

Next-Generation Sequencing-Based Monitoring of Intestinal Bacteria and Bacteriophages Following Fecal Microbiota Transplantation in Inflammatory Bowel Diseases
 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Patients and Study Design
2.3. DNA Isolation and NGS Procedures
2.4. Bioinformatic Analysis of Bacterial 16S rRNA
2.5. Classification of Detected Viral Sequences
3. Results
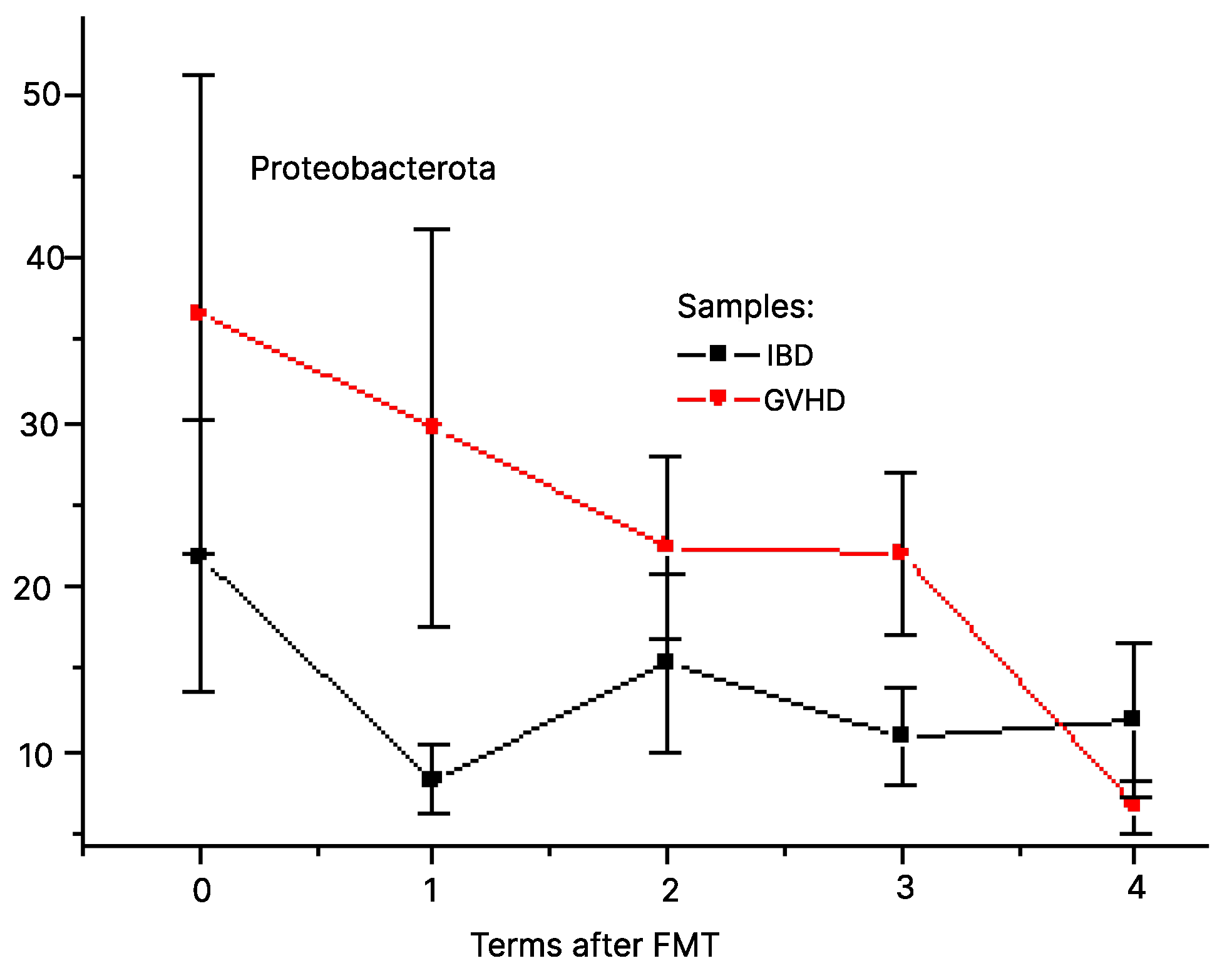
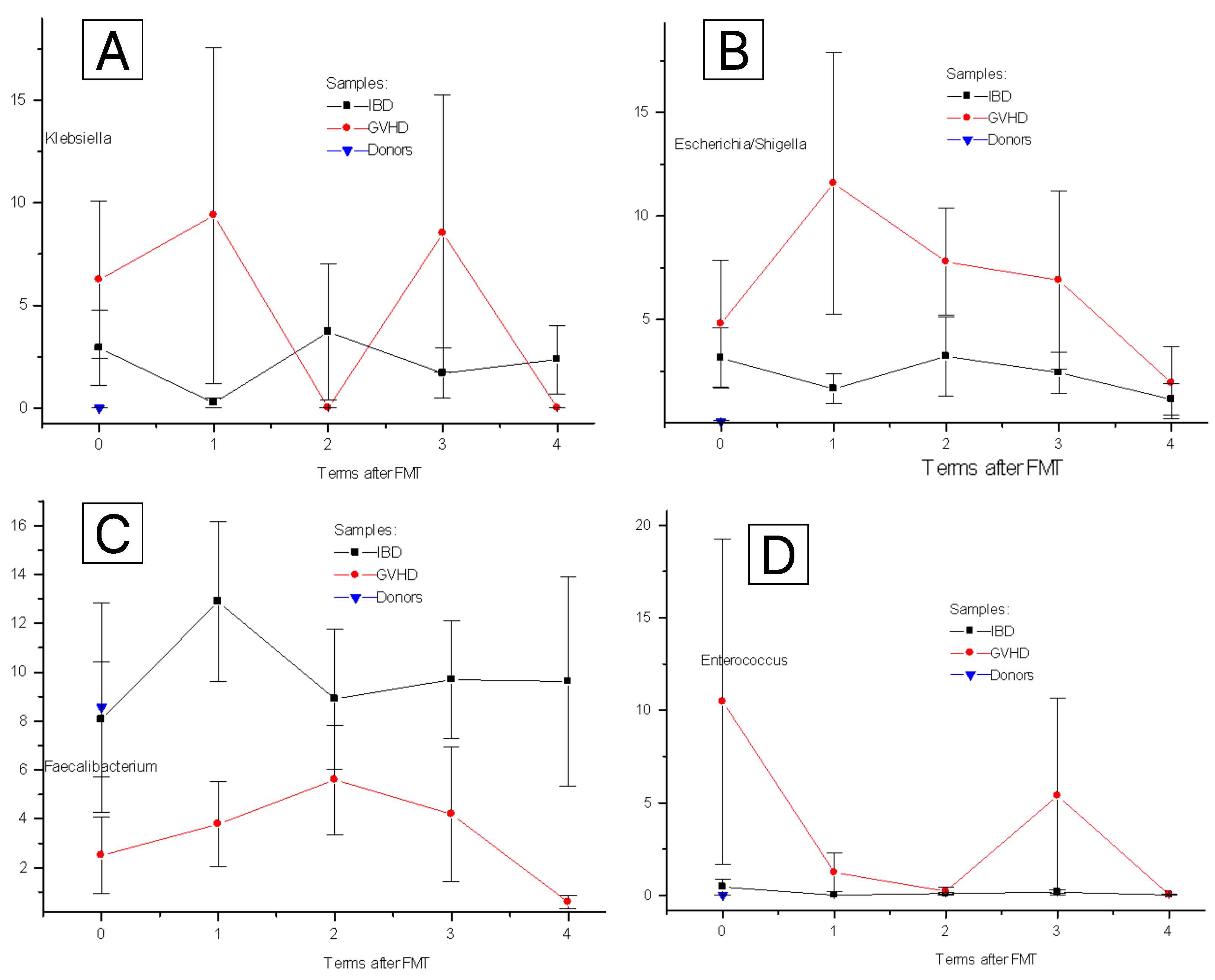
3.1. Post-FMT Changes for Clinically Relevant Bacteria
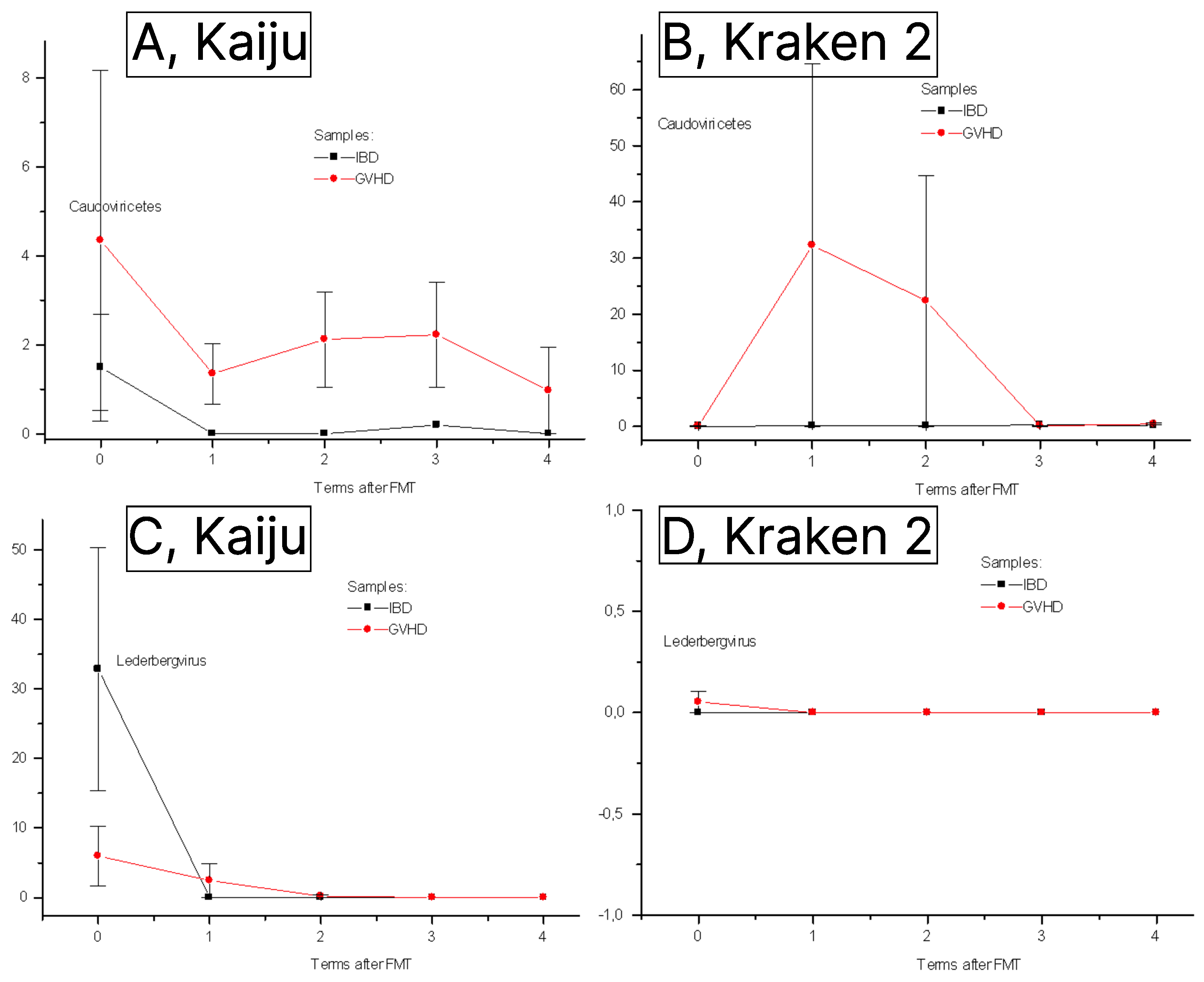
3.2. Phageome Changes Following FMT
3.3. Correlations between Bacterial Groups and Detectable Bacteriophages
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shan, Y.; Lee, M.; Chang, E.B. The Gut Microbiome and Inflammatory Bowel Diseases. Annu. Rev. Med. 2022, 73, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Gubatan, J.; Boye, T.L.; Temby, M.; Sojwal, R.S.; Holman, D.R.; Sinha, S.R.; Rogalla, S.R.; Nielsen, O.H. Gut microbiome in inflammatory bowel disease: Role in pathogenesis, dietary modulation, and colitis-associated colon cancer. Microorganisms 2022, 10, 1371. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.T. Pathophysiology of Inflammatory Bowel Diseases. N. Engl. J. Med. 2020, 383, 2652–2664. [Google Scholar] [CrossRef] [PubMed]
- Andoh, A.; Nishida, A. Alteration of the Gut Microbiome in Inflammatory Bowel Disease. Digestion 2023, 104, 16–23. [Google Scholar] [CrossRef]
- Shono, Y.; van den Brink, M.R.M. Gut microbiota injury in allogeneic haematopoietic stem cell transplantation. Nat. Rev. Cancer 2018, 18, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Airola, C.; Severino, A.; Porcari, S.; Fusco, W.; Mullish, B.H.; Gasbarrini, A.; Cammarota, G.; Ponziani, F.R.; Ianiro, G. Future Modulation of Gut Microbiota: From Eubiotics to FMT, Engineered Bacteria, and Phage Therapy. Antibiotics 2023, 12, 868. [Google Scholar] [CrossRef]
- Guzzo, G.L.; Andrews, J.M.; Weyrich, L.S. The Neglected Gut Microbiome: Fungi, Protozoa, and Bacteriophages in Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2022, 28, 1112–1122. [Google Scholar] [CrossRef]
- Cammarota, G.; Ianiro, G.; Tilg, H.; Rajilić-Stojanović, M.; Kump, P.; Satokari, R.; Sokol, H.; Arkkila, P.; Pintus, C.; Hart, A.; et al. European consensus conference on faecal microbiota transplantation in clinical practice. Gut 2017, 66, 569–580. [Google Scholar] [CrossRef]
- Goloshchapov, O.V.; Olekhnovich, E.I.; Sidorenko, S.V.; Moiseev, I.S.; Kucher, M.A.; Fedorov, D.E.; Pavlenko, A.V.; Manolov, A.I.; Gostev, V.V.; Veselovsky, V.A.; et al. Long-term impact of fecal transplantation in healthy volunteers. BMC Microbiol. 2019, 19, 312. [Google Scholar] [CrossRef]
- Ninove, L.; Nougairede, A.; Gazin, C.; Thirion, L.; Delogu, I.; Zandotti, C.; Charrel, R.N.; De Lamballerie, X. RNA and DNA bacteriophages as molecular diagnosis controls in clinical virology: A comprehensive study of more than 45,000 routine PCR tests. PLoS ONE 2011, 6, e16142. [Google Scholar] [CrossRef]
- Solovieva, E.V.; Myakinina, V.P.; Kislichkina, A.A.; Krasilnikova, V.M.; Verevkin, V.V.; Mochalov, V.V.; Lev, A.I.; Fursova, N.K.; Volozhantsev, N.V. Comparative genome analysis of novel Podoviruses lytic for hypermucoviscous Klebsiella pneumoniae of K1, K2, and K57 capsular types. Virus Res 2018, 243, 10–18. [Google Scholar] [CrossRef]
- Latka, A.; Lemire, S.; Grimon, D.; Dams, D.; Maciejewska, B.; Lu, T.; Drulis-Kawa, Z.; Briers, Y. Engineering the Modular Receptor-Binding Proteins of Klebsiella Phages Switches Their Capsule Serotype Specificity. mBio 2021, 12, e00455-21. [Google Scholar] [CrossRef] [PubMed]
- Mikalová, L.; Bosák, J.; Hříbková, H.; Dědičová, D.; Benada, O.; Šmarda, J.; Šmajs, D. Novel Temperate Phages of Salmonella enterica subsp. salamae and subsp. diarizonae and Their Activity against Pathogenic S. enterica subsp. enterica Isolates. PLoS ONE 2017, 12, e0170734. [Google Scholar] [CrossRef]
- Li, Y.; Gordon, E.; Shean, R.C.; Idle, A.; Deng, X.; Greninger, A.L.; Delwart, E. CrAssphage and its bacterial host in cat feces. Sci. Rep. 2021, 11, 815. [Google Scholar] [CrossRef] [PubMed]
- Grosjean, J.; Hantz, S.; Cotin, S.; Baclet, M.C.; Mengelle, C.; Trapes, L.; Virey, B.; Undreiner, F.; Brosset, P.; Pasquier, C.; et al. Direct genotyping of cytomegalovirus envelope glycoproteins from toddler’s saliva samples. J. Clin. Virol. 2009, 46, S43–S48. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Xu, S.; Maple, P.A.C.; Xu, W.; Brown, K.E. Differentiation between wild-type and vaccines strains of varicella zoster virus (VZV) based on four single nucleotide polymorphisms. Epidemiol. Infect. 2017, 145, 2618–2625. [Google Scholar] [CrossRef]
- Ebner, K.; Suda, M.; Watzinger, F.; Lion, T. Molecular detection and quantitative analysis of the entire spectrum of human adenoviruses by a two-reaction real-time PCR assay. J. Clin. Microbiol. 2005, 43, 3049–3053. [Google Scholar] [CrossRef] [PubMed]
- Ylihärsilä, M.; Harju, E.; Arppe, R.; Hattara, L.; Hölsä, J.; Saviranta, P.; Soukka, T.; Waris, M. Genotyping of clinically relevant human adenoviruses by array-in-well hybridization assay. Clin. Microbiol. Infect. 2013, 19, 551–557. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Cole, J.R.; Wang, Q.; Fish, J.A.; Chai, B.; McGarrell, D.M.; Sun, Y.; Brown, C.T.; Porras-Alfaro, A.; Kuske, C.R.; Tiedje, J.M. Ribosomal Database Project: Data and tools for high throughput rRNA analysis. Nucleic Acids Res. 2014, 42, D633–D642. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data; FastQC: San Diego, CA, USA, 2010. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Salzberg, S.L. Ultrafast and accurate 16S rRNA microbial community analysis using Kraken 2. Microbiome 2020, 8, 124. [Google Scholar] [CrossRef] [PubMed]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.; Goldobina, E.; Govi, B.; Shkoporov, A.N. Bacteriophages of the Order Crassvirales: What Do We Currently Know about This Keystone Component of the Human Gut Virome? Biomolecules 2023, 13, 584. [Google Scholar] [CrossRef]
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; Mcveigh, R.; O’Neill, K.; Robbertse, B.; et al. NCBI Taxonomy: A comprehensive update on curation, resources and tools. Database 2020, 2020, baaa062. [Google Scholar] [CrossRef]
- Fujimoto, K.; Kimura, Y.; Allegretti, J.R.; Yamamoto, M.; Zhang, Y.-Z.; Katayama, K.; Tremmel, G.; Kawaguchi, Y.; Shimohigoshi, M.; Hayashi, T.; et al. Functional Restoration of Bacteriomes and Viromes by Fecal Microbiota Transplantation. Gastroenterology 2021, 160, 2089–2102. [Google Scholar] [CrossRef]
- Goloshchapov, O.V.; Kucher, M.A.; Eismont, Y.A.; Chukhovin, A.B. Origin and consequences of intestinal dysfunction following cytostatic chemotherapy and hematopoietic stem cell transplantation. Cell. Ther. Transplant. 2023, 12, 4–14. [Google Scholar] [CrossRef]
- Zhou, S.; Cui, Y.; Zhang, Y.; Zhao, T.; Cong, J. Fecal microbiota transplantation for induction of remission in Crohn’s disease: A systematic review and meta-analysis. Int. J. Color. Dis. 2023, 38, 62. [Google Scholar] [CrossRef]
- Qiao, X.; Biliński, J.; Wang, L.; Yang, T.; Luo, R.; Fu, Y.; Yang, G. Safety and efficacy of fecal microbiota transplantation in the treatment of graft-versus-host disease. Bone Marrow Transplant. 2023, 58, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Metafuni, E.; Marino, L.D.; Giammarco, S.; Bellesi, S.; Limongiello, M.A.; Sorà, F.; Frioni, F.; Maggi, R.; Chiusolo, P.; Sica, S. The Role of Fecal Microbiota Transplantation in the Allogeneic Stem Cell Transplant Setting. Microorganisms 2023, 11, 2182. [Google Scholar] [CrossRef] [PubMed]
- Goloshchapov, O.V.; Bakin, E.A.; Kucher, M.A.; Stanevich, O.V.; Suvorova, M.A.; Gostev, V.V.; Glotov, O.S.; Eismont, Y.A.; Polev, D.E.; Lobenskaya, A.Y.; et al. Bacteroides fragilis is a potential marker of effective microbiota transplantation in acute graft-versus-host disease treatment. Cell. Ther. Transplant. 2020, 9, 47–59. [Google Scholar] [CrossRef]
- Campos-Madueno, E.I.; Moradi, M.; Eddoubaji, Y.; Shahi, F.; Moradi, S.; Bernasconi, O.J.; Moser, A.I.; Endimiani, A. Intestinal colonization with multidrug-resistant Enterobacterales: Screening, epidemiology, clinical impact, and strategies to decolonize carriers. Eur. J. Clin. Microbiol. Infect. Dis. 2023, 42, 229–254. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Inoue, R.; Nishida, A.; Yokota, Y.; Morishima, S.; Kawahara, M.; Kusada, H.; Tamaki, H.; Andoh, A. Features of the gut prokaryotic virome of Japanese patients with Crohn’s disease. J. Gastroenterol. 2022, 57, 559–570. [Google Scholar] [CrossRef]
- De Jonge, P.A.; Wortelboer, K.; Scheithauer, T.P.M.; van den Born, B.J.H.; Zwinderman, A.H.; Nobrega, F.L.; Dutilh, B.E.; Nieuwdorp, M.; Herrema, H. Gut virome profiling identifies a widespread bacteriophage family associated with metabolic syndrome. Nat. Commun. 2022, 13, 3594. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kitazumi, A.; Liao, Y.T.; de Los Reyes, B.G.; Wu, V.C.H. Metagenomic investigation reveals bacteriophage-mediated horizontal transfer of antibiotic resistance genes in microbial communities of an organic agricultural ecosystem. Microbiol. Spectr. 2023, 11, e00226-23. [Google Scholar] [CrossRef] [PubMed]
- Chukhlovin, A.B.; Dudurich, V.V.; Kusakin, A.V.; Polev, D.E.; Ermachenko, E.D.; Aseev, M.V.; Zakharov, Y.A.; Eismont, Y.A.; Danilov, L.G.; Glotov, O.S. Evaluation of Gut Microbiota in Healthy Persons and Type 1 Diabetes Mellitus Patients in North-Western Russia. Microorganisms 2023, 11, 1813. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Parameters | Crohn’s Disease (n = 9) | Graft-Versus-Host Disease (n = 6) |
|---|---|---|
| Gender (males/females) | 5/4 | 4/2 |
| Median age, years (min and max values) | 29 (22–59) | 27 (9–59) |
| Body mass, kg (min and max values) | 66 (42–69) | 49 (28–70) |
| Duration of the primary illness (min and max values) | 4 (1–15) | 6 (2–9) |
| Phylum | Class | Genus | Species | Bacteriophage |
|---|---|---|---|---|
| Uroviricota | Caudoviricetes | Bievrevirus | Bievrevirus bv4A7 | Escherichia phage 4A7 |
| Tequatrovirus | Tequatrovirus gee4498 | Escher phage vB_EcoM_G4498 | ||
| Traversvirus | Traversvirus II | Escherichia phage Stx2 II | ||
| Shamshuipovirus | Shamshuipovirus mEpX2 | Escherichia phage mEpX2 | ||
| Eganvirus | NC | Salmonella phage BIS20 | ||
| Brunovirus | Brunovirus SEN34 | Salmonella phage SEN34 | ||
| Goslarvirus | Goslarvirus goslar | Escherichia phage vB_EcoM_Goslar | ||
| Lambdavirus | NC | Enterobacteria phage mEp237 | ||
| Lederbergvirus | Lederbergvirus Sf6 | Shigella phage Sf6 | ||
| Lederbergvirus | NC | Enterobacter phage IME10 | ||
| Lederbergvirus | NC | Salmonella phage SEN22 | ||
| Novemvirus | Novemvirus T5282H | Enterobacter phage phiT5282H | ||
| Oengusvirus | Oengusvirus oengus | Faecalibacterium phage FP_oengus | ||
| Caudoviricetes unclassified | NC | Aeromonas phage pAEv1818 | ||
| Uroviricota | Caudoviricetes (Crassvirales) | Cohcovirus | Cohcovirus splanchnicus | uncultured phage cr30_1 |
| Kahnovirus | Kahnovirus copri | uncultured phage cr44_1 | ||
| Kahnovirus | Kahnovirus oralis | uncultured phage cr85_1 | ||
| Birpovirus | Birpovirus hominis | uncultured phage cr19_1 | ||
| Jahgtovirus | Jahgtovirus intestinihominis | uncultured phage cr107_1 | ||
| Peploviricota | Herviviricetes | Cytomegalovirus | Human betaherpesvirus 5 | NC |
| Preplasmiviricota | Tectiliviricetes | Mastadenovirus | Mastadenovirus D | Mastadenovirus 54 (serotype) |
| Bacterial Phyla, Genera | Phages (Phyla, Genera, Species) | Correlation Quotients | p Value |
|---|---|---|---|
| Proteobacteriota | Caudoviricetes | 0.667 | 0.0002 |
| Lederbergvirus | 0.477 | 0.009 | |
| Enterobacter phage IME10 | 0.552 | 0.004 | |
| E. coli phage 4A7 | 0.617 | 0.0065 | |
| Salmonella phage SEN34 | 0.538 | 0.003 | |
| Aeromonas phage pAEv1818 | 0.535 | 0.004 | |
| Genus Faecalibacteria | Bamfordviricota, Preplasmaviricota | 0.730 | 0.00003 |
| Clostridia_ss | Peploviricota | 0.660 | 0.0002 |
| Klebsiella | Lederbergvirus | 0.703 | 0.00006 |
| Enterobacter phage IME10 | 0.625 | 0.0055 | |
| E. coli phage HK106 | 0.567 | 0.002 | |
| Traversvirus | 0.526 | 0.004 | |
| Salmonella phage SEN34 | 0.564 | 0.002 | |
| Aeromonas phage pAEv1818 | 0.561 | 0.002 | |
| Enterococcus | Caudoviricetes | 0.630 | 0.0005 |
| E. coli phage 4A7 | 0.479 | 0.009 | |
| Salmonella phage SEN34 | 0.489 | 0.007 | |
| Aeromonas phage pAEv1818 | 0.504 | 0.006 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goloshchapov, O.V.; Shchukina, O.B.; Kusakin, A.V.; Tsai, V.V.; Kalinin, R.S.; Eismont, Y.A.; Glotov, O.S.; Chukhlovin, A.B. Next-Generation Sequencing-Based Monitoring of Intestinal Bacteria and Bacteriophages Following Fecal Microbiota Transplantation in Inflammatory Bowel Diseases. Pathogens 2023, 12, 1438. https://doi.org/10.3390/pathogens12121438
Goloshchapov OV, Shchukina OB, Kusakin AV, Tsai VV, Kalinin RS, Eismont YA, Glotov OS, Chukhlovin AB. Next-Generation Sequencing-Based Monitoring of Intestinal Bacteria and Bacteriophages Following Fecal Microbiota Transplantation in Inflammatory Bowel Diseases. Pathogens. 2023; 12(12):1438. https://doi.org/10.3390/pathogens12121438
Chicago/Turabian StyleGoloshchapov, Oleg V., Oksana B. Shchukina, Aleksey V. Kusakin, Viktoria V. Tsai, Roman S. Kalinin, Yury A. Eismont, Oleg S. Glotov, and Alexei B. Chukhlovin. 2023. "Next-Generation Sequencing-Based Monitoring of Intestinal Bacteria and Bacteriophages Following Fecal Microbiota Transplantation in Inflammatory Bowel Diseases" Pathogens 12, no. 12: 1438. https://doi.org/10.3390/pathogens12121438