, 8, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Clem, R.J. Defining the core apoptosis pathway in the mosquito disease vector Aedes aegypti: The roles of iap1, ark, dronc, and effector caspases. Apoptosis 2011, 16, 105–113. [Google Scholar] [CrossRef]
- Feng, L.; Liu, H.; Li, X.; Qiao, J.; Wang, S.; Guo, D.; Liu, Q. Identification of AaCASPS7, an effector caspase in Aedes albopictus. Gene 2016, 593, 117–125. [Google Scholar] [CrossRef]
- Kaneko, T.; Goldman, W.E.; Mellroth, P.; Steiner, H.; Fukase, K.; Kusumoto, S.; Harley, W.; Fox, A.; Golenbock, D.; Silverman, N. Monomeric and polymeric gram-negative peptidoglycan but not purified LPS stimulate the Drosophila IMD pathway. Immunity 2004, 20, 637–649. [Google Scholar] [CrossRef] [Green Version]
- Hsu, S.H.; Mao, W.H.; Cross, J.H. Establishment of a line of cells derived from ovarian tissue of Clex quinquefasciatus Say. J. Med. Entomol. 1970, 7, 703–707. [Google Scholar] [CrossRef]
- Turell, M.J.; Linthicum, K.J.; Patrican, L.A.; Davies, F.G.; Kairo, A.; Bailey, C.L. Vector competence of selected African mosquito (Diptera: Culicidae) species for Rift Valley fever virus. J. Med. Entomol. 2008, 45, 102–108. [Google Scholar] [CrossRef]
- Ramirez, J.L.; Souza-Neto, J.; Torres Cosme, R.; Rovira, J.; Ortiz, A.; Pascale, J.M.; Dimopoulos, G. Reciprocal tripartite interactions between the Aedes aegypti midgut microbiota, innate immune system and dengue virus influences vector competence. PLoS Negl. Trop. Dis. 2012, 6, e1561. [Google Scholar] [CrossRef]
- Hixson, B.; Bing, X.L.; Yang, X.; Bonfini, A.; Nagy, P.; Buchon, N. A transcriptomic atlas of Aedes aegypti reveals detailed functional organization of major body parts and gut regional specializations in sugar-fed and blood-fed adult females. Elife 2022, 11, e76132. [Google Scholar] [CrossRef] [PubMed]
- Moy, R.H.; Gold, B.; Molleston, J.M.; Schad, V.; Yanger, K.; Salzano, M.V.; Yagi, Y.; Fitzgerald, K.A.; Stanger, B.Z.; Soldan, S.S.; et al. Antiviral autophagy restrictsRift Valley fever virus infection and is conserved from flies to mammals. Immunity 2014, 40, 51–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikegami, T.; Hill, T.E.; Smith, J.K.; Zhang, L.; Juelich, T.L.; Gong, B.; Slack, O.A.; Ly, H.J.; Lokugamage, N.; Freiberg, A.N. Rift Valley Fever Virus MP-12 Vaccine Is Fully Attenuated by a Combination of Partial Attenuations in the S, M, and L Segments. J. Virol. 2015, 89, 7262–7276. [Google Scholar] [CrossRef] [Green Version]
- Ikegami, T. Rift Valley fever vaccines: An overview of the safety and efficacy of the live-attenuated MP-12 vaccine candidate. Expert. Rev. Vaccines. 2017, 16, 601–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikegami, T.; Narayanan, K.; Won, S.; Kamitani, W.; Peters, C.J.; Makino, S. Rift Valley fever virus NSs protein promotes post-transcriptional downregulation of protein kinase PKR and inhibits eIF2alpha phosphorylation. PLoS Pathog. 2009, 5, e1000287. [Google Scholar] [CrossRef] [Green Version]
- Ikegami, T.; Won, S.; Peters, C.J.; Makino, S. Rescue of infectious rift valley fever virus entirely from cDNA, analysis of virus lacking the NSs gene, and expression of a foreign gene. J. Virol. 2006, 80, 2933–2940. [Google Scholar] [CrossRef] [Green Version]
- Moutailler, S.; Roche, B.; Thiberge, J.M.; Caro, V.; Rougeon, F.; Failloux, A.B. Host alternation is necessary to maintain the genome stability of rift valley fever virus. PLoS. Negl. Trop. Dis. 2011, 5, e1156. [Google Scholar] [CrossRef] [Green Version]
- Crabtree, M.B.; Kent Crockett, R.J.; Bird, B.H.; Nichol, S.T.; Erickson, B.R.; Biggerstaff, B.J.; Horiuchi, K.; Miller, B.R. Infection and transmission of Rift Valley fever viruses lacking the NSs and/or NSm genes in mosquitoes: Potential role for NSm in mosquito infection. PLoS. Negl. Trop. Dis. 2012, 6, e1639. [Google Scholar] [CrossRef] [Green Version]
- Kreher, F.; Tamietti, C.; Gommet, C.; Guillemot, L.; Ermonval, M.; Failloux, A.B.; Panthier, J.J.; Bouloy, M.; Flamand, M. The Rift Valley fever accessory proteins NSm and P78/NSm-GN are distinct determinants of virus propagation in vertebrate and invertebrate hosts. Emerg. Microbes. Infect. 2014, 3, e71. [Google Scholar] [CrossRef]
- Kading, R.C.; Crabtree, M.B.; Bird, B.H.; Nichol, S.T.; Erickson, B.R.; Horiuchi, K.; Biggerstaff, B.J.; Miller, B.R. Deletion of the NSm virulence gene of Rift Valley fever virus inhibits virus replication in and dissemination from the midgut of Aedes aegypti mosquitoes. PLoS Negl. Trop. Dis. 2014, 8, e2670. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.L.; Snell, T.K.; Bennett, S.; Wyckoff, J.H., 3rd; Heaslip, D.; Flatt, J.; Harris, E.K.; Hartman, D.A.; Lian, E.; Bird, B.H.; et al. Safety study of Rift Valley Fever human vaccine candidate (DDVax) in mosquitoes. Transbound Emerg. Dis. 2022, 69, 2621–2633. [Google Scholar] [CrossRef] [PubMed]
- Fragkoudis, R.; Chi, Y.; Siu, R.W.; Barry, G.; Attarzadeh-Yazdi, G.; Merits, A.; Nash, A.A.; Fazakerley, J.K.; Kohl, A. Semliki Forest virus strongly reduces mosquito host defence signaling. Insect. Mol. Biol. 2008, 17, 647–656. [Google Scholar] [CrossRef] [Green Version]
- Michel, T.; Reichhart, J.M.; Hoffmann, J.A.; Royet, J. Drosophila Toll is activated by Gram-positive bacteria through a circulating peptidoglycan recognition protein. Nature 2001, 414, 756–759. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.L.; Muturi, E.J.; Flor-Weiler, L.B.; Vermillion, K.; Rooney, A.P. Peptidoglycan Recognition Proteins (PGRPs) Modulates Mosquito Resistance to Fungal Entomopathogens in a Fungal-Strain Specific Manner. Front. Cell Infect Microbiol. 2019, 9, 465. [Google Scholar] [CrossRef] [Green Version]
- Wimalasiri-Yapa, B.; Stassen, L.; Hu, W.; Yakob, L.; McGraw, E.A.; Pyke, A.T.; Jansen, C.C.; Devine, G.J.; Frentiu, F.D. Chikungunya Virus Transmission at Low Temperature by Aedes albopictus Mosquitoes. Pathogens 2019, 8, 149. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.; Liu, Y.; Zhang, X.; Wang, J.; Li, Z.; Pang, X.; Wang, P.; Cheng, G. Complement-related proteins control the flavivirus infection of Aedes aegypti by inducing antimicrobial peptides. PLoS Pathog. 2014, 10, e1004027. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Ryu, J.H.; Han, S.J.; Choi, K.H.; Nam, K.B.; Jang, I.H.; Lemaitre, B.; Brey, P.T.; Lee, W.J. Gram-negative bacteria-binding protein, a pattern recognition receptor for lipopolysaccharide and beta-1,3-glucan that mediates the signaling for the induction of innate immune genes in Drosophila melanogaster cells. J. Biol. Chem. 2000, 275, 32721–32727. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Weber, A.N.; Atilano, M.L.; Filipe, S.R.; Gay, N.J.; Ligoxygakis, P. Sensing of Gram-positive bacteria in Drosophila: GNBP1 is needed to process and present peptidoglycan to PGRP-SA. EMBO J. 2006, 25, 5005–5014. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Park, J.W.; Kwon, H.M.; Hwang, H.O.; Jang, I.H.; Masuda, A.; Kurokawa, K.; Nakayama, H.; Lee, W.J.; Dohmae, N.; et al. Diversity of innate immune recognition mechanism for bacterial polymeric meso-diaminopimelic acid-type peptidoglycan in insects. J. Biol. Chem. 2010, 285, 32937–32945. [Google Scholar] [CrossRef] [Green Version]
- Warr, E.; Das, S.; Dong, Y.; Dimopoulos, G. The Gram-negative bacteria-binding protein gene family: Its role in the innate immune system of anopheles gambiae and in anti-Plasmodium defence. Insect. Mol. Biol. 2008, 17, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Behura, S.K.; Franz, A.W.E. The midgut transcriptome of Aedes aegypti fed with saline or protein meals containing chikungunya virus reveals genes potentially involved in viral midgut escape. BMC Genomics 2017, 18, 382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Licciardi, S.; Loire, E.; Cardinale, E.; Gislard, M.; Dubois, E.; Cetre-Sossah, C. In vitro shared transcriptomic responses of Aedes aegypti to arboviral infections: Example of dengue and Rift Valley fever viruses. Parasit Vectors 2020, 13, 395. [Google Scholar] [CrossRef] [PubMed]
- Dziarski, R.; Gupta, D. A Balancing Act: PGRPs Preserve and Protect. Cell Host. Microbe. 2018, 23, 149–151. [Google Scholar] [CrossRef] [Green Version]
- Dziarski, R.; Gupta, D. The peptidoglycan recognition proteins (PGRPs). Genome Biol. 2006, 7, 232. [Google Scholar] [CrossRef] [Green Version]
- Weng, S.C.; Li, H.H.; Li, J.C.; Liu, W.L.; Chen, C.H.; Shiao, S.H. A Thioester-Containing Protein Controls Dengue Virus Infection in Aedes aegypti Through Modulating Immune Response. Front. Immunol. 2021, 12, 670122. [Google Scholar] [CrossRef]
- Dong, Y.; Dong, S.; Dizaji, N.B.; Rutkowski, N.; Pohlenz, T.; Myles, K.; Dimopoulos, G. The Aedes aegypti siRNA pathway mediates broad-spectrum defense against human pathogenic viruses and modulates antibacterial and antifungal defenses. PLoS Biol. 2022, 20, e3001668. [Google Scholar] [CrossRef]
- Merkling, S.H.; Crist, A.B.; Henrion-Lacritick, A.; Frangeul, L.; Gausson, V.; Blanc, H.; Baidaliuk, A.; Saleh, M.C.; Lambrechts, L. Multifaceted contributions of Dicer2 to arbovirus transmission by Aedes aegypti. bioRxiv 2022, 11, 516413. [Google Scholar]
- Nunez, A.I.; Esteve-Codina, A.; Gomez-Garrido, J.; Brustolin, M.; Talavera, S.; Berdugo, M.; Dabad, M.; Alioto, T.; Bensaid, A.; Busquets, N. Alteration in the Culex pipiens transcriptome reveals diverse mechanisms of the mosquito immune system implicated upon Rift Valley fever phlebovirus exposure. PLoS Negl. Trop. Dis. 2020, 14, e0008870. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol * | Pathway | Vectorbase Gene ID | Forward | Reverse | Reference |
|---|---|---|---|---|---|
| Housekeeping Gene and Antimicrobial Peptides (AMPs) | |||||
| RPS7 | AAEL009496 | ccaggctatcctggagttg | gacgtgcttgccggagaac | [44] | |
| ATT | Toll [45], IMD [46] | AAEL003389 | aacaaaggaagaaatagcgccg | ccttttggccgctgaacag | This study |
| CECE | IMD [9,46] | AAEL000611 | ctcgttctgctcatcgggtt | tccttccaatttcttccccagc | |
| CECN | Toll [46], IMD [46] | AAEL000621 | tcttggttcttgtggccgtt | cttgccgaatttccacctgg | |
| DEFA | Toll [46], IMD [46] | AAEL003841 | ctctgtgtaccgtggccatc | ctccggcagttcatcgaaaaga | |
| DIPT | IMD | AAEL004833 | gaagtggaaccagcagtgtcc | tcgtcctgttgatgggtagct | |
| GAMB | Toll [46], IMD [46], Jak-STAT [46] | AAEL004522 | agctgcctataccgatgctt | caataccgggctccatatgc | |
| TEP20 | Toll [8], IMD [47], Jak-STAT [48,49], JNK [48] | AAEL001794 | ggatcttgccgctactgatttg | cggtccaatcactgaaaagcc | |
| Transcription factors (TFs) | |||||
| REL1 | Toll | AAEL007696/ AAEL006930 | tctgcccaacaacctcatagt | tggtggcatttcttggtcga | This study |
| REL2 | IMD | AAEL007624 | acttatctcggccctctgga | tgatgttgcgtcgttcaatcg | |
| STAT | Jak-STAT | AAEL009692 | ggcaacagttttccaatcgagg | Gactgggacgtttagcaatcg | |
| Negative regulators (NRs) | |||||
| CAC | Toll | AAEL000709 | gaagtccaaggagcaacaacag | acggcaaggtgtaggtaagtat | This study |
| CAS | IMD | AAEL027860 | gccagtgtgaagttttccagg | atgtccgacgcttccatcag | |
| PIAS | Jak-STAT | AAEL026694 | acgacgagttctgcaatgact | tgtcgatggtggggatgga | |
| PUC | JNK | AAEL010411 | cctggagtacaagcagatccc | gggcgtcctcaatgaattcga | |
| Pattern recognition receptors (PRRs) | |||||
| PGRP-LC | IMD | AAEL014640 | cagttcgaagcagttaccgg | ccccgatgtgagcttgtaga | This study |
| PGRP-LE | IMD | AAEL027982 | acgttaactccatcaccggt | gtccgccatttgacactatct | |
| PGRP-S1 | Toll/IMD | AAEL009474 | caagtggagcgacattggtt | aactcgatgccagccaattc | |
| GNBP-A1 | Toll?/IMD [47] | AAEL007626 | agtgaattatgtctcggcaca | cgaacagctttattccgggaa | |
| GNBP-A2 | Toll? | AAEL000652 | tggaaagatattgattcgcgc | cgcattagacccgaagcata | |
| GNBP-B1 | Toll [47] | AAEL003889 | gcaccctttacattcgtccc | ggtgcattgttcaacagggt | |
| GNBP-B3 | Toll? | AAEL009176 | accaacaaccgagcgaattc | agcaaccaatgtaggacgga | |
| GNBP-B4 | Toll? | AAEL009178 | agctgatgaaactggtgagga | ctttcacaaccatcccacgc | |
| GNBP-B5 | Toll? | AAEL003894 | accggtcaaattccttcgtg | tcgccaaattcatcaaccga | |
| GNBP-B6 | Toll? | AAEL007064 | tcaatctcaacgagggtccc | gtggatctgttctgcccaac | |
| SCRC2 | Jak-STAT [14] | AAEL006361 | tcccaagttccgatttgtgtca | aaatccgttatccacagccgat | |
| Viruses | |||||
| CFAV | tgatgcgtggtgattgacatg | tgcaagtagtctgtccggttc | This study | ||
| PCLPV N | tcccacgtcagatgcaaact | ttgtgttccttgggtgcctc | |||
| CYV segment A | acctccaaaacaccgaacaag | cttccccataatgccacgttt | |||
| RVFV N | tgccacgagtyagagcca | gtgggtccgagagtytgc | [5] | ||
| BUNV M | tcagacggtatagaaggggca | atcaaggagtgggaagccatc | This study | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laureti, M.; Lee, R.-X.; Bennett, A.; Wilson, L.A.; Sy, V.E.; Kohl, A.; Dietrich, I. Rift Valley Fever Virus Primes Immune Responses in Aedes aegypti Cells. Pathogens 2023, 12, 563. https://doi.org/10.3390/pathogens12040563
Laureti M, Lee R-X, Bennett A, Wilson LA, Sy VE, Kohl A, Dietrich I. Rift Valley Fever Virus Primes Immune Responses in Aedes aegypti Cells. Pathogens. 2023; 12(4):563. https://doi.org/10.3390/pathogens12040563
Chicago/Turabian StyleLaureti, Mathilde, Rui-Xue Lee, Amelia Bennett, Lucas Aladar Wilson, Victoria Elena Sy, Alain Kohl, and Isabelle Dietrich. 2023. "Rift Valley Fever Virus Primes Immune Responses in Aedes aegypti Cells" Pathogens 12, no. 4: 563. https://doi.org/10.3390/pathogens12040563