
Designing a Next-Generation Multiepitope-Based Vaccine against Staphylococcus aureus Using Reverse Vaccinology Approaches
 ,
,
Abstract
:1. Introduction
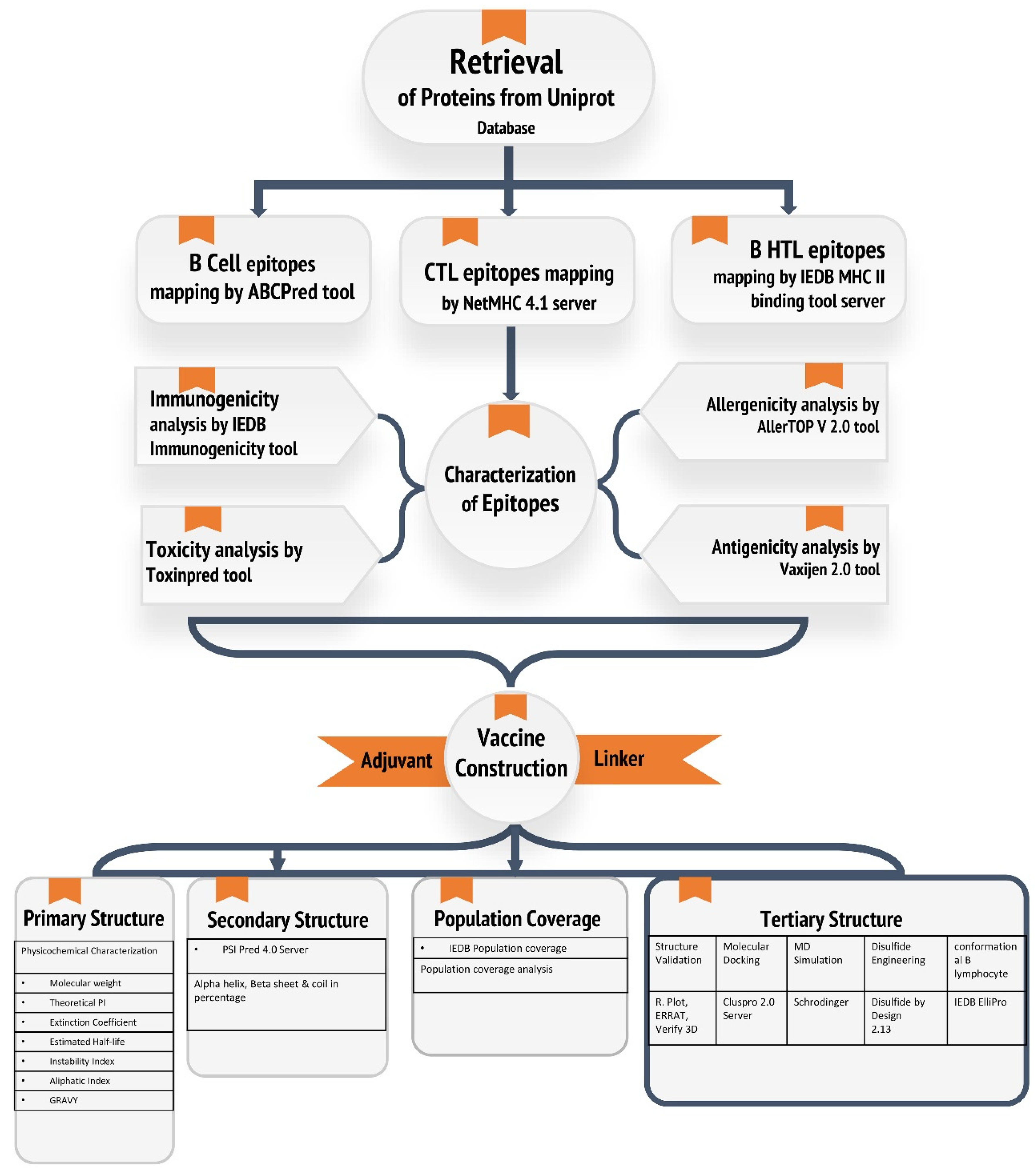
2. Materials and Methods
2.1. Retrieval of Protein Sequence
2.2. Linear B Cell Epitope Prediction
2.3. T Cell Epitope Prediction
2.3.1. CTL Epitope Prediction
2.3.2. HTL Epitope Prediction
2.4. Screened Epitopes Characterization
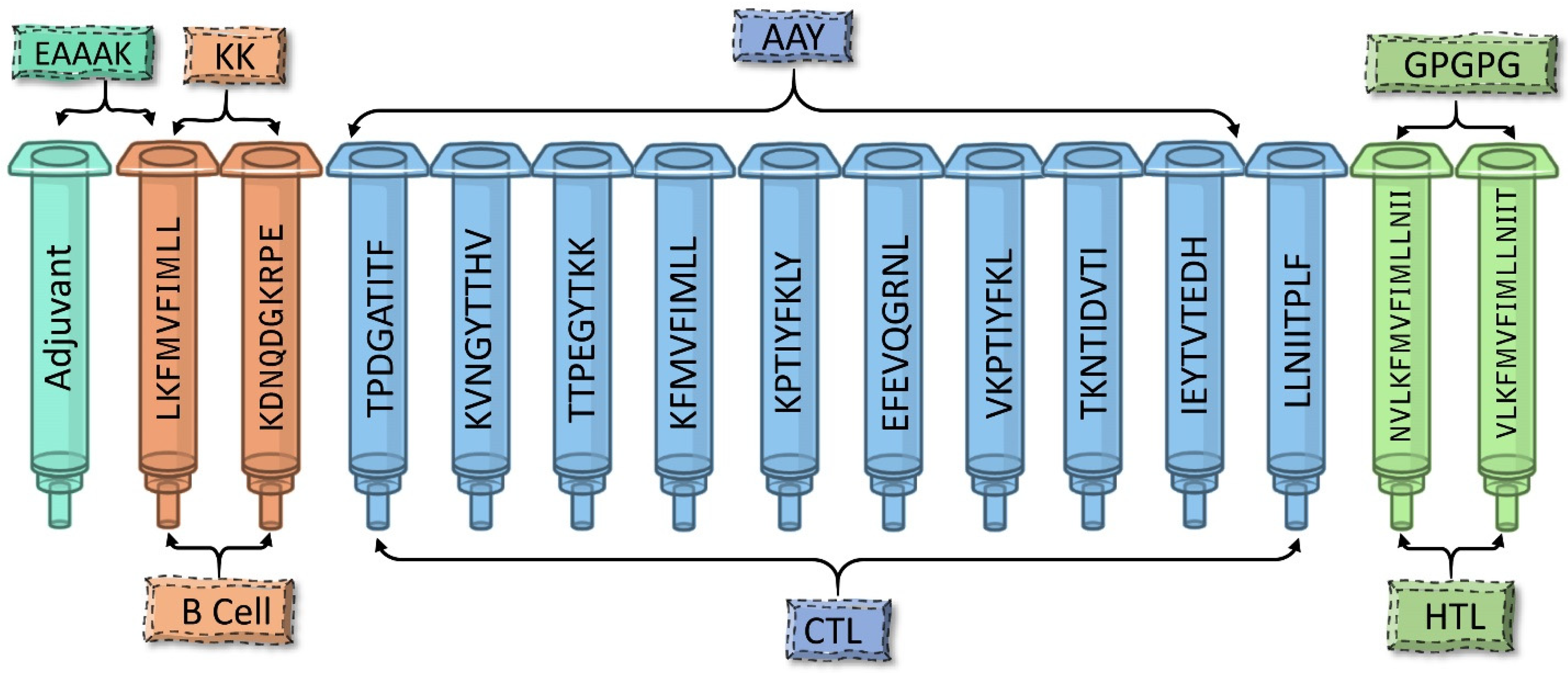
2.5. Assembly of the Multi-Epitope Vaccine
2.6. Population Coverage Analysis of Vaccine Construct
2.7. Physicochemical Features and Secondary Structure and Solubility Prediction of the Vaccine Construct
2.8. Tertiary Structure Prediction and Validation of the Multi-Epitope Vaccine
2.9. Disulfide Engineering of the Vaccine Protein
2.10. Discontinuous B Cell Epitope Prediction
2.11. Prediction of IFN-γ Inducing Epitopes
2.12. Studies on Molecular Docking of Vaccine Constructs with TLR-2
2.13. Molecular Dynamics Simulation
2.14. MMPBSA Binding Free Energy Analysis
2.15. Codon Adaptation and In Silico Cloning
2.16. Immune Simulation of Vaccine Constructs
3. Results
3.1. Epitope Prediction for B Cell Lymphocytes
3.2. Epitope Prediction for T Cells
3.2.1. Epitope Prediction for Cytotoxic T-Lymphocyte (CTL)
3.2.2. Epitope Prediction for Helper T-Lymphocyte (HTL)
3.3. Designing Multi-Epitope Subunit Vaccine Construct
3.4. Allergenicity and Antigenicity Prediction of the Vaccine Construct
3.5. Prediction of Population Coverage
3.6. Physicochemical Characterization, Secondary Structure, and Solubility of Designed Vaccine Construct
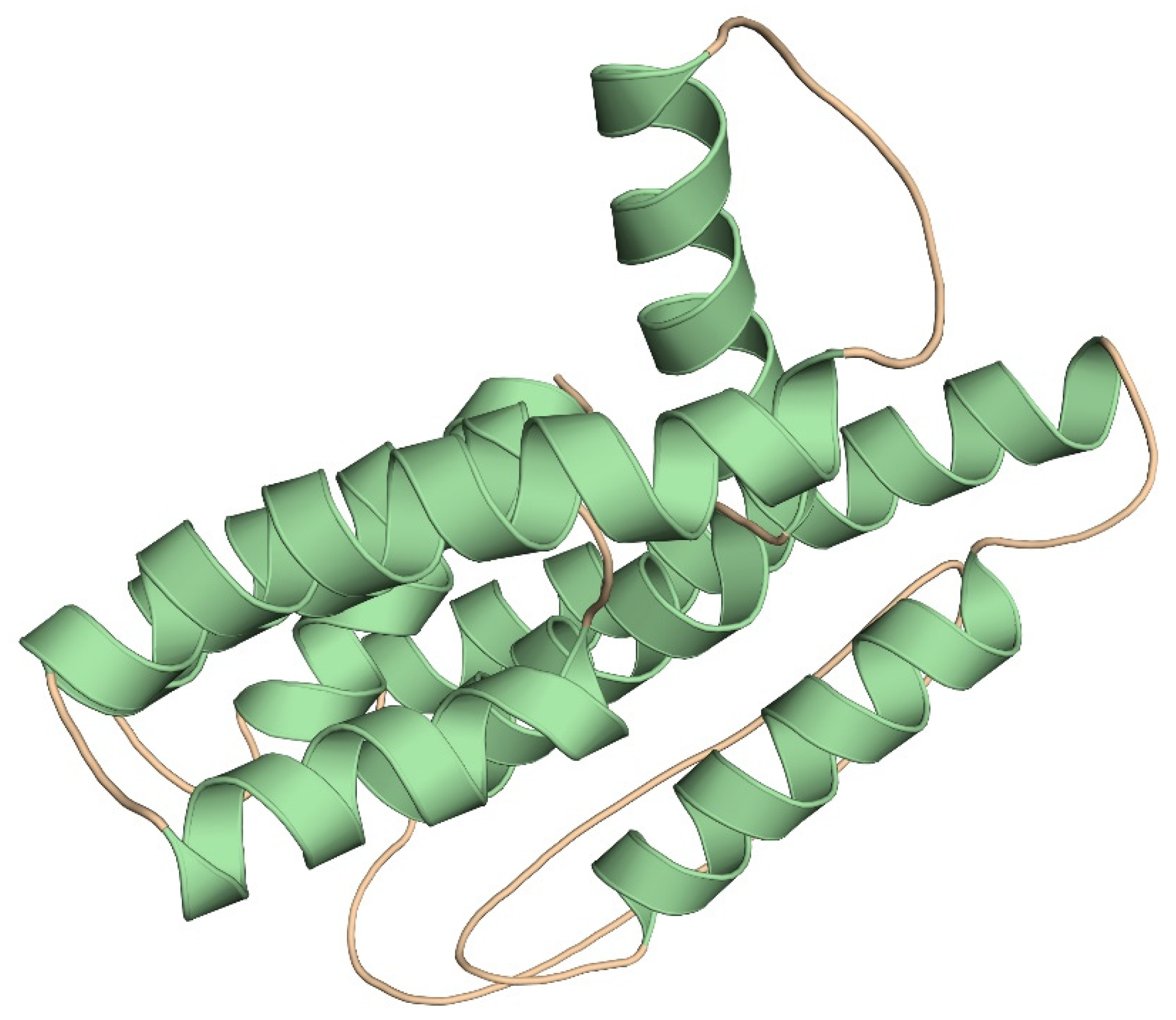
3.7. Prediction and Validation of Tertiary Structure
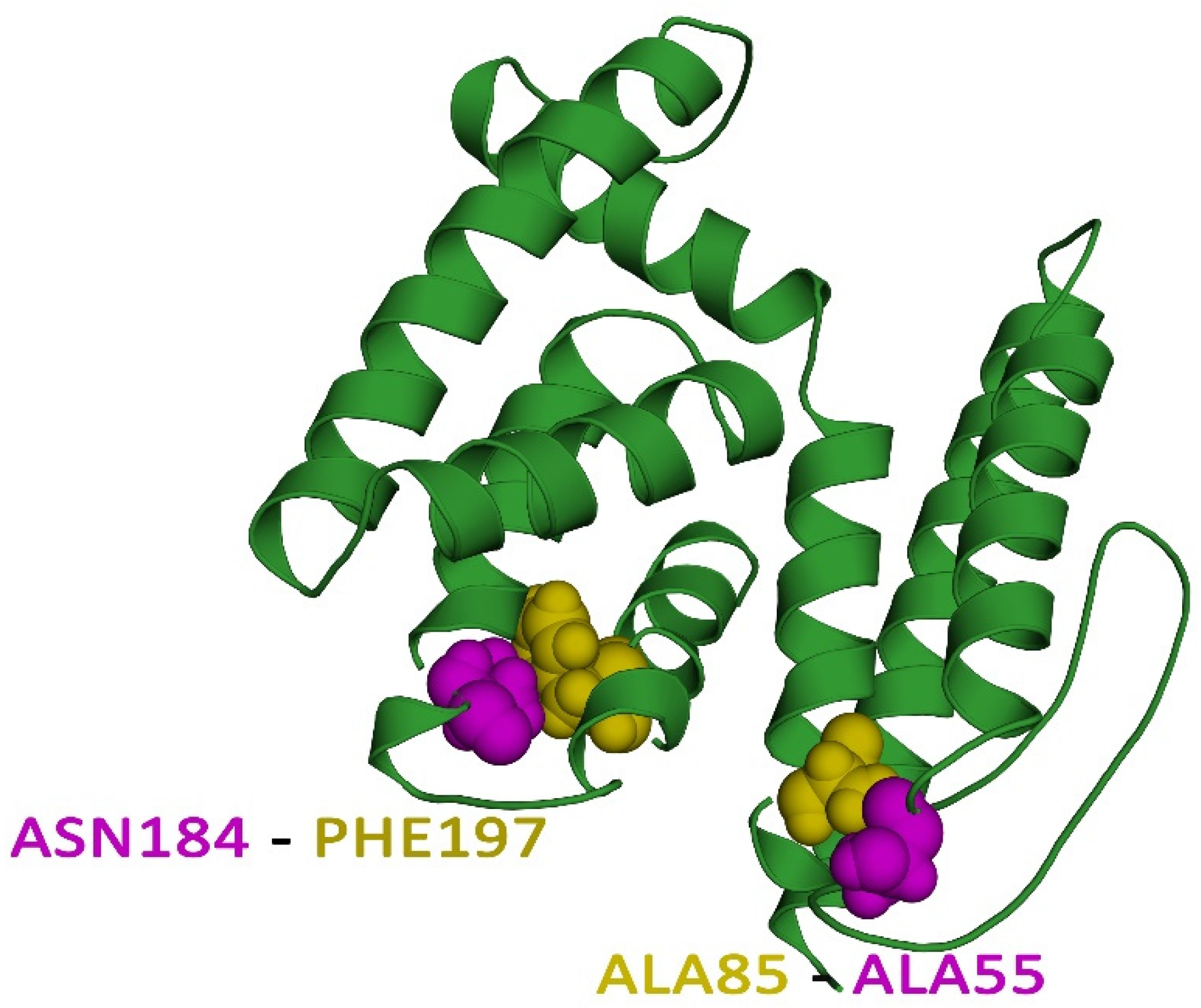
3.8. Vaccine Protein Disulfide Engineering
3.9. Prediction of Discontinuous B Cell Epitopes
3.10. IFN-γ Inducing Epitope Analysis
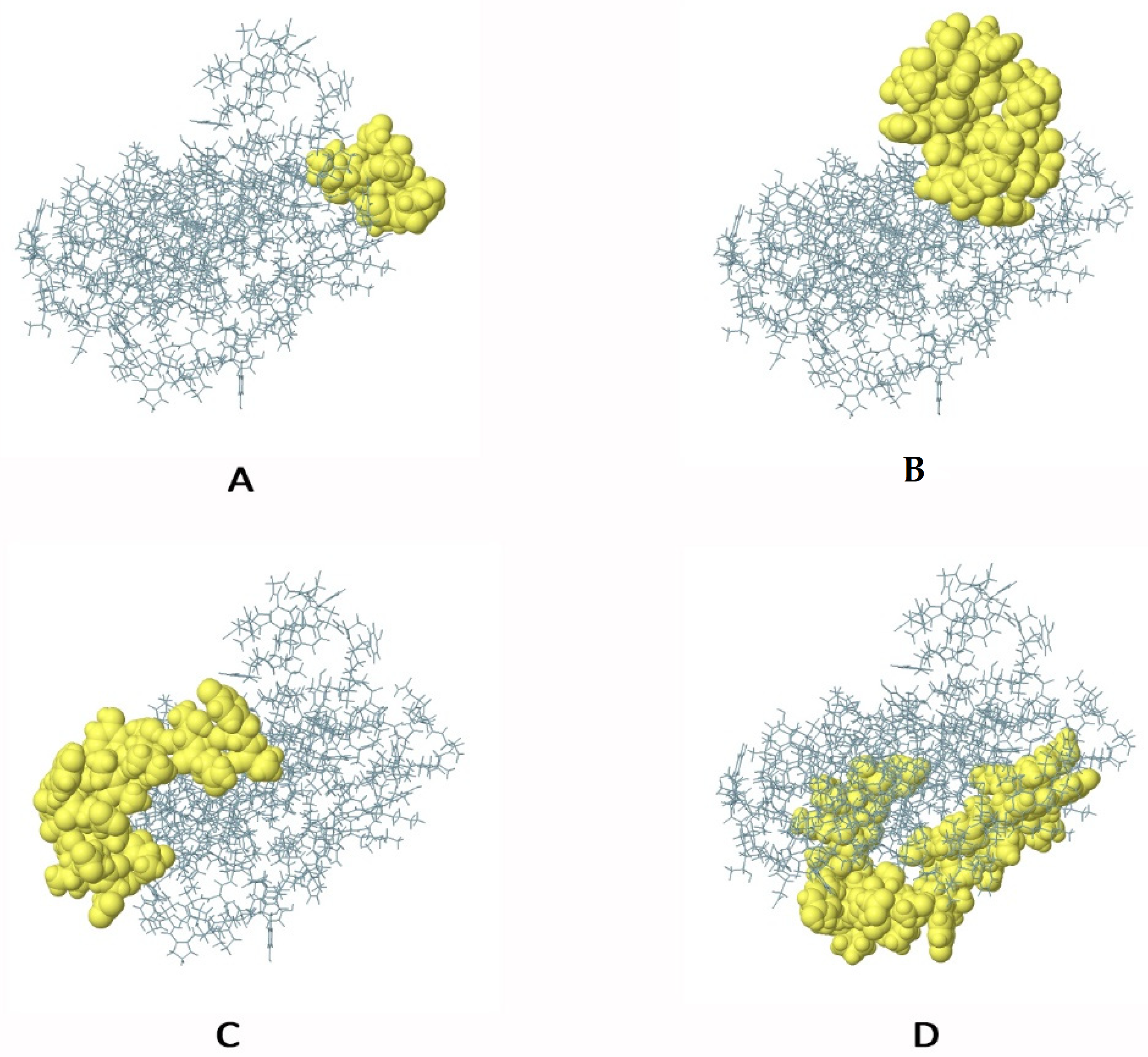
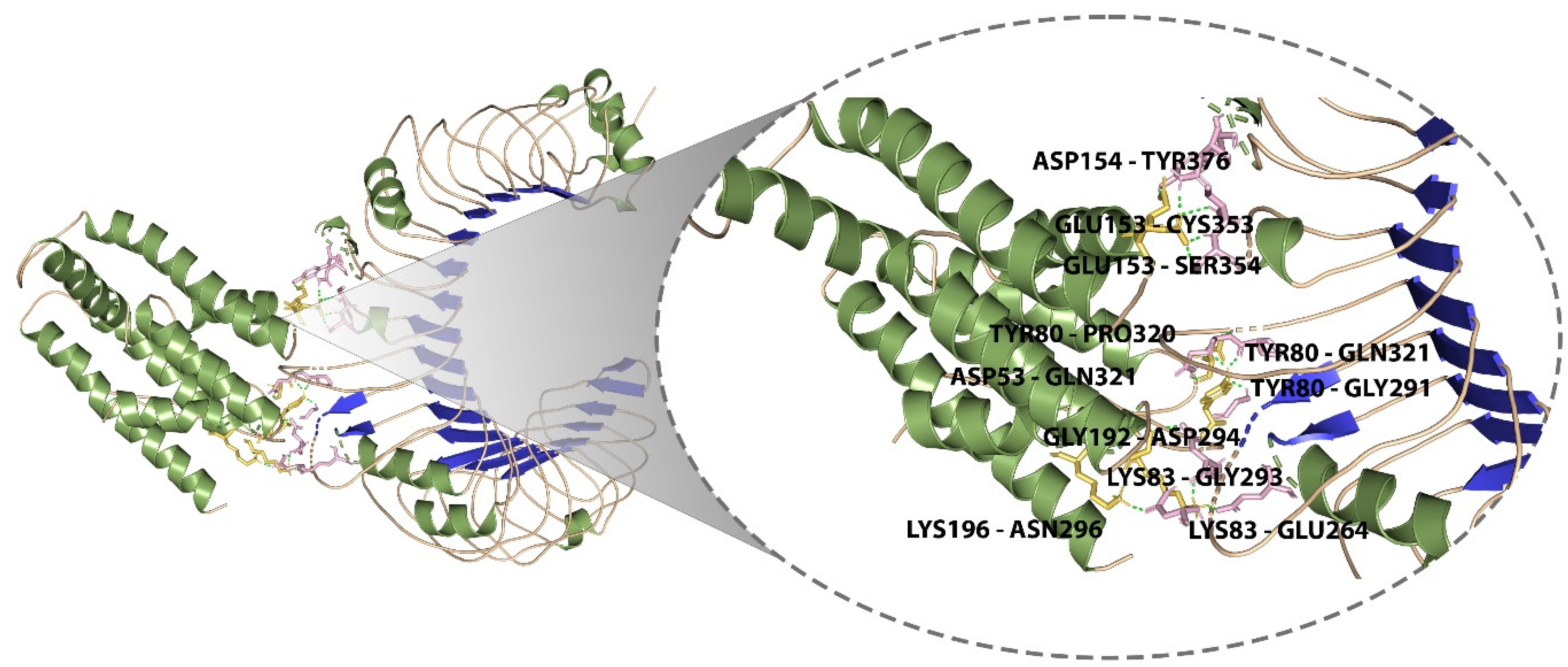
3.11. Molecular Docking of the Designed Vaccine with Immunological Receptor
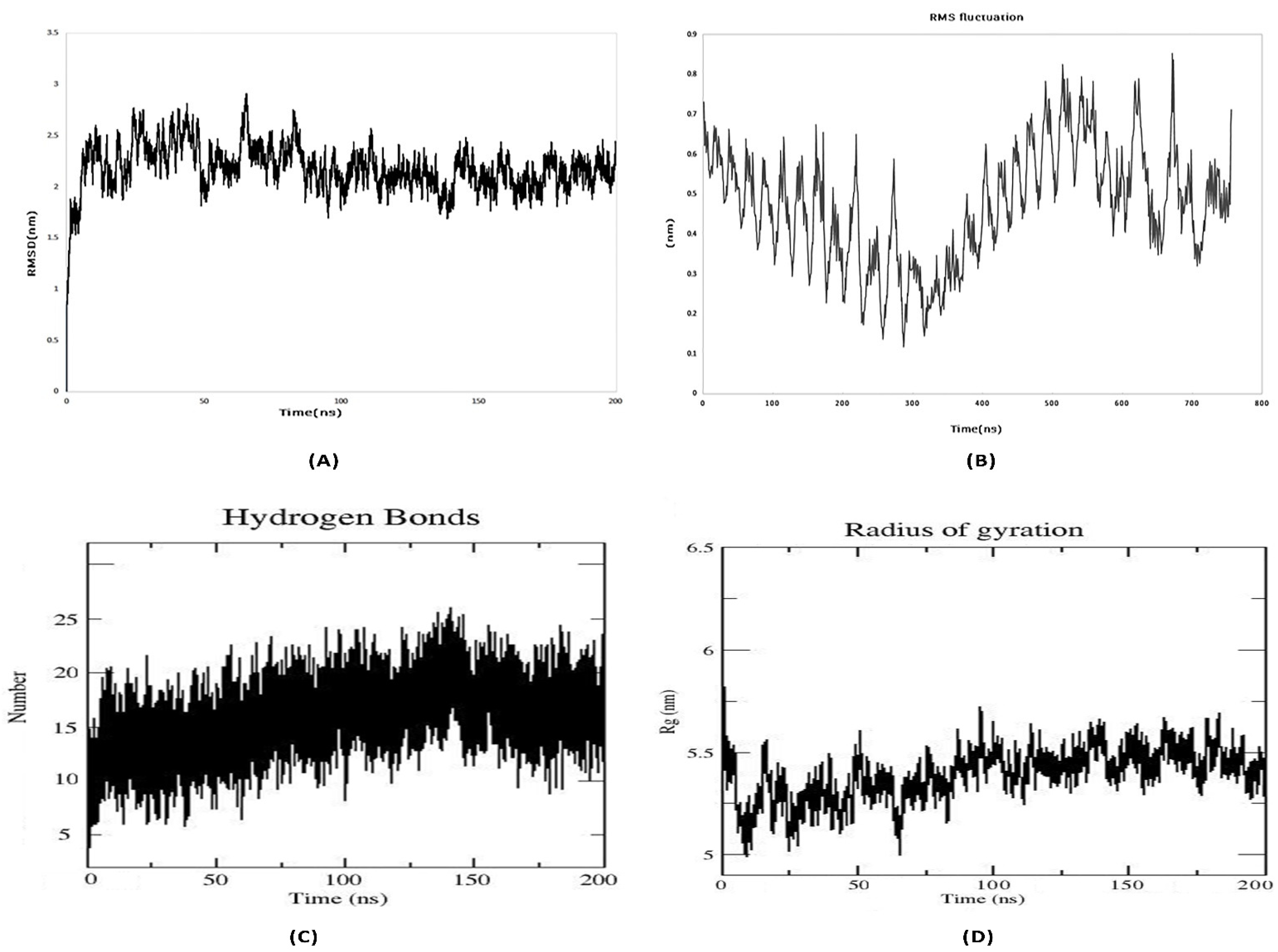
3.12. Molecular Dynamics Simulation
3.13. MMPBSA Binding Free Energy Analysis
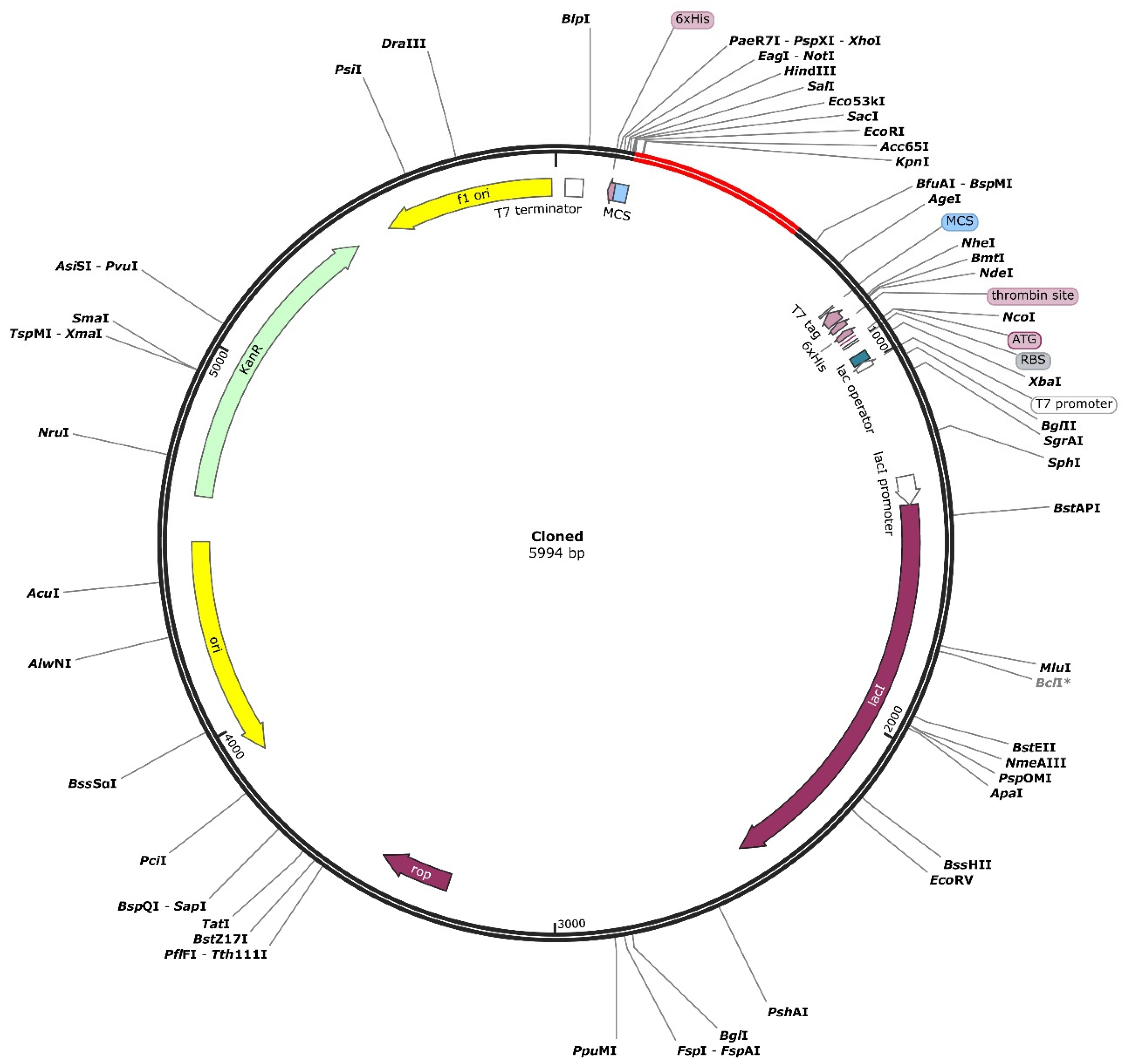
3.14. Codon Optimization and In Silico Cloning
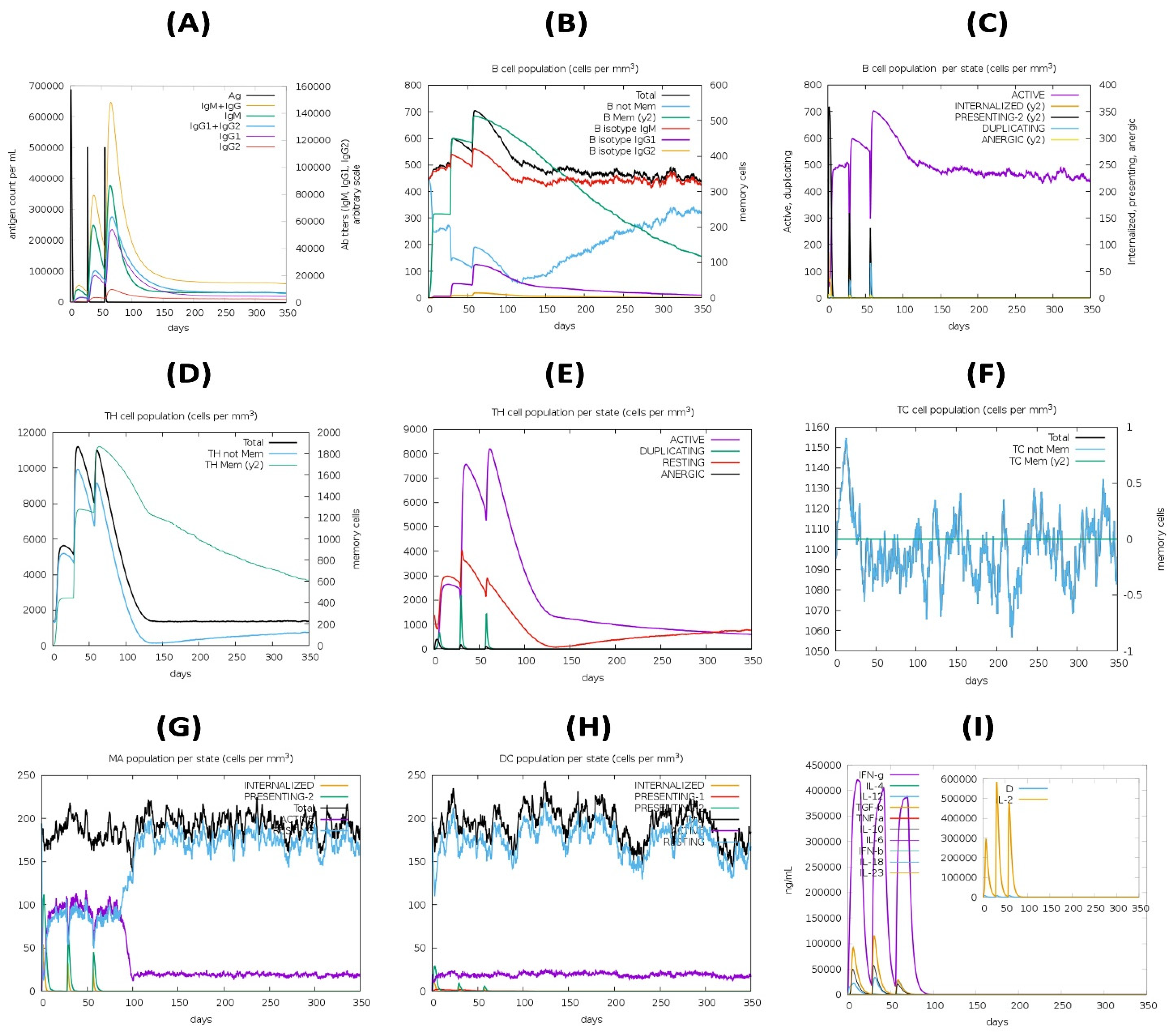
3.15. Immune Simulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Santajit, S.; Indrawattana, N. Mechanisms of Antimicrobial Resistance in ESKAPE Pathogens. BioMed Res. Int. 2016, 2016, 2475067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Hal, S.J.; Jensen, S.O.; Vaska, V.L.; Espedido, B.A.; Paterson, D.L.; Gosbell, I.B. Predictors of Mortality in Staphylococcus aureus Bacteremia. Clin. Microbiol. Rev. 2012, 25, 362–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foster, T.J. Antibiotic resistance in Staphylococcus aureus. Current status and future prospects. FEMS Microbiol. Rev. 2017, 41, 430–449. [Google Scholar] [CrossRef] [PubMed]
- Stranger-Jones, Y.K.; Bae, T.; Schneewind, O. Vaccine assembly from surface proteins of Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2006, 103, 16942–16947. [Google Scholar] [CrossRef] [Green Version]
- Herman-Bausier, P.; Valotteau, C.; Pietrocola, G.; Rindi, S.; Alsteens, D.; Foster, T.J.; Speziale, P.; Dufrêne, Y.F. Mechanical Strength and Inhibition of the Staphylococcus aureus Collagen-Binding Protein Cna. Mbio 2016, 7, e01529-16. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.Y. Molecular Pathogenesis of Staphylococcus aureus Infection. Pediatr. Res. 2009, 65, 71R–77R. [Google Scholar] [CrossRef]
- Nilsson, I.M.; Patti, J.M.; Bremell, T.; Höök, M.; Tarkowski, A. Vaccination with a recombinant fragment of collagen adhesin provides protection against Staphylococcus aureus-mediated septic death. J. Clin. Investig. 1998, 101, 2640–2649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patti, J.M.; Bremell, T.; Krajewska-Pietrasik, D.; Abdelnour, A.; Tarkowski, A.; Rydén, C.; Höök, M. The Staphylococcus aureus collagen adhesin is a virulence determinant in experimental septic arthritis. Infect. Immun. 1994, 62, 152–161. [Google Scholar] [CrossRef] [Green Version]
- Espeziale, P.; Pietrocola, G.; Foster, T.J.; Geoghegan, J.A. Protein-based biofilm matrices in Staphylococci. Front. Cell. Infect. Microbiol. 2014, 4, 171. [Google Scholar] [CrossRef] [Green Version]
- Rohde, H.; Burdelski, C.; Bartscht, K.; Hussain, M.; Buck, F.; Horstkotte, M.A.; Knobloch, J.K.-M.; Heilmann, C.; Herrmann, M.; Mack, D. Induction of Staphylococcus epidermidis biofilm formation via proteolytic processing of the accumulation-associated protein by staphylococcal and host proteases. Mol. Microbiol. 2005, 55, 1883–1895. [Google Scholar] [CrossRef]
- Fattom, A.I.; Horwith, G.; Fuller, S.; Propst, M.; Naso, R. Development of StaphVAX™, a polysaccharide conjugate vaccine against S. aureus infection: From the lab bench to phase III clinical trials. Vaccine 2004, 22, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Botelho-Nevers, E.; Verhoeven, P.; Paul, S.; Grattard, F.; Pozzetto, B.; Berthelot, P.; Lucht, F. Staphylococcal vaccine development: Review of past failures and plea for a future evaluation of vaccine efficacy not only on staphylococcal infections but also on mucosal carriage. Expert Rev. Vaccines 2013, 12, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Giersing, B.K.; Dastgheyb, S.S.; Modjarrad, K.; Moorthy, V. Status of vaccine research and development of vaccines for Staphylococcus aureus. Vaccine 2016, 34, 2962–2966. [Google Scholar] [CrossRef] [Green Version]
- Faezi, S.; Bahrmand, A.R.; Sardari, S.; Nikokar, I.; Khanaki, K.; Siadat, S.D.; Goudarzi, G.; Elmi, A.; Mahdavi, M. Epitope-based immunoinformatics study of a novel PilQ380–706-PilA fusion protein from Pseudomonas aeruginosa. Gene Rep. 2019, 15, 100385. [Google Scholar] [CrossRef]
- Mehla, K.; Ramana, J. Identification of epitope-based peptide vaccine candidates against enterotoxigenic Escherichia coli: A comparative genomics and immunoinformatics approach. Mol. BioSystems 2016, 12, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Bröker, B.M.; Mrochen, D.; Péton, V. The T Cell Response to Staphylococcus aureus. Pathogens 2016, 5, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daum, R.S.; Spellberg, B. Progress Toward a Staphylococcus aureus Vaccine. Clin. Infect. Dis. 2012, 54, 560–567. [Google Scholar] [CrossRef] [Green Version]
- Gjertsson, I.; Hultgren, O.H.; Stenson, M.; Holmdahl, R.; Tarkowski, A. Are B Lymphocytes of Importance in Severe Staphylococcus aureus Infections? Infect. Immun. 2000, 68, 2431–2434. [Google Scholar] [CrossRef] [Green Version]
- Hansson, M.; Nygren, P.A.K.; Sta˚hl, S. Design and production of recombinant subunit vaccines. Biotechnol. Appl. Biochem. 2000, 32, 95–107. [Google Scholar] [CrossRef]
- Testa, J.S.; Philip, R. Role of T-cell epitope-based vaccine in prophylactic and therapeutic applications. Futur. Virol. 2012, 7, 1077–1088. [Google Scholar] [CrossRef] [Green Version]
- Qamar, M.T.U.; Ahmad, S.; Fatima, I.; Ahmad, F.; Shahid, F.; Naz, A.; Abbasi, S.W.; Khan, A.; Mirza, M.U.; Ashfaq, U.A.; et al. Designing multi-epitope vaccine against Staphylococcus aureus by employing subtractive proteomics, reverse vaccinology and immuno-informatics approaches. Comput. Biol. Med. 2021, 132, 104389. [Google Scholar] [CrossRef] [PubMed]
- Kolla, H.B.; Tirumalasetty, C.; Sreerama, K.; Ayyagari, V.S. An immunoinformatics approach for the design of a multi-epitope vaccine targeting super antigen TSST-1 of Staphylococcus aureus. J. Genet. Eng. Biotechnol. 2021, 19, 69. [Google Scholar] [CrossRef]
- Teymournejad, O.; Montgomery, C.P. Evasion of Immunological Memory by S. aureus Infection: Implications for Vaccine Design. Front. Immunol. 2021, 12, 633672. [Google Scholar] [CrossRef]
- Hajighahramani, N.; Nezafat, N.; Eslami, M.; Negahdaripour, M.; Rahmatabadi, S.S.; Ghasemi, Y. Immunoinformatics analysis and in silico designing of a novel multi-epitope peptide vaccine against Staphylococcus aureus. Infect. Genet. Evol. 2017, 48, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Soltan, M.A.; Magdy, D.; Solyman, S.M.; Hanora, A. Design of Staphylococcus aureus New Vaccine Candidates with B and T Cell Epitope Mapping, Reverse Vaccinology, and Immunoinformatics. OMICS A J. Integr. Biol. 2020, 24, 195–204. [Google Scholar] [CrossRef]
- Glowalla, E.; Tosetti, B.; Krönke, M.; Krut, O. Proteomics-Based Identification of Anchorless Cell Wall Proteins as Vaccine Candidates against Staphylococcus aureus. Infect. Immun. 2009, 77, 2719–2729. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Raghava, G.P.S. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins Struct. Funct. Bioinform. 2006, 65, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved predictions of MHC antigen presentation by concurrent motif deconvolution and integration of MS MHC eluted ligand data. Nucleic Acids Res. 2020, 48, W449–W454. [Google Scholar] [CrossRef] [PubMed]
- Calis, J.J.A.; Maybeno, M.; Greenbaum, J.A.; Weiskopf, D.; De Silva, A.D.; Sette, A.; Keşmir, C.; Peters, B. Properties of MHC Class I Presented Peptides That Enhance Immunogenicity. PLOS Comput. Biol. 2013, 9, e1003266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, M.; Lund, O.; Buus, S.; Lundegaard, C. MHC Class II epitope predictive algorithms. Immunology 2010, 130, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [Green Version]
- Dimitrov, I.; Bangov, I.; Flower, D.R.; Doytchinova, I. AllerTOP v. 2—A server for in silico prediction of allergens. J. Mol. Model. 2014, 20, 2278. [Google Scholar] [CrossRef]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.S.; Open Source Drug Discovery Consortium. In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bui, H.-H.; Sidney, J.; Dinh, K.; Southwood, S.; Newman, M.J.; Sette, A. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform. 2006, 7, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- McGuffin, L.J.; Bryson, K.; Jones, D.T.J.B. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnan, C.N.; Randall, A.; Baldi, P. SOLpro: Accurate sequence-based prediction of protein solubility. Bioinformatics 2009, 25, 2200–2207. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, D.; Lüthy, R.; Bowie, J.U. VERIFY3D: Assessment of protein models with three-dimensional profiles. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1997; Volume 277, pp. 396–404. [Google Scholar] [CrossRef]
- Craig, D.B.; Dombkowski, A.A. Disulfide by Design 2.0: A web-based tool for disulfide engineering in proteins. BMC Bioinform. 2013, 14, 346. [Google Scholar] [CrossRef] [Green Version]
- Ponomarenko, J.V.; Bui, H.-H.; Li, W.; Fusseder, N.; Bourne, P.E.; Sette, A.; Peters, B. ElliPro: A new structure-based tool for the prediction of antibody epitopes. BMC Bioinform. 2008, 9, 514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhanda, S.K.; Vir, P.; Raghava, G.P. Designing of interferon-gamma inducing MHC class-II binders. Biol. Direct 2013, 8, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; Boelens, R.; Bonvin, A.M.J.J. HADDOCK: A Protein−Protein Docking Approach Based on Biochemical or Biophysical Information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khatoon, N.; Pandey, R.K.; Prajapati, V.K. Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci. Rep. 2017, 7, 8285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pian, Y.; Chen, S.; Hao, H.; Zheng, Y.; Zhu, L.; Xu, B.; Liu, K.; Li, M.; Jiang, H.; Jiang, Y. Phenol-soluble modulin α4 mediates Staphylococcus aureus-associated vascular leakage by stimulating heparin-binding protein release from neutrophils. Sci. Rep. 2016, 6, 29373. [Google Scholar]
- Peschel, A.; Otto, M. Phenol-soluble modulins and staphylococcal infection. Nat. Rev. Microbiol. 2013, 11, 667–673. [Google Scholar] [CrossRef]
- Mahapatra, S.R.; Sahoo, S.; Dehury, B.; Raina, V.; Patro, S.; Misra, N.; Suar, M. Designing an efficient multi-epitope vaccine displaying interactions with diverse HLA molecules for an efficient humoral and cellular immune response to prevent COVID-19 infection. Expert Rev. Vaccines 2020, 19, 871–885. [Google Scholar] [CrossRef] [PubMed]
- Deller, M.C.; Kong, L.; Rupp, B. Protein stability: A crystallographer’s perspective. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016, 72, 72–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dombkowski, A.A.; Sultana, K.Z.; Craig, D.B. Protein disulfide engineering. FEBS Lett. 2014, 588, 206–212. [Google Scholar] [CrossRef] [Green Version]
- Yimin; Kohanawa, M.; Zhao, S.; Ozaki, M.; Haga, S.; Nan, G.; Kuge, Y.; Tamaki, N. Contribution of Toll-Like Receptor 2 to the Innate Response against Staphylococcus aureus Infection in Mice. PLoS ONE 2013, 8, e74287. [Google Scholar] [CrossRef]
- Kielian, T.; Haney, A.; Mayes, P.M.; Garg, S.; Esen, N. Toll-Like Receptor 2 Modulates the Proinflammatory Milieu in Staphylococcus aureus -Induced Brain Abscess. Infect. Immun. 2005, 73, 7428–7435. [Google Scholar] [CrossRef] [Green Version]
- Musilova, J.; Mulcahy, M.E.; Kuijk, M.M.; McLoughlin, R.M.; Bowie, A.G. Toll-like receptor 2–dependent endosomal signaling by Staphylococcus aureus in monocytes induces type I interferon and promotes intracellular survival. J. Biol. Chem. 2019, 294, 17031–17042. [Google Scholar] [CrossRef] [Green Version]
- Efournier, B. The function of TLR2 during staphylococcal diseases. Front. Cell. Infect. Microbiol. 2013, 2, 167. [Google Scholar] [CrossRef] [Green Version]
- Tong, S.Y.C.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G., Jr. Staphylococcus aureus Infections: Epidemiology, Pathophysiology, Clinical Manifestations, and Management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, J.; Mahapatra, S.R.; Singh, P.; Patro, S.; Kushwaha, G.S.; Misra, N.; Suar, M. B and T cell epitope-based peptides predicted from clumping factor protein of Staphylococcus aureus as vaccine targets. Microb. Pathog. 2021, 160, 105171. [Google Scholar] [CrossRef] [PubMed]
- Chambers, H.F.; DeLeo, F.R. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 2009, 7, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, R.; Sahoo, P.; Mahapatra, S.R.; Dey, J.; Ghosh, M.; Kushwaha, G.S.; Misra, N.; Suar, M.; Raina, V.; Son, Y.-O. Development of a Conserved Chimeric Vaccine for Induction of Strong Immune Response against Staphylococcus aureus Using Immunoinformatics Approaches. Vaccines 2021, 9, 1038. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Davies, D. Origins and Evolution of Antibiotic Resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [Green Version]
- Serruto, D.; Rappuoli, R. Post-genomic vaccine development. FEBS Lett. 2006, 580, 2985–2992. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.; Junaid, M.; Kaushik, A.C.; Ali, A.; Ali, S.S.; Mehmood, A.; Wei, D.-Q. Computational identification, characterization and validation of potential antigenic peptide vaccines from hrHPVs E6 proteins using immunoinformatics and computational systems biology approaches. PLoS ONE 2018, 13, e0196484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.; Khan, A.; Rehman, A.U.; Ahmad, I.; Ullah, S.; Khan, A.A.; Ali, S.S.; Afridi, S.G.; Wei, D.-Q. Immunoinformatics and structural vaccinology driven prediction of multi-epitope vaccine against Mayaro virus and validation through in-silico expression. Infect. Genet. Evol. 2019, 73, 390–400. [Google Scholar] [CrossRef] [PubMed]
- Dey, J.; Mahapatra, S.R.; Raj, T.K.; Kaur, T.; Jain, P.; Tiwari, A.; Patro, S.; Misra, N.; Suar, M. Designing a novel multi-epitope vaccine to evoke a robust immune response against pathogenic multidrug-resistant Enterococcus faecium bacterium. Gut Pathog. 2022, 14, 21. [Google Scholar] [CrossRef] [PubMed]
- Dey, J.; Mahapatra, S.R.; Patnaik, S.; Lata, S.; Kushwaha, G.S.; Panda, R.K.; Misra, N.; Suar, M. Molecular Characterization and Designing of a Novel Multiepitope Vaccine Construct Against Pseudomonas aeruginosa. Int. J. Pept. Res. Ther. 2022, 28, 49. [Google Scholar] [CrossRef] [PubMed]
- Oli, A.N.; Obialor, W.O.; Ifeanyichukwu, M.O.; Odimegwu, D.C.; Okoyeh, J.N.; Emechebe, G.O.; Adejumo, S.A.; Ibeanu, G.C. Immunoinformatics and Vaccine Development: An Overview. ImmunoTargets Ther. 2020, 9, 13–30. [Google Scholar] [CrossRef] [Green Version]
- Dey, J.; Mahapatra, S.R.; Lata, S.; Patro, S.; Misra, N.; Suar, M. Exploring Klebsiella pneumoniae capsule polysaccharide proteins to design multiepitope subunit vaccine to fight against pneumonia. Expert Rev. Vaccines 2022, 21, 569–587. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, S.R.; Dey, J.; Kaur, T.; Sarangi, R.; Bajoria, A.A.; Kushwaha, G.S.; Misra, N.; Suar, M. Immunoinformatics and molecular docking studies reveal a novel Multi-Epitope peptide vaccine against pneumonia infection. Vaccine 2021, 39, 6221–6237. [Google Scholar] [CrossRef] [PubMed]
- Reed, S.G.; Hsu, F.-C.; Carter, D.; Orr, M.T. The science of vaccine adjuvants: Advances in TLR4 ligand adjuvants. Curr. Opin. Immunol. 2016, 41, 85–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahapatra, S.R.; Dey, J.; Kushwaha, G.S.; Puhan, P.; Mohakud, N.K.; Panda, S.K.; Lata, S.; Misra, N.; Suar, M. Immunoinformatic approach employing modeling and simulation to design a novel vaccine construct targeting MDR efflux pumps to confer wide protection against typhoidal Salmonella serovars. J. Biomol. Struct. Dyn. 2021, 40, 11809–11821. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Uniprot_ID | B Cell Epitope | Position | Score | Antigencity Score | Toxicity | Hydrophobicity | Hydropathicity | Hydrophilicity | Charge | Mol wt. |
|---|---|---|---|---|---|---|---|---|---|---|
| Q53654 | LKFMVFIMLL | 6 | 0.69 | 2.1966 | NON-TOXIN | 0.35 | 2.56 | −1.33 | 1 | 1254.86 |
| KDNQDGKRPE | 546 | 0.67 | 2.3986 | NON-TOXIN | −0.73 | −3.18 | 1.84 | 0 | 1186.38 |
| Uniprot_ID | CTL Epitope | Alleles | Position | Score | Antigencity Score | Immunogenicity | Toxicity | Hydrophobicity | Hydropathicity | Hydrophilicity | Charge | Mol wt. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Q53654 | TPDGATITF | HLA-A*01:01, HLA-A*02:01, HLA-A*03:01, HLA-A*24:02, HLA-A*26:01, HLA-B*07:02, HLA-B*08:01, HLA-B*27:05, HLA-B*39:01, HLA-B*40:01, HLA-B*58:01, HLA-B*15:01 | 107 | 1.279 | 0.9339 | 0.25094 | NON-TOXIN | 0.05 | 0.17 | −0.33 | −1 | 922.12 |
| KVNGYTTHV | HLA-A*02:01, HLA-A*01:01, HLA-A*03:01, HLA-A*24:02, HLA-A*26:01, HLA-B*07:02, HLA-B*08:01, HLA-B*27:05, HLA-B*39:01, HLA-B*40:01, HLA-B*58:01, HLA-B*15:01 | 1069 | 0.226 | 1.5081 | 0.1166 | NON-TOXIN | −0.14 | −0.59 | −0.38 | 1.5 | 1018.27 | |
| TTPEGYTKK | HLA-A*03:01, HLA-A*01:01, HLA-A*02:01, HLA-A*24:02, HLA-A*26:01, HLA-B*07:02, HLA-B*08:01, HLA-B*27:05, HLA-B*39:01, HLA-B*40:01, HLA-B*58:01, HLA-B*15:01 | 511 | 0.996 | 0.8267 | 0.03343 | NON-TOXIN | −0.36 | −1.86 | 0.61 | 1 | 1024.26 | |
| KFMVFIMLL | HLA-A*24:02, HLA-A*01:01, HLA-A*02:01, HLA-A*03:01, HLA-A*26:01, HLA-B*07:02, HLA-B*08:01, HLA-B*27:05, HLA-B*39:01, HLA-B*40:01, HLA-B*58:01, HLA-B*15:01 | 7 | 0.635 | 1.7858 | 0.06914 | NON-TOXIN | 0.33 | 2.42 | −1.28 | 1 | 1141.68 | |
| KPTIYFKLY | HLA-B*07:02, HLA-A*01:01, HLA-A*02:01, HLA-A*03:01, HLA-A*24:02, HLA-A*26:01, HLA-B*08:01, HLA-B*27:05, HLA-B*39:01, HLA-B*40:01, HLA-B*58:01, HLA-B*15:01 | 448 | 2.837 | 1.6477 | 0.06464 | NON-TOXIN | −0.06 | −0.18 | −0.57 | 2 | 1172.56 | |
| EFEVQGRNL | HLA-B*08:01, HLA-A*01:01, HLA-A*02:01, HLA-A*03:01, HLA-A*24:02, HLA-A*26:01, HLA-B*07:02, HLA-B*27:05, HLA-B*39:01, HLA-B*40:01, HLA-B*58:01, HLA-B*15:01 | 130 | 1.717 | 1.6709 | 0.03304 | NON-TOXIN | −0.28 | −0.9 | 0.4 | −1 | 1091.32 | |
| VKPTIYFKL | HLA-B*27:05, HLA-A*01:01, HLA-A*02:01, HLA-A*03:01, HLA-A*24:02, HLA-A*26:01, HLA-B*07:02, HLA-B*08:01, HLA-B*39:01, HLA-B*40:01, HLA-B*58:01, HLA-B*15:01 | 447 | 3.894 | 1.6698 | 0.13438 | NON-TOXIN | 0 | 0.43 | −0.48 | 2 | 1108.52 | |
| TKNTIDVTI | HLA-B*39:01, HLA-A*01:01, HLA-A*02:01, HLA-A*03:01, HLA-A*24:02, HLA-A*26:01, HLA-B*07:02, HLA-B*08:01, HLA-B*27:05, HLA-B*40:01, HLA-B*58:01, HLA-B*15:01 | 257 | 0.65 | 0.9440 | 0.24496 | NON-TOXIN | −0.11 | 0.02 | −0.01 | 0 | 1004.28 | |
| IEYTVTEDH | HLA-B*40:01, HLA-A*01:01, HLA-A*02:01, HLA-A*03:01, HLA-A*24:02, HLA-A*26:01, HLA-B*07:02, HLA-B*08:01, HLA-B*27:05, HLA-B*39:01, HLA-B*58:01, HLA-B*15:01 | 969 | 2.678 | 1.1265 | 0.21206 | NON-TOXIN | −0.16 | −0.86 | 0.23 | −2.5 | 1106.28 | |
| LLNIITPLF | HLA-B*15:01, HLA-A*01:01, HLA-A*02:01, HLA-A*03:01, HLA-A*24:02, HLA-A*26:01, HLA-B*07:02, HLA-B*08:01, HLA-B*27:05, HLA-B*39:01, HLA-B*40:01, HLA-B*58:01 | 14 | 0.567 | 0.6552 | 0.28212 | NON-TOXIN | 0.31 | 1.93 | −1.3 | 0 | 1043.46 |
| Uniprot_ID | MHC II Epitope | Alleles | Pos | IC50 Value | Percentile_Rank | Antigencity Score | Toxicity | Hydrophobicity | Hydropathicity | Hydrophilicity | Charge | Mol wt. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Q53654 | NVLKFMVFIMLLNII | HLA-DPA1*01:03, HLA-DPB1*02:01, HLA-DPB1*01:01, HLA-DRB1*01:01, HLA-DRB1*09:01,HLA-DRB3*02:02, HLA-DRB1*13:02, HLA-DRB1*11:01, HLA-DRB1*04:01, HLA-DRB1*12:01, HLA-DPA1*03:01, HLA-DPB1*04:02, HLA-DRB1*04:05, HLA-DRB1*15:01, HLA-DQA1*01:01, HLA-DQB1*05:01, HLA-DRB1*08:02, HLA-DPA1*02:01, HLA-DPB1*14:01, HLA-DPB1*04:01, HLA-DQA1*05:01, HLA-DQB1*03:01, HLA-DQA1*04:01, HLA-DQB1*04:02, HLA-DPA1*02:01, HLA-DPA1*02:01, HLA-DPB1*05:01, HLA-DPA1*01:03, HLA-DQA1*05:01, HLA-DQB1*02:01, HLA-DQA1*03:01, HLA-DQB1*03:02, HLA-DQA1*01:02, HLA-DQB1*06:02, HLA-DRB3*01:01, HLA-DRB5*01:01, HLA-DRB1*07:01, HLA-DRB4*01:01, HLA-DRB1*03:01 | 4–18 | 36 | 0.21 | 1.2706 | NON-TOXIN | 0.28 | 2.12 | −1.2 | 1 | 1808.61 |
| VLKFMVFIMLLNIIT | HLA-DRB1*15:01, HLA-DRB3*01:01, HLA-DPA1*03:01, HLA-DPB1*04:02, HLA-DPA1*01:03, HLA-DPB1*02:01,HLA-DRB1*01:01, HLA-DRB1*09:01,HLA-DRB3*02:02, HLA-DRB1*13:02, HLA-DRB1*11:01, HLA-DRB1*04:01, HLA-DRB1*12:01,HLA-DRB1*04:05, HLA-DQA1*01:01, HLA-DQB1*05:01, HLA-DRB1*08:02, HLA-DPA1*02:01, HLA-DPB1*14:01, HLA-DPA1*01:03, HLA-DPB1*04:01, HLA-DQA1*05:01, HLA-DQB1*03:01, HLA-DQA1*04:01, HLA-DQB1*04:02, HLA-DPA1*02:01, HLA-DPB1*01:01, HLA-DPA1*02:01, HLA-DPB1*05:01,HLA-DQA1*05:01, HLA-DQB1*02:01, HLA-DQA1*03:01, HLA-DQB1*03:02, HLA-DQA1*01:02, HLA-DQB1*06:02, HLA-DRB5*01:01, HLA-DRB1*07:01, HLA-DRB4*01:01, HLA-DRB1*03:01 | 5–19 | 50 | 0.42 | 1.3372 | Non toxin | 0.31 | 2.31 | −1.24 | 1 | 1795.61 |
| Energies | Energies (KJ/mol) | Std. Dev (KJ/mol) |
|---|---|---|
| van der Waal energy | −179.773 | +/−56.735 |
| Electrostatic energy | −26.076 | +/−17.625 |
| Polar solvation energy | 116.241 | +/−34.842 |
| SASA energy | −20.333 | +/−3.782 |
| Total Binding energy | −109.941 | +/−15.971 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mahapatra, S.R.; Dey, J.; Raj, T.K.; Misra, N.; Suar, M. Designing a Next-Generation Multiepitope-Based Vaccine against Staphylococcus aureus Using Reverse Vaccinology Approaches. Pathogens 2023, 12, 376. https://doi.org/10.3390/pathogens12030376
Mahapatra SR, Dey J, Raj TK, Misra N, Suar M. Designing a Next-Generation Multiepitope-Based Vaccine against Staphylococcus aureus Using Reverse Vaccinology Approaches. Pathogens. 2023; 12(3):376. https://doi.org/10.3390/pathogens12030376
Chicago/Turabian StyleMahapatra, Soumya Ranjan, Jyotirmayee Dey, T. Kiran Raj, Namrata Misra, and Mrutyunjay Suar. 2023. "Designing a Next-Generation Multiepitope-Based Vaccine against Staphylococcus aureus Using Reverse Vaccinology Approaches" Pathogens 12, no. 3: 376. https://doi.org/10.3390/pathogens12030376