
Complete Genome Sequence and Analysis of a ST573 Multidrug-Resistant Methicillin-Resistant Staphylococcus aureus SauR3 Clinical Isolate from Terengganu, Malaysia
 ,
,  , , , , ,
, , , , ,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. Background of the S. aureus SauR3 Isolate
2.2. Genome Features and Molecular Typing of SauR3
2.3. Prediction of Antimicrobial Resistance Genes from the SauR3 Genome

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antimicrobial Class | Resistance Phenotype | Detected Resistance Phenotype * | Resistance Genotype | Nucleotide Coordinates ‡ | Location of the Resistance Gene | Mechanism of Resistance |
|---|---|---|---|---|---|---|
| β-lactams | Penicillin | R | blaZ family | 24,826–23,981 | pSauR3-1 | Antibiotic inactivation enzyme |
| Cefoxitin, oxacillin, cefoperazone | R | mecA | 1,987,326–1,985,317 | Chromosomal/SCCmec | Antibiotic target alteration | |
| Fluoroquinolones | Ciprofloxacin, moxifloxacin | R | norA, norC, sdrM | 1,284,437–1,283,271 1,927,502–1,926,114 2,609,957–2,611,300 | Chromosomal | MFS efflux pump |
| Macrolides | Erythromycin | R (iMLSB) | ermC | 1407–2141 | pSauR3-3 | Antibiotic target alteration |
| lmrS | 2,612,195–2,613,637 | Chromosomal | Efflux pump | |||
| msrA | 33,114–31,648 | pSauR3-1 | Antibiotic target protection | |||
| mphC | 31,549–30,650 | pSauR3-1 | Antibiotic inactivation enzyme | |||
| Lincosamide | Clindamycin | R (iMLSB) | ermC | 1407–2141 | pSauR3-3 | Antibiotic target alteration |
| Aminoglycosides | Gentamicin | R | aph(3′)-IIIa, aadE | 35,670–36,464 | pSauR3-1 | Antibiotic inactivation enzyme |
| aac(6′)Ie-aph(2″)Ia | 1,961,711–1,963,150 | Chromosomal/SCCmec | Antibiotic inactivation enzyme | |||
| Fosfomycin | Fosfomycin | S | fosB | 2,465,206–2,465,625 | Chromosomal | Antibiotic inactivation enzyme |
| Atypical aminoglycoside | Streptothricin | ND | SAT-4 | 35,035–35,577 | pSauR3-1 | Antibiotic inactivation enzyme |
2.4. Prediction of Virulence Genes in the SauR3 Genome
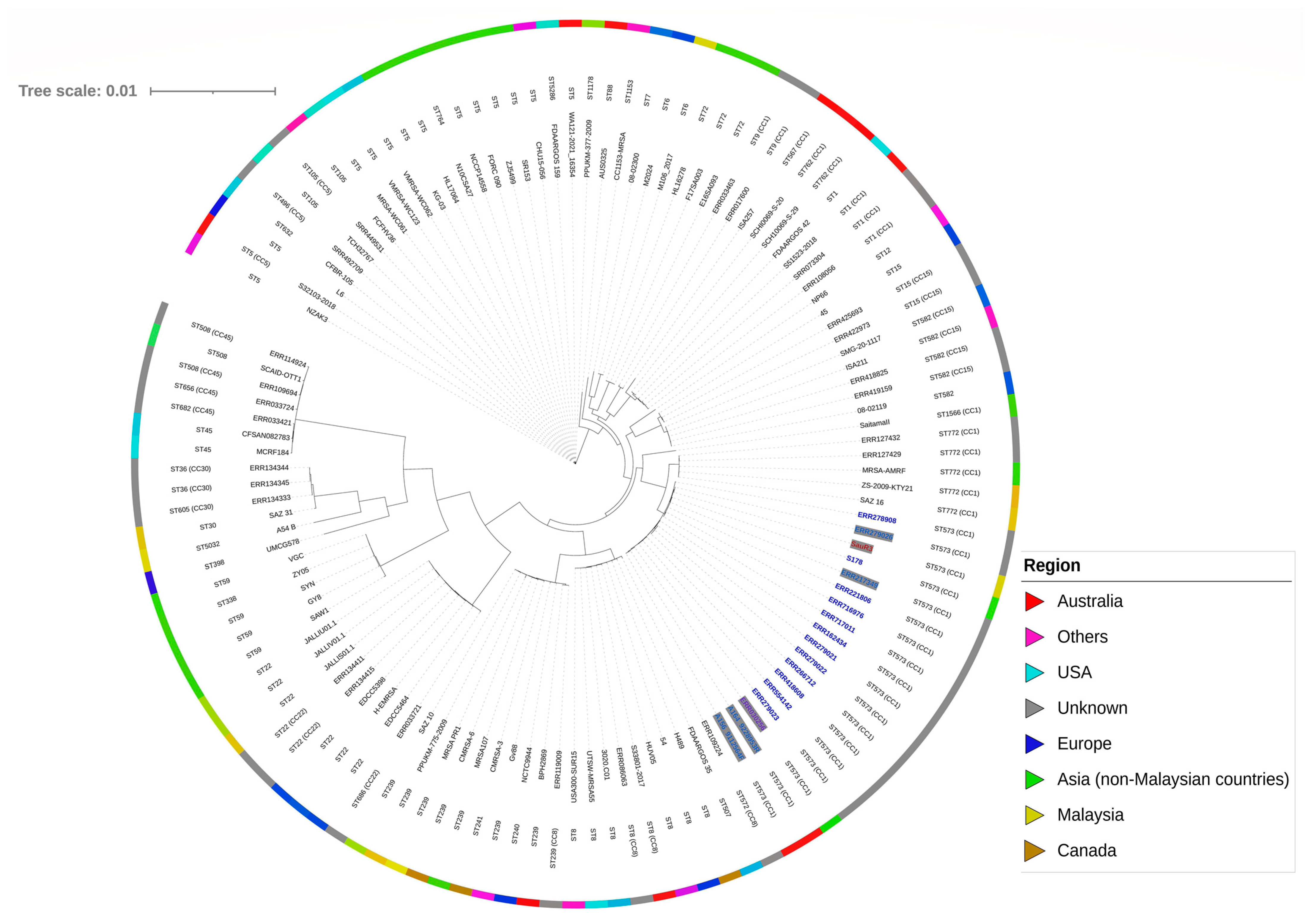
2.5. Phylogenetic Analysis of SauR3
2.6. The SCCmec Element in SauR3
2.7. Genomic Islands in SauR3
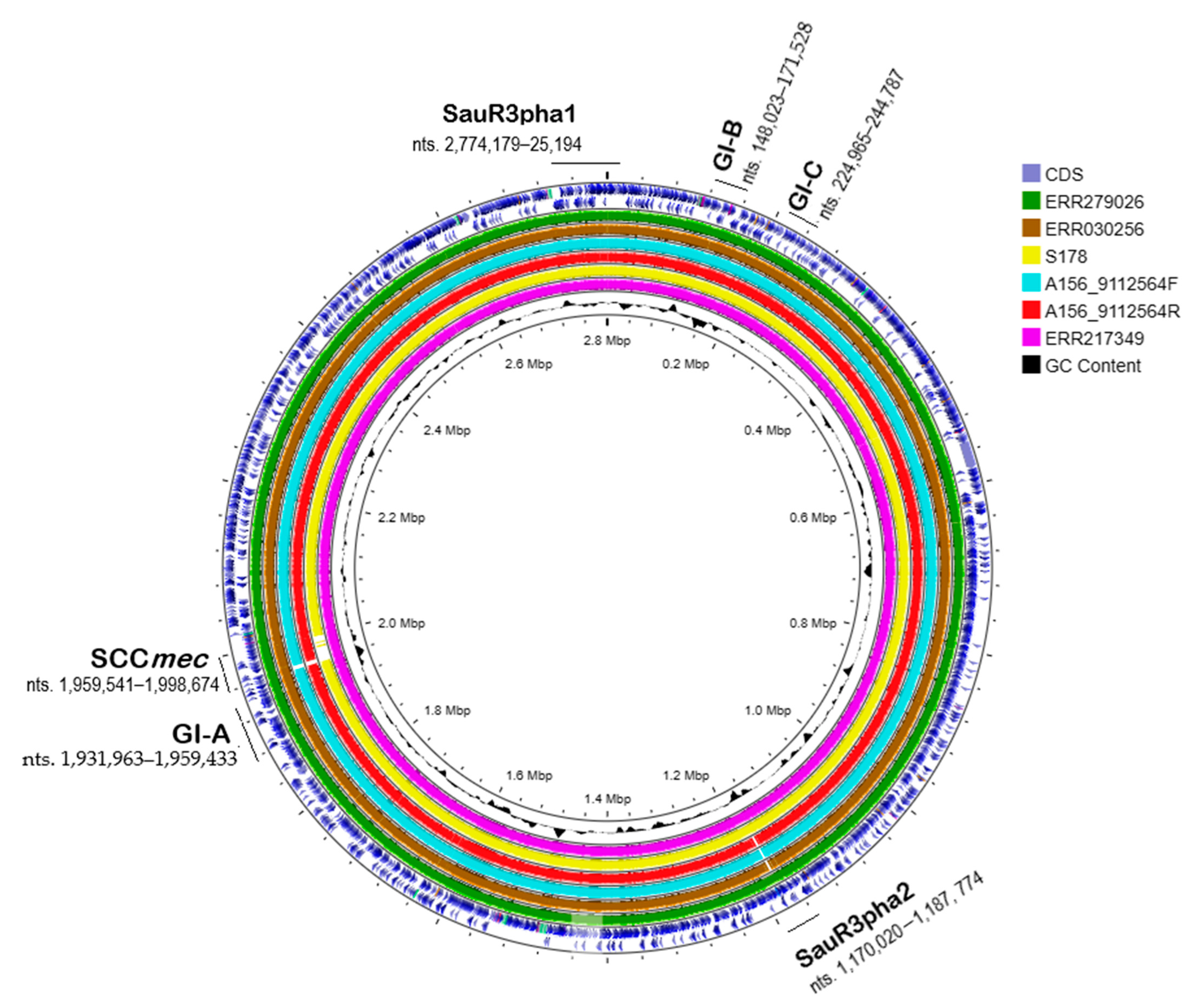
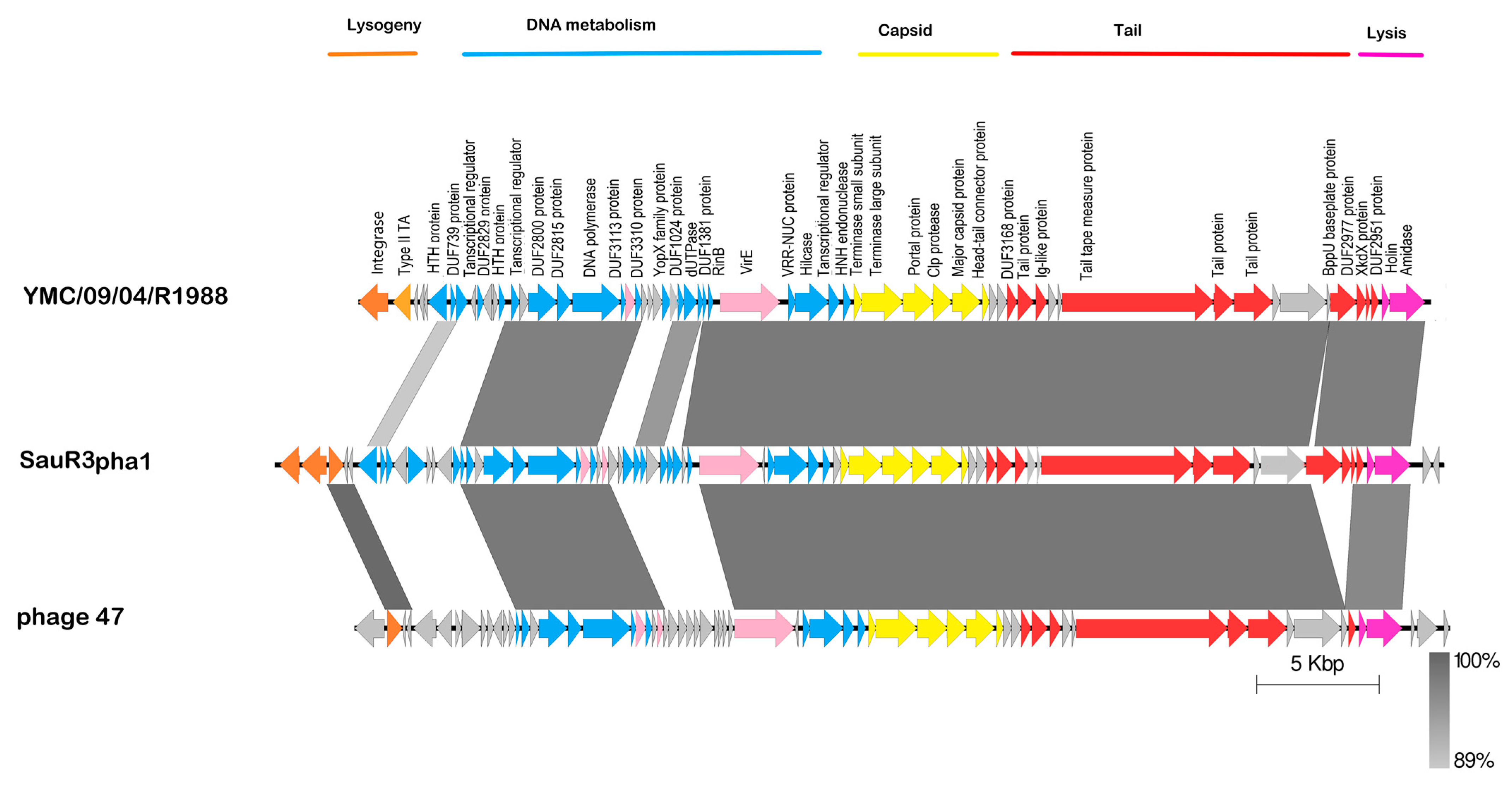
2.8. Prophages in SauR3
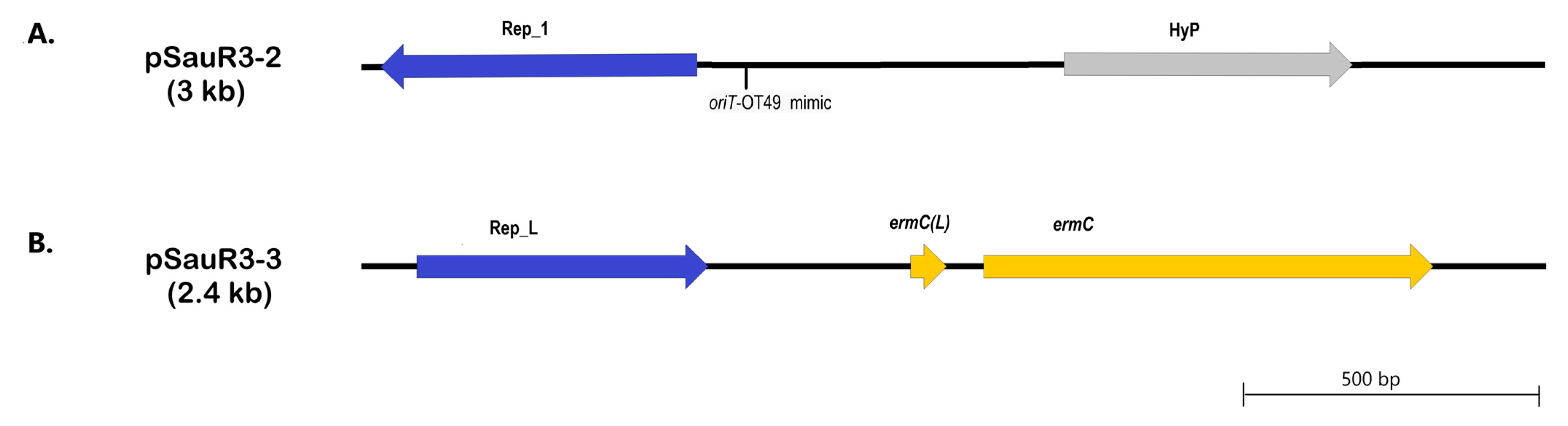
2.9. Plasmids in SauR3
3. Concluding Remarks
4. Materials and Methods
4.1. Ethical Approval
4.2. Growth of S. aureus SauR3
4.3. Genomic DNA (gDNA) Extraction
4.4. Genome Sequencing, Assembly and Annotation
4.5. Computational Genome Analysis
4.6. Phylogenetic Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Niek, W.K.; Teh, C.S.J.; Idris, N.; Sit, P.S.; Lee, Y.Q.; Thong, K.L.; Sri La Sri Ponnampalavanar, S. Methicillin-Resistant Staphylococcus aureus Bacteraemia, 2003–2015: Comparative Evaluation of Changing Trends in Molecular Epidemiology and Clinical Outcomes of Infections. Infect. Genet. Evol. 2020, 85, 104567. [Google Scholar] [CrossRef]
- Sit, P.S.; Teh, C.S.J.; Idris, N.; Ponnampalavanar, S. Methicillin-Resistant Staphylococcus aureus (MRSA) Bacteremia: Correlations between Clinical, Phenotypic, Genotypic Characteristics and Mortality in a Tertiary Teaching Hospital in Malaysia. Infect. Genet. Evol. 2018, 59, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, D.; Borges, A.; Simões, M. Staphylococcus aureus Toxins and Their Molecular Activity in Infectious Diseases. Toxins 2018, 10, 252. [Google Scholar] [CrossRef] [Green Version]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, Research, and Development of New Antibiotics: The WHO Priority List of Antibiotic-Resistant Bacteria and Tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Che Hamzah, A.M.; Yeo, C.C.; Puah, S.M.; Chua, K.H.; Rahman, N.I.A.; Abdullah, F.H.; Othman, N.; Chew, C.H. Tigecycline and Inducible Clindamycin Resistance in Clinical Isolates of Methicillin-Resistant Staphylococcus aureus from Terengganu, Malaysia. J. Med. Microbiol. 2019, 68, 1299–1305. [Google Scholar] [CrossRef]
- Lee, A.S.; De Lencastre, H.; Garau, J.; Kluytmans, J.; Malhotra-Kumar, S.; Peschel, A.; Harbarth, S. Methicillin-Resistant Staphylococcus aureus. Nat. Rev. 2018, 4, 18033. [Google Scholar] [CrossRef] [Green Version]
- Uehara, Y. Current Status of Staphylococcal Cassette Chromosome Mec (SCCmec). Antibiotics 2022, 11, 86. [Google Scholar] [CrossRef]
- Wang, W.; Hu, Y.; Baker, M.; Dottorini, T.; Li, H.; Dong, Y.; Bai, Y.; Fanning, S.; Li, F. Novel SCCmec Type XV (7A) and Two Pseudo-SCCmec Variants in Foodborne MRSA in China. J. Antimicrob. Chemother. 2022, 77, 903–909. [Google Scholar] [CrossRef]
- Malachowa, N.; Deleo, F.R. Mobile Genetic Elements of Staphylococcus aureus. Cell. Mol. Life Sci. 2010, 67, 3057–3071. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.J.; Huang, Y.C. New Epidemiology of Staphylococcus aureus Infection in Asia. Clin. Microbiol. Infect. 2014, 20, 605–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Qiu, L.; Zheng, X.; Liu, J. Trend in Antimicrobial Resistance of Staphylococcus aureus: Results from the China Antimicrobial Surveillance Network (CHINET) in the Last 15-Year-Period Reports (2005–2019). Infect. Drug Resist. 2021, 14, 2179–2181. [Google Scholar] [CrossRef] [PubMed]
- Ministry of Health Malaysia Ministry of Health Malaysia. National Antibiotic Resistance Surveillance Report 2021. Available online: https://myohar.moh.gov.my/reports-human-health/ (accessed on 1 December 2022). [CrossRef]
- Cunningham, S.A.; Chia, N.; Jeraldo, P.R.; Quest, D.J.; Johnson, J.A.; Boxrud, D.J.; Taylor, A.J.; Chen, J.; Jenkins, G.D.; Drucker, T.M.; et al. Comparison of Whole-Genome Sequencing Methods for Analysis of Staphylococcus aureus Outbreaks. J. Clin. Microbiol. 2017, 55, 1946–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Talib, H.; Samsudin, S.; Adnan, A.; Murugaiah, C. Genetic Diversity among Methicillin-Resistant Staphylococcus aureus in Malaysia (2002–2020). Trop. Med. Infect. Dis. 2022, 7, 360. [Google Scholar] [CrossRef]
- Che Hamzah, A.M.; Yeo, C.C.; Puah, S.M.; Chua, K.H.; Chew, C.H. Staphylococcus aureus aphylococcus Aureus Infections in Malaysia: A Review of Antimicrobial Resistance and Characteristics of the Clinical Isolates, 1990–2017. Antibiotics 2019, 8, 128. [Google Scholar] [CrossRef] [Green Version]
- Che Hamzah, A.M.; Al-Trad, E.; Puah, S.M.; Rahman, N.I.A.; Ismail, S.; Yeo, C.C.; Chew, C. PGP-005—Whole Genome Sequence Analysis of Methicillin-Resistant Staphylococcus aureus Indicates Predominance of the EMRSA-15 (ST22-SCCmec IV [2b]) Clone in Terengganu, Malaysia. Int. J. Antimicrob. Agents 2021, 58, 21004039. [Google Scholar] [CrossRef]
- Hsu, L.Y.; Harris, S.R.; Chlebowicz, M.A.; Lindsay, J.A.; Koh, T.H.; Krishnan, P.; Tan, T.Y.; Hon, P.Y.; Grubb, W.B.; Bentley, S.D.; et al. Evolutionary Dynamics of Methicillin-Resistant Staphylococcus aureus within a Healthcare System. Genome Biol. 2015, 16, 81. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.T.; Hanifah, Y.A.; Yusof, M.Y.M.; Goering, R.V.; Thong, K.L. Temporal Changes in the Genotypes of Methicillin-Resistant Staphylococcus aureus Strains Isolated from a Tertiary Malaysian Hospital Based on MLST, Spa, and Mec-Associated Dru Typing. Diagn. Microbiol. Infect. Dis. 2012, 74, 106–112. [Google Scholar] [CrossRef]
- Magiorakos, A.P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-Resistant, Extensively Drug-Resistant and Pandrug-Resistant Bacteria: An International Expert Proposal for Interim Standard Definitions for Acquired Resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef] [Green Version]
- Miklasińska-Majdanik, M. Mechanisms of Resistance to Macrolide Antibiotics among Staphylococcus aureus. Antibiotics 2021, 10, 1406. [Google Scholar] [CrossRef]
- Ali, M.S.; Isa, N.M.; Abedelrhman, F.M.; Alyas, T.B.; Mohammed, S.E.; Ahmed, A.E.; Ahmed, Z.S.A.; Lau, N.S.; Garbi, M.I.; Amirul, A.A.A.; et al. Genomic Analysis of Methicillin-Resistant Staphylococcus aureus Strain SO-1977 from Sudan. BMC Microbiol. 2019, 19, 126. [Google Scholar] [CrossRef] [PubMed]
- Ngoi, S.T.; Niek, W.K.; Lee, Y.W.; AbuBakar, S.; Teh, C.S.J. Genomic Analysis Revealed a Novel Genotype of Methicillin-Susceptible Staphylococcus aureus Isolated from a Fatal Sepsis Case in Dengue Patient. Sci. Rep. 2021, 11, 4228. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.U.; Chua, K.H.; Chew, C.H.; Yeo, C.C.; Abdullah, F.H.; Othman, N.; Kee, B.P.; Puah, S.M. Spa Diversity of Methicillin-Resistant and -Susceptible Staphylococcus aureus in Clinical Strains from Malaysia: A High Prevalence of Invasive European Spa-Type T032. PeerJ 2021, 9, e11195. [Google Scholar] [CrossRef] [PubMed]
- Nimmo, G.R.; Coombs, G.W. Community-Associated Methicillin-Resistant Staphylococcus aureus (MRSA) in Australia. Int. J. Antimicrob. Agents 2008, 31, 401–410. [Google Scholar] [CrossRef]
- Sheng, W.H.; Wang, J.T.; Lauderdale, T.L.; Weng, C.M.; Chen, D.; Chang, S.C. Epidemiology and Susceptibilities of Methicillin-Resistant Staphylococcus aureus in Taiwan: Emphasis on Chlorhexidine Susceptibility. Diagn. Microbiol. Infect. Dis. 2009, 63, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Costa, S.S.; Viveiros, M.; Amaral, L.; Couto, I. Multidrug Efflux Pumps in Staphylococcus aureus: An Update. Open Microbiol. J. 2013, 7, 59–71. [Google Scholar] [CrossRef] [Green Version]
- Hassanzadeh, S.; Ganjloo, S.; Pourmand, M.R.; Mashhadi, R.; Ghazvini, K. Epidemiology of Efflux Pumps Genes Mediating Resistance among Staphylococcus aureus: A Systematic Review. Microb. Pathog. 2020, 139, 103850. [Google Scholar] [CrossRef] [PubMed]
- Bruce, S.A.; Smith, J.T.; Mydosh, J.L.; Ball, J.; Needle, D.B.; Gibson, R.; Andam, C.P. Shared Antibiotic Resistance and Virulence Genes in Staphylococcus aureus from Diverse Animal Hosts. Sci. Rep. 2022, 12, 4413. [Google Scholar] [CrossRef] [PubMed]
- Floyd, J.L.; Smith, K.P.; Kumar, S.H.; Floyd, J.T.; Varela, M.F. LmrS Is a Multidrug Efflux Pump of the Major Facilitator Superfamily from Staphylococcus aureus. Antimicrob. Agents Chemother. 2010, 54, 5406–5412. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, C.; Hooper, D.C. Transcriptional Regulator TetR21 Controls the Expression of the Staphylococcus aureus LmrS Efflux Pump. Antimicrob. Agents Chemother. 2017, 61, e00649-17. [Google Scholar]
- Lina, G.; Quaglia, A.; Reverdy, M.E.; Leclercq, R.; Vandenesch, F.; Etienne, J. Distribution of Genes Encoding Resistance to Macrolides, Lincosamides, and Streptogramins among Staphylococci. Antimicrob. Agents Chemother. 1999, 43, 1062–1066. [Google Scholar] [CrossRef] [Green Version]
- Jarajreh, D.; Aqel, A.; Alzoubi, H.; Al-Zereini, W. Prevalence of Inducible Clindamycin Resistance in Methicillin-Resistant Staphylococcus aureus: The First Study in Jordan. J. Infect. Dev. Ctries. 2017, 11, 350–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calderwood, M.S.; Desjardins, C.A.; Sakoulas, G.; Nicol, R.; Dubois, A.; Delaney, M.L.; Kleinman, K.; Cosimi, L.A.; Feldgarden, M.; Onderdonk, A.B.; et al. Staphylococcal Enterotoxin P Predicts Bacteremia in Hospitalized Patients Colonized with Methicillin-Resistant Staphylococcus aureus. J. Infect. Dis. 2014, 209, 571–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujol, M.; Peña, C.; Pallares, R.; Ariza, J.; Ayats, J.; Dominguez, M.A.; Gudiol, F. Nosocomial Staphylococcus aureus Bacteremia among Nasal Carriers of Methicillin-Resistant and Methicillin-Susceptible Strains. Am. J. Med. 1996, 100, 509–516. [Google Scholar] [CrossRef] [Green Version]
- Munro, P.; Benchetrit, M.; Nahori, M.A.; Stefani, C.; Clément, R.; Michiels, J.F.; Landraud, L.; Dussurget, O.; Lemichez, E. The Staphylococcus aureus Epidermal Cell Differentiation Inhibitor Toxin Promotes Formation of Infection Foci in a Mouse Model of Bacteremia. Infect. Immun. 2010, 78, 3404–3411. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.K.; Dhasmana, N.; Dubey, N.; Kumar, N.; Gangwal, A.; Gupta, M.; Singh, Y. Bacterial Virulence Factors: Secreted for Survival. Indian J. Microbiol. 2017, 57, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Otto, M. MRSA Virulence and Spread. Cell. Microbiol. 2012, 14, 1513–1521. [Google Scholar] [CrossRef] [Green Version]
- McClure, J.A.M.; Lakhundi, S.; Kashif, A.; Conly, J.M.; Zhang, K. Genomic Comparison of Highly Virulent, Moderately Virulent, and Avirulent Strains from a Genetically Closely-Related MRSA ST239 Sub-Lineage Provides Insights into Pathogenesis. Front. Microbiol. 2018, 9, 1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monecke, S.; Baier, V.; Coombs, G.W.; Slickers, P.; Ziegler, A.; Ehricht, R. Genome Sequencing and Molecular Characterisation of Staphylococcus aureus ST772-MRSA-V, “Bengal Bay Clone”. BMC Res. Notes 2013, 6, 548. [Google Scholar] [CrossRef] [Green Version]
- Blomfeldt, A.; Larssen, K.W.; Moghen, A.; Gabrielsen, C.; Elstrøm, P.; Aamot, H.V.; Jørgensen, S.B. Emerging Multidrug-Resistant Bengal Bay Clone ST772-MRSA-V in Norway: Molecular Epidemiology 2004–2014. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 1911–1921. [Google Scholar] [CrossRef]
- Earls, M.R.; Steinig, E.J.; Monecke, S.; Samaniego Castruita, J.A.; Simbeck, A.; Schneider-Brachert, W.; Vremerǎ, T.; Dorneanu, O.S.; Loncaric, I.; Bes, M.; et al. Exploring the Evolution and Epidemiology of European CC1-MRSA-IV: Tracking a Multidrug-Resistant Community-Associated Meticillin-Resistant Staphylococcus aureus Clone. Microb. Genomics 2021, 7, 000601. [Google Scholar] [CrossRef]
- Zhou, W.; Jin, Y.; Liu, X.; Chen, Y.; Shen, P.; Xiao, Y. Comparison of Genetic Features and Evolution of Global and Chinese Strains of Community-Associated Methicillin-Resistant Staphylococcus aureus ST22. Microbiol. Spectr. 2022, 10, e02037-21. [Google Scholar] [CrossRef] [PubMed]
- Balakuntla, J.; Prabhakara, S.; Arakere, G. Novel Rearrangements in the Staphylococcal Cassette Chromosome Mec Type V Elements of Indian ST772 and ST672 Methicillin Resistant Staphylococcus aureus Strains. PLoS ONE 2014, 9, e94293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boundy, S.; Safo, M.K.; Wang, L.; Musayev, F.N.; O’Farrell, H.C.; Rife, J.P.; Archer, G.L. Characterization of the Staphylococcus aureus RRNA Methyltransferase Encoded by Orfx, the Gene Containing the Staphylococcal Chromosome Cassette Mec (SCCmec) Insertion Site. J. Biol. Chem. 2013, 288, 132–140. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Skov, R.L.; Han, X.; Larsen, A.R.; Larsen, J.; Sørum, M.; Wulf, M.; Voss, A.; Hiramatsu, K.; Ito, T. Novel Types of Staphylococcal Cassette Chromosome Mec Elements Identified in Clonal Complex 398 Methicillin-Resistant Staphylococcus aureus Strains. Antimicrob. Agents Chemother. 2011, 55, 3046–3050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimonaco, N.J.; Aubrey, W.; Kenobi, K.; Clare, A.; Creevey, C.J. No One Tool to Rule Them All: Prokaryotic Gene Prediction Tool Annotations Are Highly Dependent on the Organism of Study. Bioinformatics 2022, 38, 1198–1207. [Google Scholar] [CrossRef]
- Liu, J.; Chen, D.; Peters, B.M.; Li, L.; Li, B.; Xu, Z.; Shirliff, M.E. Staphylococcal Chromosomal Cassettes Mec (SCCmec): A Mobile Genetic Element in Methicillin-Resistant Staphylococcus aureus. Microb. Pathog. 2016, 101, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Waldron, D.E.; Lindsay, J.A. Sau1: A Novel Lineage-Specific Type I Restriction-Modification System That Blocks Horizontal Gene Transfer into Staphylococcus aureus and between S. Aureus Isolates of Different Lineages. J. Bacteriol. 2006, 188, 5578–5585. [Google Scholar] [CrossRef] [Green Version]
- Argudín, M.Á.; Mendoza, M.C.; Rodicio, M.R. Food Poisoning and Staphylococcus aureus Enterotoxins. Toxins 2010, 2, 1751–1773. [Google Scholar] [CrossRef]
- Cockfield, J.D.; Pathak, S.; Edgeworth, J.D.; Lindsay, J.A. Rapid Determination of Hospital-Acquired Meticillin-Resistant Staphylococcus aureus Lineages. J. Med. Microbiol. 2007, 56, 614–619. [Google Scholar] [CrossRef]
- Murray, N.E. Type I Restriction Systems: Sophisticated Molecular Machines (a Legacy of Bertani and Weigle). Microbiol. Mol. Biol. Rev. 2000, 64, 412–434. [Google Scholar] [CrossRef] [Green Version]
- Vasu, K.; Nagaraja, V. Diverse Functions of Restriction-Modification Systems in Addition to Cellular Defense. Microbiol. Mol. Biol. Rev. 2013, 77, 53–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Katayama, Y.; Asada, K.; Mori, N.; Tsutsumimoto, K.; Tiensasitorn, C.; Hiramatsu, K. Structural Comparison of Three Types of Staphylococcal Cassette Chromosome Mec Integrated in the Chromosome in Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2001, 45, 1323–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noto, M.J.; Kreiswirth, B.N.; Monk, A.B.; Archer, G.L. Gene Acquisition at the Insertion Site for SCCmec, the Genomic Island Conferring Methicillin Resistance in Staphylococcus aureus. J. Bacteriol. 2008, 190, 1276–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, Y.; Ito, T.; Hiramatsu, K. A New Mobile Genetic Element, Staphylococcal Cassette Chromosome Mec, Encodes Methicillin Resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 2000, 44, 1549–1555. [Google Scholar] [CrossRef] [Green Version]
- Penna, B.; Silva, M.B.; Soares, A.E.R.; Vasconcelos, A.T.R.; Ramundo, M.S.; Ferreira, F.A.; Silva-Carvalho, M.C.; de Sousa, V.S.; Rabello, R.F.; Bandeira, P.T.; et al. Comparative Genomics of MRSA Strains from Human and Canine Origins Reveals Similar Virulence Gene Repertoire. Sci. Rep. 2021, 11, 4724. [Google Scholar] [CrossRef]
- Alibayov, B.; Baba-Moussa, L.; Sina, H.; Zdeňková, K.; Demnerová, K. Staphylococcus aureus Mobile Genetic Elements. Mol. Biol. Rep. 2014, 41, 5005–5018. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Kraft, B.; Yu, W.; Demicrioglu, D.D.; Hertlein, T.; Burian, M.; Schmaler, M.; Boller, K.; Bekeredjian-Ding, I.; Ohlsen, K.; et al. The ΝSaα Specific Lipoprotein Like Cluster (Lpl) of S. Aureus USA300 Contributes to Immune Stimulation and Invasion in Human Cells. PLoS Pathog. 2015, 11, e1004984. [Google Scholar] [CrossRef] [Green Version]
- Aswani, V.; Najar, F.; Pantrangi, M.; Mau, B.; Schwan, W.R.; Shukla, S.K. Virulence Factor Landscape of a Staphylococcus aureus equence Type 45 Strain, MCRF184. BMC Genomics 2019, 20, 123. [Google Scholar] [CrossRef]
- Côrtes, M.F.; Costa, M.O.C.; Lima, N.C.B.; Souza, R.C.; Almeida, L.G.P.; Guedes, L.P.C.; Vasconcelos, A.T.R.; Nicolás, M.F.; Figueiredo, A.M.S. Complete Genome Sequence of Community-Associated Methicillin-Resistant Staphylococcus aureus (Strain USA400-0051), A Prototype of the USA400 Clone. Mem. Inst. Oswaldo Cruz 2017, 112, 790–792. [Google Scholar] [CrossRef]
- Ji, X.; Krüger, H.; Tao, J.; Wang, Y.; Feßler, A.T.; Bai, R.; Wang, S.; Dong, Y.; Shen, J.; Wang, Y.; et al. Comparative Analysis of Genomic Characteristics, Fitness and Virulence of MRSA ST398 and ST9 Isolated from China and Germany. Emerg. Microbes Infect. 2021, 10, 1481–1494. [Google Scholar] [CrossRef]
- Stentzel, S.; Teufelberger, A.; Nordengrün, M.; Kolata, J.; Schmidt, F.; van Crombruggen, K.; Michalik, S.; Kumpfmüller, J.; Tischer, S.; Schweder, T.; et al. Staphylococcal Serine Protease–like Proteins Are Pacemakers of Allergic Airway Reactions to Staphylococcus aureus. J. Allergy Clin. Immunol. 2017, 139, 492–500.e8. [Google Scholar] [CrossRef] [Green Version]
- Paharik, A.E.; Salgado-pabon, W.; Meyerholz, D.K.; White, M.J.; Schlievert, P.M.; Horswill, R. Staphylococcus aureus Protein Production and Virulence in a Rabbit Model of Pneumonia. mSphere 2016, 1, e00208-16. [Google Scholar] [CrossRef] [Green Version]
- Jeon, J.; D’Souza, R.; Hong, S.K.; Lee, Y.; Yong, D.; Choi, J.; Lee, K.; Chong, Y. Complete Genome Sequence of the Bacteriophage YMC/09/04/R1988 MRSA BP: A Lytic Phage from a Methicillin-Resistant Staphylococcus aureus Isolate. FEMS Microbiol. Lett. 2014, 359, 144–146. [Google Scholar] [CrossRef] [Green Version]
- Laumay, F.; Benchetrit, H.; Corvaglia, A.R.; van der Mee-Marquet, N.; François, P. The Staphylococcus aureus Cc398 Lineage: An Evolution Driven by the Acquisition of Prophages and Other Mobile Genetic Elements. Genes 2021, 12, 1752. [Google Scholar] [CrossRef] [PubMed]
- McMillan, E.A.; Gupta, S.K.; Williams, L.E.; Jové, T.; Hiott, L.M.; Woodley, T.A.; Barrett, J.B.; Jackson, C.R.; Wasilenko, J.L.; Simmons, M.; et al. Antimicrobial Resistance Genes, Cassettes, and Plasmids Present in Salmonella Enterica Associated with United States Food Animals. Front. Microbiol. 2019, 10, 832. [Google Scholar] [CrossRef] [Green Version]
- Douarre, P.E.; Mallet, L.; Radomski, N.; Felten, A.; Mistou, M.Y. Analysis of COMPASS, a New Comprehensive Plasmid Database Revealed Prevalence of Multireplicon and Extensive Diversity of IncF Plasmids. Front. Microbiol. 2020, 11, 483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, A.J.; Lindsay, J.A. The Distribution of Plasmids That Carry Virulence and Resistance Genes inStaphylococcus aureus Is Lineage Associated. BMC Microbiol. 2012, 12, 104. [Google Scholar] [CrossRef] [Green Version]
- Carattoli, A.; Miriagou, V.; Bertini, A.; Loli, A.; Colinon, C.; Villa, L.; Whichard, J.M.; Rossolini, G.M. Replicon Typing of Plasmids Encoding Resistance to Newer β-Lactams. Emerg. Infect. Dis. 2006, 12, 1145–1148. [Google Scholar] [CrossRef]
- Hopkins, K.L.; Liebana, E.; Villa, L.; Batchelor, M.; Threlfall, E.J.; Carattoli, A. Replicon Typing of Plasmids Carrying CTX-M or CMY β-Lactamases Circulating among Salmonella and Escherichia Coli Isolates. Antimicrob. Agents Chemother. 2006, 50, 3203–3206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, T.J.; Wannemuehler, Y.M.; Johnson, S.J.; Logue, C.M.; White, D.G.; Doetkott, C.; Nolan, L.K. Plasmid Replicon Typing of Commensal and Pathogenic Escherichia Coli Isolates. Appl. Environ. Microbiol. 2007, 73, 1976–1983. [Google Scholar] [CrossRef] [Green Version]
- Weaver, K. The Fst/Ldr Family of Type I TA System Toxins: Potential Roles in Stress Response, Metabolism and Pathogenesis. Toxins 2020, 12, 474. [Google Scholar] [CrossRef]
- Andresen, L.; Martínez-Burgo, Y.; Zangelin, J.N.; Rizvanovic, A.; Holmqvist, E. The Small Toxic Salmonella Protein Timp Targets the Cytoplasmic Membrane and Is Repressed by the Small RNA TimR. MBio 2020, 11, e01659-20. [Google Scholar] [CrossRef] [PubMed]
- Su, M.; Ling, Y.; Yu, J.; Wu, J.; Xiao, J. Small Proteins: Untapped Area of Potential Biological Importance. Front. Genet. 2013, 4, 286. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, F.G.; Yui Eto, K.; Murphy, R.J.T.; Fairhurst, H.M.; Coombs, G.W.; Grubb, W.B.; Ramsay, J.P. Origin-of-Transfer Sequences Facilitate Mobilisation of Non-Conjugative Antimicrobial-Resistance Plasmids in Staphylococcus aureus. Nucleic Acids Res. 2015, 43, 7971–7983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, T.W.; Khokhlova, O.E.; Iwao, Y.; Higuchi, W.; Hung, W.C.; Reva, I.V.; Singur, O.A.; Gostev, V.V.; Sidorenko, S.V.; Peryanova, O.V.; et al. Complete Circular Genome Sequence of Successful ST8/SCCmecIV Community-Associated Methicillin-Resistant Staphylococcus aureus (OC8) in Russia: One-Megabase Genomic Inversion, IS256′s Spread, and Evolution of Russia ST8-IV. PLoS ONE 2016, 11, e0164168. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.K.; Kislow, J.; Briska, A.; Henkhaus, J.; Dykes, C. Optical Mapping Reveals a Large Genetic Inversion between Two Methicillin-Resistant Staphylococcus aureus Strains. J. Bacteriol. 2009, 191, 5717–5723. [Google Scholar] [CrossRef] [Green Version]
- Grindley, N.D.F.; Whiteson, K.L.; Rice, P.A. Mechanisms of Site-Specific Recombination. Annu. Rev. Biochem. 2006, 75, 567–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.A.; Thomas, J.; Grossman, A.D. The Bacillus Subtilis Conjugative Transposon ICEBs1 Mobilizes Plasmids Lacking Dedicated Mobilization Functions. J. Bacteriol. 2012, 194, 3165–3172. [Google Scholar] [CrossRef] [Green Version]
- Ramsay, J.P.; Firth, N. Diverse Mobilization Strategies Facilitate Transfer of Non-Conjugative Mobile Genetic Elements. Curr. Opin. Microbiol. 2017, 38, 1–9. [Google Scholar] [CrossRef]
- Ramsay, J.P.; Kwong, S.M.; Murphy, R.J.T.; Yui Eto, K.; Price, K.J.; Nguyen, Q.T.; O’Brien, F.G.; Grubb, W.B.; Coombs, G.W.; Firth, N. An Updated View of Plasmid Conjugation and Mobilization in Staphylococcus. Mob. Genet. Elem. 2016, 6, e1208317. [Google Scholar] [CrossRef] [Green Version]
- Catchpole, I.; Thomas, C.; Davies, A.; Dyke, K.G.H. The Nucleotide Sequence of Staphylococcus aureus Plasmid PT48 Conferring Inducible Macrolide-Lincosamide-Streptogramin B Resistance and Comparison with Similar Plasmids Expressing Constitutive Resistance. J. Gen. Microbiol. 1988, 134, 697–709. [Google Scholar] [CrossRef] [Green Version]
- Kwong, S.M.; Ramsay, J.P.; Jensen, S.O.; Firth, N. Replication of Staphylococcal Resistance Plasmids. Front. Microbiol. 2017, 8, 2279. [Google Scholar] [CrossRef] [Green Version]
- Lanza, V.F.; Tedim, A.P.; Martínez, J.L.; Baquero, F.; Coque, T.M. The Plasmidome of Firmicutes: Impact on the Emergence and the Spread of Resistance to Antimicrobials. Microbiol. Spectr. 2015, 3, PLAS-0039-2014. [Google Scholar] [CrossRef] [Green Version]
- Firth, N.; Jensen, S.O.; Kwong, S.M.; Skurray, R.A.; Ramsay, J.P. Staphylococcal Plasmids, Transposable and Integrative Elements. Microbiol. Spectr. 2018, 6, 6. [Google Scholar] [CrossRef]
- Green, M.R.; Sambrook, J. Isolating DNA from Gram-Negative Bacteria. Cold Spring Harb. Protoc. 2017, 2017, 83–84. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving Bacterial Genome Assemblies from Short and Long Sequencing Reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatusova, T.; Dicuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI Prokaryotic Genome Annotation Pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-Access Bacterial Population Genomics: BIGSdb Software, the PubMLST.Org Website and Their Applications. Wellcome Open Res. 2018, 3, 1–20. [Google Scholar] [CrossRef]
- Carattoli, A.; Hasman, H. PlasmidFinder and In Silico PMLST: Identification and Typing of Plasmid Replicons in Whole-Genome Sequencing (WGS). Methods Mol. Biol. 2020, 2075, 285–294. [Google Scholar] [CrossRef]
- Carattoli, A.; Zankari, E.; Garciá-Fernández, A.; Larsen, M.V.; Lund, O.; Villa, L.; Aarestrup, F.M.; Hasman, H. In Silico Detection and Typing of Plasmids Using Plasmidfinder and Plasmid Multilocus Sequence Typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and Model-Centric Curation of the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A Comparative Pathogenomic Platform with an Interactive Web Interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xiong, Z.; Sun, L.; Yang, J.; Jin, Q. VFDB 2012 Update: Toward the Genetic Diversity and Molecular Evolution of Bacterial Virulence Factors. Nucleic Acids Res. 2012, 40, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A Fast Phage Search Tool. Nucleic Acids Res. 2011, 39, 347–352. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A Better, Faster Version of the PHAST Phage Search Tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [Green Version]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded Prediction of Genomic Islands for Larger-Scale Datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid Large-Scale Prokaryote Pan Genome Analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. Fasttree: Computing Large Minimum Evolution Trees with Profiles Instead of a Distance Matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Trad, E.I.; Che Hamzah, A.M.; Puah, S.M.; Chua, K.H.; Hanifah, M.Z.; Ayub, Q.; Palittapongarnpim, P.; Kwong, S.M.; Chew, C.H.; Yeo, C.C. Complete Genome Sequence and Analysis of a ST573 Multidrug-Resistant Methicillin-Resistant Staphylococcus aureus SauR3 Clinical Isolate from Terengganu, Malaysia. Pathogens 2023, 12, 502. https://doi.org/10.3390/pathogens12030502
Al-Trad EI, Che Hamzah AM, Puah SM, Chua KH, Hanifah MZ, Ayub Q, Palittapongarnpim P, Kwong SM, Chew CH, Yeo CC. Complete Genome Sequence and Analysis of a ST573 Multidrug-Resistant Methicillin-Resistant Staphylococcus aureus SauR3 Clinical Isolate from Terengganu, Malaysia. Pathogens. 2023; 12(3):502. https://doi.org/10.3390/pathogens12030502
Chicago/Turabian StyleAl-Trad, Esra’a I., Ainal Mardziah Che Hamzah, Suat Moi Puah, Kek Heng Chua, Muhamad Zarul Hanifah, Qasim Ayub, Prasit Palittapongarnpim, Stephen M. Kwong, Ching Hoong Chew, and Chew Chieng Yeo. 2023. "Complete Genome Sequence and Analysis of a ST573 Multidrug-Resistant Methicillin-Resistant Staphylococcus aureus SauR3 Clinical Isolate from Terengganu, Malaysia" Pathogens 12, no. 3: 502. https://doi.org/10.3390/pathogens12030502