
Host-Directed Therapies for Tuberculosis

Abstract
:1. Introduction
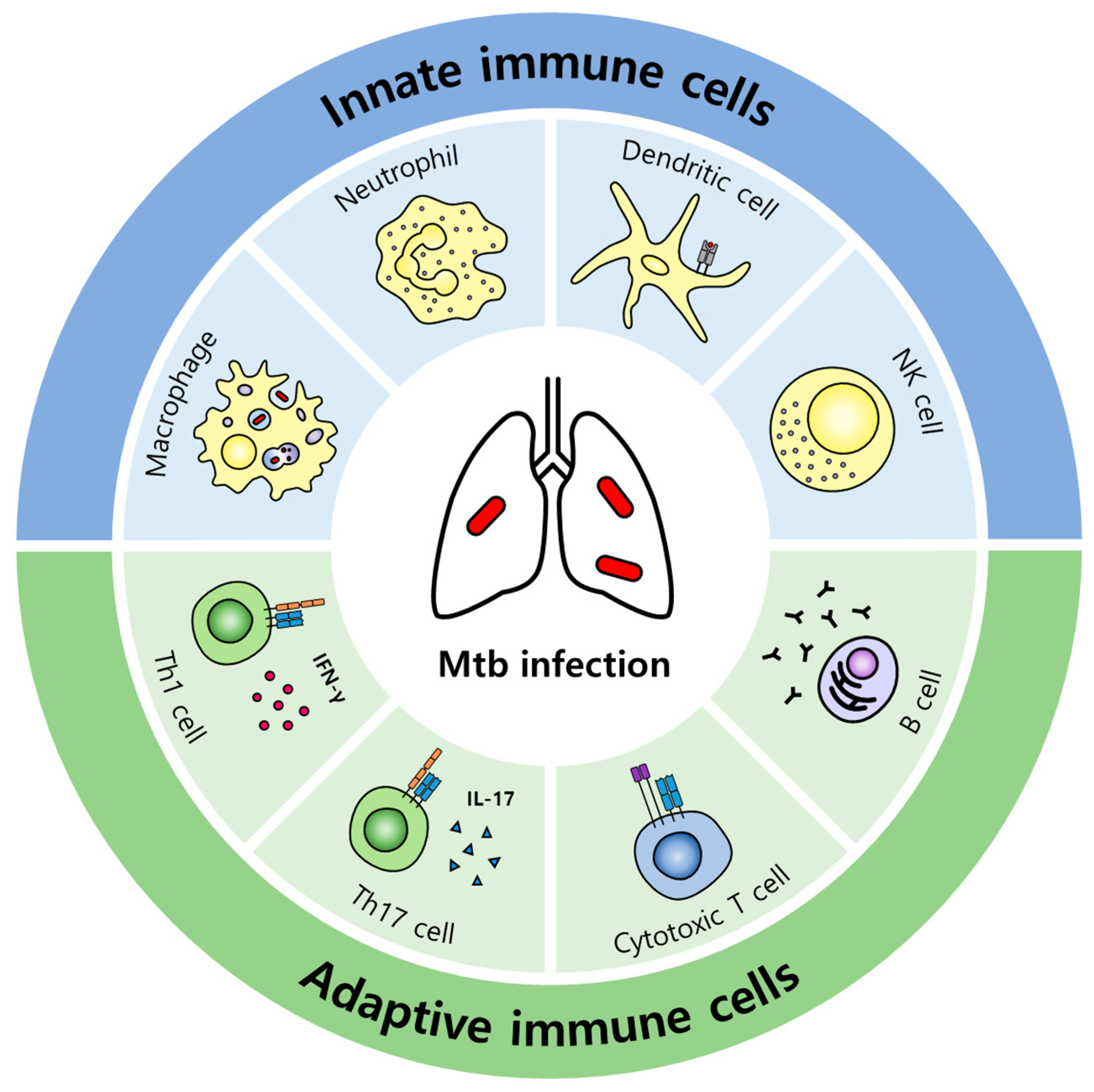
2. Host Immune Responses against Mtb
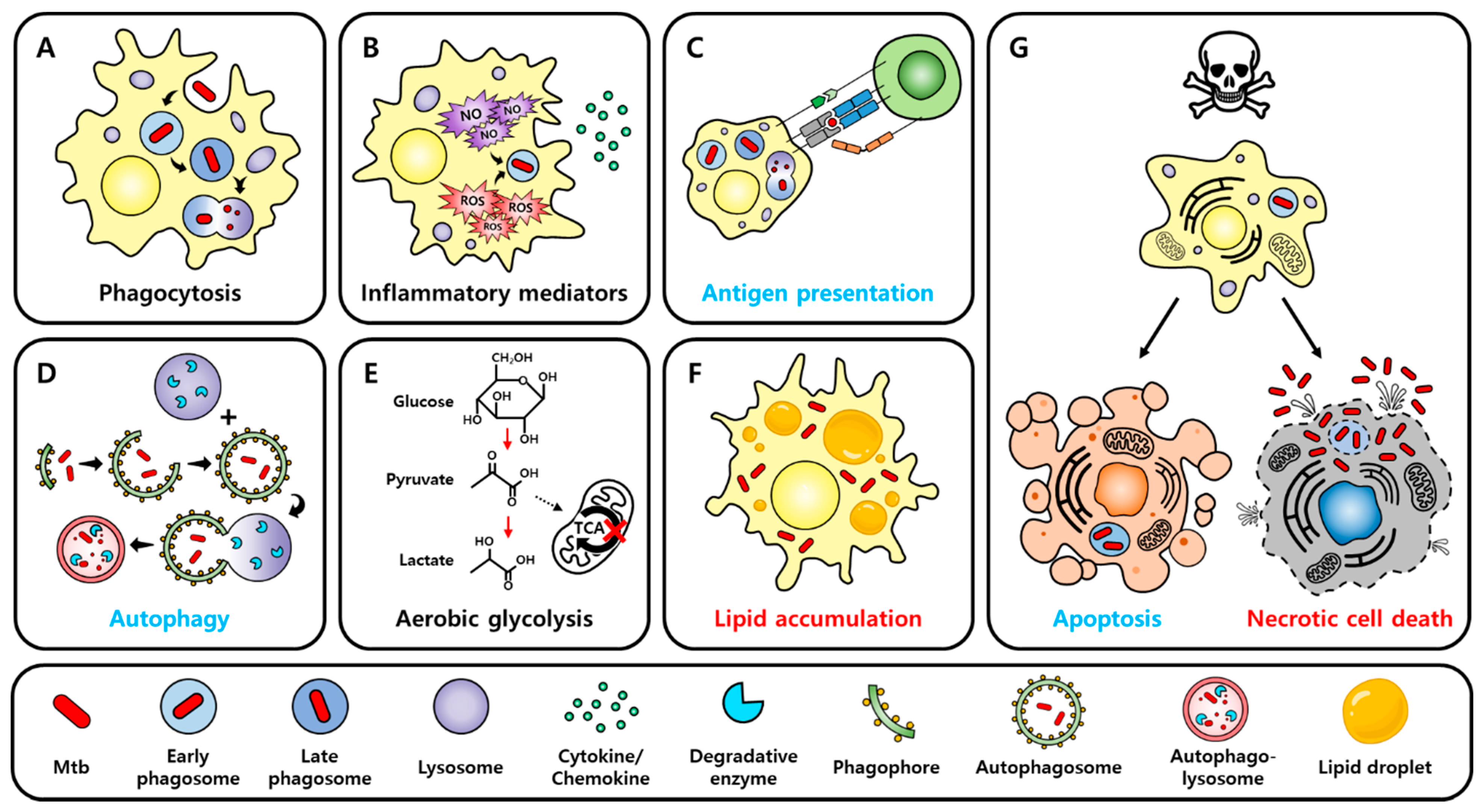
2.1. Innate Immune Responses
2.2. Adaptive Immune Responses
2.3. Cell Death
2.4. Metabolism
3. Potent HDT Drugs for TB
3.1. Modulating Innate Immune Responses
3.2. Modulating Adaptive Immune Responses
3.3. Targeting Cell Death
3.4. Regulating Metabolism
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Hancock, R.E.; Nijnik, A.; Philpott, D.J. Modulating immunity as a therapy for bacterial infections. Nat. Rev. Microbiol. 2012, 10, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Liu, J.; Zhang, D.; Xu, Z.; Ji, J.; Wen, C. Cytokine Storm in COVID-19: The Current Evidence and Treatment Strategies. Front. Immunol. 2020, 11, 1708. [Google Scholar] [CrossRef] [PubMed]
- Dunne, P.J.; Fletcher, J.M. Recent advances in regulatory T cell therapy of autoimmunity, graft rejection and cancer. Recent Pat. Inflamm. Allergy Drug Discov. 2010, 4, 231–243. [Google Scholar] [CrossRef]
- Dinarello, C.A.; van der Meer, J.W. Treating inflammation by blocking interleukin-1 in humans. Semin. Immunol. 2013, 25, 469–484. [Google Scholar] [CrossRef] [Green Version]
- Kubli, S.P.; Berger, T.; Araujo, D.V.; Siu, L.L.; Mak, T.W. Beyond immune checkpoint blockade: Emerging immunological strategies. Nat. Rev. Drug Discov. 2021, 20, 899–919. [Google Scholar] [CrossRef] [PubMed]
- Antimicrobial Resistance Collaborators. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Hoffman, P.S. Antibacterial Discovery: 21st Century Challenges. Antibiotics 2020, 9, 213. [Google Scholar] [CrossRef]
- Kaufmann, S.H.E.; Dorhoi, A.; Hotchkiss, R.S.; Bartenschlager, R. Host-directed therapies for bacterial and viral infections. Nat. Rev. Drug Discov. 2018, 17, 35–56. [Google Scholar] [CrossRef]
- World Health Organization. Global Tuberculosis Report 2021; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- World Health Organization. WHO Consolidated Guidelines on Tuberculosis. Module 4: Treatment-Drug-Resistant Tuberculosis Treatment; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Liang, L.; Wu, Q.; Gao, L.; Hao, Y.; Liu, C.; Xie, Y.; Sun, H.; Yan, X.; Li, F.; Li, H.; et al. Factors contributing to the high prevalence of multidrug-resistant tuberculosis: A study from China. Thorax 2012, 67, 632–638. [Google Scholar] [CrossRef] [Green Version]
- Ndjeka, N.; Schnippel, K.; Master, I.; Meintjes, G.; Maartens, G.; Romero, R.; Padanilam, X.; Enwerem, M.; Chotoo, S.; Singh, N.; et al. High treatment success rate for multidrug-resistant and extensively drug-resistant tuberculosis using a bedaquiline-containing treatment regimen. Eur. Respir. J. 2018, 52, 1801528. [Google Scholar] [CrossRef]
- Dheda, K.; Gumbo, T.; Maartens, G.; Dooley, K.E.; McNerney, R.; Murray, M.; Furin, J.; Nardell, E.A.; London, L.; Lessem, E.; et al. The epidemiology, pathogenesis, transmission, diagnosis, and management of multidrug-resistant, extensively drug-resistant, and incurable tuberculosis. Lancet Respir. Med. 2017, 5, 291–360. [Google Scholar] [CrossRef]
- Flannagan, R.S.; Cosio, G.; Grinstein, S. Antimicrobial mechanisms of phagocytes and bacterial evasion strategies. Nat. Rev. Microbiol. 2009, 7, 355–366. [Google Scholar] [CrossRef]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barber, D.L.; Mayer-Barber, K.D.; Feng, C.G.; Sharpe, A.H.; Sher, A. CD4 T cells promote rather than control tuberculosis in the absence of PD-1-mediated inhibition. J. Immunol. 2011, 186, 1598–1607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, S.; Huynh, J.P.; Lampropoulou, V.; Loginicheva, E.; Esaulova, E.; Gounder, A.P.; Boon, A.C.M.; Schwarzkopf, E.A.; Bradstreet, T.R.; Edelson, B.T.; et al. Irg1 expression in myeloid cells prevents immunopathology during M. tuberculosis infection. J. Exp. Med. 2018, 215, 1035–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arango Duque, G.; Descoteaux, A. Macrophage cytokines: Involvement in immunity and infectious diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussi, C.; Gutierrez, M.G. Mycobacterium tuberculosis infection of host cells in space and time. FEMS Microbiol. Rev. 2019, 43, 341–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravesloot-Chavez, M.M.; Van Dis, E.; Stanley, S.A. The Innate Immune Response to Mycobacterium tuberculosis Infection. Annu. Rev. Immunol. 2021, 39, 611–637. [Google Scholar] [CrossRef]
- Flynn, J.L.; Goldstein, M.M.; Chan, J.; Triebold, K.J.; Pfeffer, K.; Lowenstein, C.J.; Schreiber, R.; Mak, T.W.; Bloom, B.R. Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity 1995, 2, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Li, F.; Chen, J.W.; Wang, J. Risk of tuberculosis infection in anti-TNF-alpha biological therapy: From bench to bedside. J. Microbiol. Immunol. Infect. 2014, 47, 268–274. [Google Scholar] [CrossRef]
- Mayer-Barber, K.D.; Andrade, B.B.; Barber, D.L.; Hieny, S.; Feng, C.G.; Caspar, P.; Oland, S.; Gordon, S.; Sher, A. Innate and adaptive interferons suppress IL-1alpha and IL-1beta production by distinct pulmonary myeloid subsets during Mycobacterium tuberculosis infection. Immunity 2011, 35, 1023–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juffermans, N.P.; Florquin, S.; Camoglio, L.; Verbon, A.; Kolk, A.H.; Speelman, P.; van Deventer, S.J.; van Der Poll, T. Interleukin-1 signaling is essential for host defense during murine pulmonary tuberculosis. J. Infect. Dis. 2000, 182, 902–908. [Google Scholar] [CrossRef] [Green Version]
- Queval, C.J.; Brosch, R.; Simeone, R. The Macrophage: A Disputed Fortress in the Battle against Mycobacterium tuberculosis. Front. Microbiol. 2017, 8, 2284. [Google Scholar] [CrossRef] [PubMed]
- Augenstreich, J.; Arbues, A.; Simeone, R.; Haanappel, E.; Wegener, A.; Sayes, F.; Le Chevalier, F.; Chalut, C.; Malaga, W.; Guilhot, C.; et al. ESX-1 and phthiocerol dimycocerosates of Mycobacterium tuberculosis act in concert to cause phagosomal rupture and host cell apoptosis. Cell Microbiol. 2017, 19, e12726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, R.O.; Manzanillo, P.S.; Cox, J.S. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 2012, 150, 803–815. [Google Scholar] [CrossRef] [Green Version]
- Manzanillo, P.S.; Ayres, J.S.; Watson, R.O.; Collins, A.C.; Souza, G.; Rae, C.S.; Schneider, D.S.; Nakamura, K.; Shiloh, M.U.; Cox, J.S. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature 2013, 501, 512–516. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.S.; Silwal, P.; Kim, S.Y.; Yoshimori, T.; Jo, E.K. Autophagy-activating strategies to promote innate defense against mycobacteria. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Silwal, P.; Paik, S.; Kim, J.K.; Yoshimori, T.; Jo, E.K. Regulatory Mechanisms of Autophagy-Targeted Antimicrobial Therapeutics against Mycobacterial Infection. Front. Cell. Infect. Microbiol. 2021, 11, 633360. [Google Scholar] [CrossRef]
- Rivas-Santiago, B.; Rivas Santiago, C.E.; Castaneda-Delgado, J.E.; Leon-Contreras, J.C.; Hancock, R.E.; Hernandez-Pando, R. Activity of LL-37, CRAMP and antimicrobial peptide-derived compounds E2, E6 and CP26 against Mycobacterium tuberculosis. Int. J. Antimicrob. Agents 2013, 41, 143–148. [Google Scholar] [CrossRef]
- Deffert, C.; Cachat, J.; Krause, K.H. Phagocyte NADPH oxidase, chronic granulomatous disease and mycobacterial infections. Cell Microbiol. 2014, 16, 1168–1178. [Google Scholar] [CrossRef]
- MacMicking, J.D.; North, R.J.; LaCourse, R.; Mudgett, J.S.; Shah, S.K.; Nathan, C.F. Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc. Natl. Acad. Sci. USA 1997, 94, 5243–5248. [Google Scholar] [CrossRef] [Green Version]
- Segal, A.W. How neutrophils kill microbes. Annu. Rev. Immunol. 2005, 23, 197–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jena, P.; Mohanty, S.; Mohanty, T.; Kallert, S.; Morgelin, M.; Lindstrom, T.; Borregaard, N.; Stenger, S.; Sonawane, A.; Sorensen, O.E. Azurophil granule proteins constitute the major mycobactericidal proteins in human neutrophils and enhance the killing of mycobacteria in macrophages. PLoS ONE 2012, 7, e50345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korbel, D.S.; Schneider, B.E.; Schaible, U.E. Innate immunity in tuberculosis: Myths and truth. Microbes Infect. 2008, 10, 995–1004. [Google Scholar] [CrossRef]
- North, R.J.; Jung, Y.J. Immunity to tuberculosis. Annu. Rev. Immunol. 2004, 22, 599–623. [Google Scholar] [CrossRef] [PubMed]
- Lyadova, I.V.; Panteleev, A.V. Th1 and Th17 Cells in Tuberculosis: Protection, Pathology, and Biomarkers. Mediators Inflamm. 2015, 2015, 854507. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Chen, T.; Mandelin, J.; Ceponis, A.; Miller, N.E.; Hukkanen, M.; Ma, G.F.; Konttinen, Y.T. Regulation of macrophage activation. Cell. Mol. Life Sci. 2003, 60, 2334–2346. [Google Scholar] [CrossRef]
- Schaible, U.E.; Sturgill-Koszycki, S.; Schlesinger, P.H.; Russell, D.G. Cytokine activation leads to acidification and increases maturation of Mycobacterium avium-containing phagosomes in murine macrophages. J. Immunol. 1998, 160, 1290–1296. [Google Scholar]
- Torrado, E.; Cooper, A.M. IL-17 and Th17 cells in tuberculosis. Cytokine Growth Factor Rev. 2010, 21, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Lin, P.L.; Flynn, J.L. CD8 T cells and Mycobacterium tuberculosis infection. Semin. Immunopathol. 2015, 37, 239–249. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.; Raqib, R.; Guethmundsson, G.H.; Bergman, P.; Agerberth, B.; Rekha, R.S. Host-Directed Therapy as a Novel Treatment Strategy to Overcome Tuberculosis: Targeting Immune Modulation. Antibiotics 2020, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.; Reljic, R.; Naylor, I.; Clark, S.O.; Falero-Diaz, G.; Singh, M.; Challacombe, S.; Marsh, P.D.; Ivanyi, J. Passive protection with immunoglobulin A antibodies against tuberculous early infection of the lungs. Immunology 2004, 111, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Maglione, P.J.; Xu, J.; Chan, J. B cells moderate inflammatory progression and enhance bacterial containment upon pulmonary challenge with Mycobacterium tuberculosis. J. Immunol. 2007, 178, 7222–7234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, Y.; Lee, H.J.; Jung, Y.M.; Jung, Y.J. Regulated Necrotic Cell Death in Alternative Tumor Therapeutic Strategies. Cells 2020, 9, 2709. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, X.; Gueydan, C.; Han, J. Plasma membrane changes during programmed cell deaths. Cell Res. 2018, 28, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Szondy, Z.; Sarang, Z.; Kiss, B.; Garabuczi, E.; Koroskenyi, K. Anti-inflammatory Mechanisms Triggered by Apoptotic Cells during Their Clearance. Front. Immunol. 2017, 8, 909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Lam, A.; Prabhu, R.; Gross, C.M.; Riesenberg, L.A.; Singh, V.; Aggarwal, S. Role of apoptosis and autophagy in tuberculosis. Am J. Physiol. Lung Cell. Mol. Physiol. 2017, 313, L218–L229. [Google Scholar] [CrossRef] [Green Version]
- Winau, F.; Weber, S.; Sad, S.; de Diego, J.; Hoops, S.L.; Breiden, B.; Sandhoff, K.; Brinkmann, V.; Kaufmann, S.H.; Schaible, U.E. Apoptotic vesicles crossprime CD8 T cells and protect against tuberculosis. Immunity 2006, 24, 105–117. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.J.; Lee, J.; Choi, J.A.; Cho, S.N.; Son, S.H.; Kwon, S.J.; Son, J.W.; Song, C.H. M1 macrophage dependent-p53 regulates the intracellular survival of mycobacteria. Apoptosis 2020, 25, 42–55. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.H.; Liu, H.; Ge, B. Innate immunity in tuberculosis: Host defense vs. pathogen evasion. Cell. Mol. Immunol. 2017, 14, 963–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckwith, K.S.; Beckwith, M.S.; Ullmann, S.; Saetra, R.S.; Kim, H.; Marstad, A.; Asberg, S.E.; Strand, T.A.; Haug, M.; Niederweis, M.; et al. Plasma membrane damage causes NLRP3 activation and pyroptosis during Mycobacterium tuberculosis infection. Nat. Commun. 2020, 11, 2270. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Khan, N.; Gan, H.; Tzelepis, F.; Nishimura, T.; Park, S.Y.; Divangahi, M.; Remold, H.G. Bcl-xL mediates RIPK3-dependent necrosis in M. tuberculosis-infected macrophages. Mucosal Immunol. 2017, 10, 1553–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stutz, M.D.; Ojaimi, S.; Allison, C.; Preston, S.; Arandjelovic, P.; Hildebrand, J.M.; Sandow, J.J.; Webb, A.I.; Silke, J.; Alexander, W.S.; et al. Necroptotic signaling is primed in Mycobacterium tuberculosis-infected macrophages, but its pathophysiological consequence in disease is restricted. Cell Death Differ. 2018, 25, 951–965. [Google Scholar] [CrossRef] [Green Version]
- Pajuelo, D.; Gonzalez-Juarbe, N.; Tak, U.; Sun, J.; Orihuela, C.J.; Niederweis, M. NAD(+) Depletion Triggers Macrophage Necroptosis, a Cell Death Pathway Exploited by Mycobacterium tuberculosis. Cell Rep. 2018, 24, 429–440. [Google Scholar] [CrossRef] [Green Version]
- Amaral, E.P.; Costa, D.L.; Namasivayam, S.; Riteau, N.; Kamenyeva, O.; Mittereder, L.; Mayer-Barber, K.D.; Andrade, B.B.; Sher, A. A major role for ferroptosis in Mycobacterium tuberculosis-induced cell death and tissue necrosis. J. Exp. Med. 2019, 216, 556–570. [Google Scholar] [CrossRef]
- Martin, C.J.; Booty, M.G.; Rosebrock, T.R.; Nunes-Alves, C.; Desjardins, D.M.; Keren, I.; Fortune, S.M.; Remold, H.G.; Behar, S.M. Efferocytosis is an innate antibacterial mechanism. Cell Host Microbe 2012, 12, 289–300. [Google Scholar] [CrossRef] [Green Version]
- Vorobjeva, N.V.; Chernyak, B.V. NETosis: Molecular Mechanisms, Role in Physiology and Pathology. Biochemistry 2020, 85, 1178–1190. [Google Scholar] [CrossRef]
- Braian, C.; Hogea, V.; Stendahl, O. Mycobacterium tuberculosis- induced neutrophil extracellular traps activate human macrophages. J. Innate Immun. 2013, 5, 591–602. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [Green Version]
- Braverman, J.; Sogi, K.M.; Benjamin, D.; Nomura, D.K.; Stanley, S.A. HIF-1alpha Is an Essential Mediator of IFN-gamma-Dependent Immunity to Mycobacterium tuberculosis. J. Immunol. 2016, 197, 1287–1297. [Google Scholar] [CrossRef] [PubMed]
- Agard, M.; Asakrah, S.; Morici, L.A. PGE(2) suppression of innate immunity during mucosal bacterial infection. Front. Cell. Infect. Microbiol. 2013, 3, 45. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Divangahi, M.; Gan, H.; Shin, D.S.; Hong, S.; Lee, D.M.; Serhan, C.N.; Behar, S.M.; Remold, H.G. Lipid mediators in innate immunity against tuberculosis: Opposing roles of PGE2 and LXA4 in the induction of macrophage death. J. Exp. Med. 2008, 205, 2791–2801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Divangahi, M.; Chen, M.; Gan, H.; Desjardins, D.; Hickman, T.T.; Lee, D.M.; Fortune, S.; Behar, S.M.; Remold, H.G. Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat. Immunol. 2009, 10, 899–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Lin, F.; Chen, L.; Chen, W.; Pan, X.; Bai, Y.; Cai, Y.; Lu, H. Lipoxin A4 regulates M1/M2 macrophage polarization via FPR2-IRF pathway. Inflammopharmacology 2022, 30, 487–498. [Google Scholar] [CrossRef]
- Bafica, A.; Scanga, C.A.; Serhan, C.; Machado, F.; White, S.; Sher, A.; Aliberti, J. Host control of Mycobacterium tuberculosis is regulated by 5-lipoxygenase-dependent lipoxin production. J. Clin. Investig. 2005, 115, 1601–1606. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Xu, R.; Gu, H.; Zhang, E.; Qu, J.; Cao, W.; Huang, X.; Yan, H.; He, J.; Cai, Z. Metabolic reprogramming in macrophage responses. Biomark. Res. 2021, 9, 1. [Google Scholar] [CrossRef]
- Howard, N.C.; Khader, S.A. Immunometabolism during Mycobacterium tuberculosis Infection. Trends Microbiol. 2020, 28, 832–850. [Google Scholar] [CrossRef]
- Brandenburg, J.; Marwitz, S.; Tazoll, S.C.; Waldow, F.; Kalsdorf, B.; Vierbuchen, T.; Scholzen, T.; Gross, A.; Goldenbaum, S.; Holscher, A.; et al. WNT6/ACC2-induced storage of triacylglycerols in macrophages is exploited by Mycobacterium tuberculosis. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Asalla, S.; Mohareer, K.; Banerjee, S. Small Molecule Mediated Restoration of Mitochondrial Function Augments Anti-Mycobacterial Activity of Human Macrophages Subjected to Cholesterol Induced Asymptomatic Dyslipidemia. Front. Cell. Infect. Microbiol. 2017, 7, 439. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.T.; Stenger, S.; Li, H.; Wenzel, L.; Tan, B.H.; Krutzik, S.R.; Ochoa, M.T.; Schauber, J.; Wu, K.; Meinken, C.; et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 2006, 311, 1770–1773. [Google Scholar] [CrossRef] [PubMed]
- Campbell, G.R.; Spector, S.A. Vitamin D inhibits human immunodeficiency virus type 1 and Mycobacterium tuberculosis infection in macrophages through the induction of autophagy. PLoS Pathog. 2012, 8, e1002689. [Google Scholar] [CrossRef] [Green Version]
- Wheelwright, M.; Kim, E.W.; Inkeles, M.S.; De Leon, A.; Pellegrini, M.; Krutzik, S.R.; Liu, P.T. All-trans retinoic acid-triggered antimicrobial activity against Mycobacterium tuberculosis is dependent on NPC2. J. Immunol. 2014, 192, 2280–2290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, M.M.; Basdeo, S.A.; Coleman, A.M.; Cheallaigh, C.N.; Peral de Castro, C.; McLaughlin, A.M.; Dunne, P.J.; Harris, J.; Keane, J. All-trans Retinoic Acid Augments Autophagy during Intracellular Bacterial Infection. Am. J. Respir. Cell Mol. Biol. 2018, 59, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Vilcheze, C.; Hartman, T.; Weinrick, B.; Jacobs, W.R., Jr. Mycobacterium tuberculosis is extraordinarily sensitive to killing by a vitamin C-induced Fenton reaction. Nat. Commun. 2013, 4, 1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; He, W.; Du, X.; Huang, Y.; Fu, Y.; Yang, Y.; Hu, C.; Li, S.; Wang, Q.; Wen, Q.; et al. Vitamin B1 Helps to Limit Mycobacterium tuberculosis Growth via Regulating Innate Immunity in a Peroxisome Proliferator-Activated Receptor-gamma-Dependent Manner. Front. Immunol. 2018, 9, 1778. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Hu, S.; Du, X.; Wen, Q.; Zhong, X.P.; Zhou, X.; Zhou, C.; Xiong, W.; Gao, Y.; Zhang, S.; et al. Vitamin B5 Reduces Bacterial Growth via Regulating Innate Immunity and Adaptive Immunity in Mice Infected with Mycobacterium tuberculosis. Front. Immunol. 2018, 9, 365. [Google Scholar] [CrossRef] [Green Version]
- Chaurasiya, S.K.; Srivastava, K.K. Downregulation of protein kinase C-alpha enhances intracellular survival of Mycobacteria: Role of PknG. BMC Microbiol. 2009, 9, 271. [Google Scholar] [CrossRef] [Green Version]
- Wong, D.; Bach, H.; Sun, J.; Hmama, Z.; Av-Gay, Y. Mycobacterium tuberculosis protein tyrosine phosphatase (PtpA) excludes host vacuolar-H+-ATPase to inhibit phagosome acidification. Proc. Natl. Acad. Sci. USA 2011, 108, 19371–19376. [Google Scholar] [CrossRef] [Green Version]
- Yogalingam, G.; Pendergast, A.M. Abl kinases regulate autophagy by promoting the trafficking and function of lysosomal components. J. Biol. Chem. 2008, 283, 35941–35953. [Google Scholar] [CrossRef] [Green Version]
- Bruns, H.; Stegelmann, F.; Fabri, M.; Dohner, K.; van Zandbergen, G.; Wagner, M.; Skinner, M.; Modlin, R.L.; Stenger, S. Abelson tyrosine kinase controls phagosomal acidification required for killing of Mycobacterium tuberculosis in human macrophages. J. Immunol. 2012, 189, 4069–4078. [Google Scholar] [CrossRef]
- Cho, J.H.; Lee, H.J.; Ko, H.J.; Yoon, B.I.; Choe, J.; Kim, K.C.; Hahn, T.W.; Han, J.A.; Choi, S.S.; Jung, Y.M.; et al. The TLR7 agonist imiquimod induces anti-cancer effects via autophagic cell death and enhances anti-tumoral and systemic immunity during radiotherapy for melanoma. Oncotarget 2017, 8, 24932–24948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Kang, S.J.; Woo, Y.; Hahn, T.W.; Ko, H.J.; Jung, Y.J. TLR7 Stimulation With Imiquimod Induces Selective Autophagy and Controls Mycobacterium tuberculosis Growth in Mouse Macrophages. Front. Microbiol. 2020, 11, 1684. [Google Scholar] [CrossRef]
- Kim, J.J.; Lee, H.M.; Shin, D.M.; Kim, W.; Yuk, J.M.; Jin, H.S.; Lee, S.H.; Cha, G.H.; Kim, J.M.; Lee, Z.W.; et al. Host cell autophagy activated by antibiotics is required for their effective antimycobacterial drug action. Cell Host Microbe 2012, 11, 457–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiebler, M.; Brown, K.; Hegyi, K.; Newton, S.M.; Renna, M.; Hepburn, L.; Klapholz, C.; Coulter, S.; Obregon-Henao, A.; Henao Tamayo, M.; et al. Functional drug screening reveals anticonvulsants as enhancers of mTOR-independent autophagic killing of Mycobacterium tuberculosis through inositol depletion. EMBO Mol. Med. 2015, 7, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Ko, H.J.; Kim, S.H.; Jung, Y.J. Pasakbumin A controls the growth of Mycobacterium tuberculosis by enhancing the autophagy and production of antibacterial mediators in mouse macrophages. PLoS ONE 2019, 14, e0199799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.Y.; Gutierrez, N.M.; Marzuki, M.B.; Lu, X.; Foreman, T.W.; Paleja, B.; Lee, B.; Balachander, A.; Chen, J.; Tsenova, L.; et al. Host sirtuin 1 regulates mycobacterial immunopathogenesis and represents a therapeutic target against tuberculosis. Sci. Immunol. 2017, 2, eaaj1789. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.S.; Jin, Y.B.; Kim, Y.S.; Kim, S.; Kim, J.K.; Lee, H.M.; Suh, H.W.; Choe, J.H.; Kim, Y.J.; Koo, B.S.; et al. SIRT3 promotes antimycobacterial defenses by coordinating mitochondrial and autophagic functions. Autophagy 2019, 15, 1356–1375. [Google Scholar] [CrossRef]
- Subbian, S.; Koo, M.S.; Tsenova, L.; Khetani, V.; Zeldis, J.B.; Fallows, D.; Kaplan, G. Pharmacologic Inhibition of Host Phosphodiesterase-4 Improves Isoniazid-Mediated Clearance of Mycobacterium tuberculosis. Front. Immunol. 2016, 7, 238. [Google Scholar] [CrossRef] [Green Version]
- Subbian, S.; Tsenova, L.; Holloway, J.; Peixoto, B.; O’Brien, P.; Dartois, V.; Khetani, V.; Zeldis, J.B.; Kaplan, G. Adjunctive Phosphodiesterase-4 Inhibitor Therapy Improves Antibiotic Response to Pulmonary Tuberculosis in a Rabbit Model. eBioMedicine 2016, 4, 104–114. [Google Scholar] [CrossRef] [Green Version]
- Critchley, J.A.; Young, F.; Orton, L.; Garner, P. Corticosteroids for prevention of mortality in people with tuberculosis: A systematic review and meta-analysis. Lancet Infect. Dis. 2013, 13, 223–237. [Google Scholar] [CrossRef]
- Gupta, P.K.; Chakraborty, P.; Kumar, S.; Singh, P.K.; Rajan, M.G.; Sainis, K.B.; Kulkarni, S. G1-4A, a Polysaccharide from Tinospora cordifolia Inhibits the Survival of Mycobacterium tuberculosis by Modulating Host Immune Responses in TLR4 Dependent Manner. PLoS ONE 2016, 11, e0154725. [Google Scholar] [CrossRef] [PubMed]
- Jurado, J.O.; Alvarez, I.B.; Pasquinelli, V.; Martinez, G.J.; Quiroga, M.F.; Abbate, E.; Musella, R.M.; Chuluyan, H.E.; Garcia, V.E. Programmed death (PD)-1:PD-ligand 1/PD-ligand 2 pathway inhibits T cell effector functions during human tuberculosis. J. Immunol. 2008, 181, 116–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roca, F.J.; Ramakrishnan, L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell 2013, 153, 521–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 2014, 136, 4551–4556. [Google Scholar] [CrossRef]
- Singhal, A.; Jie, L.; Kumar, P.; Hong, G.S.; Leow, M.K.; Paleja, B.; Tsenova, L.; Kurepina, N.; Chen, J.; Zolezzi, F.; et al. Metformin as adjunct antituberculosis therapy. Sci. Transl. Med. 2014, 6, 263ra159. [Google Scholar] [CrossRef]
- Padmapriydarsini, C.; Mamulwar, M.; Mohan, A.; Shanmugam, P.; Gomathy, N.S.; Mane, A.; Singh, U.B.; Pavankumar, N.; Kadam, A.; Kumar, H.; et al. Randomized Trial of Metformin With Anti-Tuberculosis Drugs for Early Sputum Conversion in Adults With Pulmonary Tuberculosis. Clin. Infect. Dis. 2022, 75, 425–434. [Google Scholar] [CrossRef]
- Parihar, S.P.; Guler, R.; Khutlang, R.; Lang, D.M.; Hurdayal, R.; Mhlanga, M.M.; Suzuki, H.; Marais, A.D.; Brombacher, F. Statin therapy reduces the Mycobacterium tuberculosis burden in human macrophages and in mice by enhancing autophagy and phagosome maturation. J. Infect. Dis. 2014, 209, 754–763. [Google Scholar] [CrossRef] [Green Version]
- Mayer-Barber, K.D.; Andrade, B.B.; Oland, S.D.; Amaral, E.P.; Barber, D.L.; Gonzales, J.; Derrick, S.C.; Shi, R.; Kumar, N.P.; Wei, W.; et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature 2014, 511, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Byrne, S.T.; Denkin, S.M.; Zhang, Y. Aspirin and ibuprofen enhance pyrazinamide treatment of murine tuberculosis. J. Antimicrob. Chemother. 2007, 59, 313–316. [Google Scholar] [CrossRef] [Green Version]
- Adikesavalu, H.; Gopalaswamy, R.; Kumar, A.; Ranganathan, U.D.; Shanmugam, S. Autophagy Induction as a Host-Directed Therapeutic Strategy against Mycobacterium tuberculosis Infection. Medicina 2021, 57, 522. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.E.; Lee, H.K. Pattern recognition receptors and autophagy. Front. Immunol. 2014, 5, 300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howitz, K.T.; Bitterman, K.J.; Cohen, H.Y.; Lamming, D.W.; Lavu, S.; Wood, J.G.; Zipkin, R.E.; Chung, P.; Kisielewski, A.; Zhang, L.L.; et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003, 425, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Sarrica, A.; Kirika, N.; Romeo, M.; Salmona, M.; Diomede, L. Safety and Toxicology of Magnolol and Honokiol. Planta Med. 2018, 84, 1151–1164. [Google Scholar] [CrossRef] [Green Version]
- Krug, S.; Parveen, S.; Bishai, W.R. Host-Directed Therapies: Modulating Inflammation to Treat Tuberculosis. Front. Immunol. 2021, 12, 660916. [Google Scholar] [CrossRef]
- Cohen, S.B.; Gern, B.H.; Delahaye, J.L.; Adams, K.N.; Plumlee, C.R.; Winkler, J.K.; Sherman, D.R.; Gerner, M.Y.; Urdahl, K.B. Alveolar Macrophages Provide an Early Mycobacterium tuberculosis Niche and Initiate Dissemination. Cell Host Microbe 2018, 24, 439–446.e4. [Google Scholar] [CrossRef] [Green Version]
- Pennini, M.E.; Liu, Y.; Yang, J.; Croniger, C.M.; Boom, W.H.; Harding, C.V. CCAAT/enhancer-binding protein beta and delta binding to CIITA promoters is associated with the inhibition of CIITA expression in response to Mycobacterium tuberculosis 19-kDa lipoprotein. J. Immunol. 2007, 179, 6910–6918. [Google Scholar] [CrossRef] [Green Version]
- Ankley, L.; Thomas, S.; Olive, A.J. Fighting Persistence: How Chronic Infections with Mycobacterium tuberculosis Evade T Cell-Mediated Clearance and New Strategies To Defeat Them. Infect. Immun. 2020, 88, e00916-19. [Google Scholar] [CrossRef]
- Briken, V.; Miller, J.L. Living on the edge: Inhibition of host cell apoptosis by Mycobacterium tuberculosis. Future Microbiol. 2008, 3, 415–422. [Google Scholar] [CrossRef] [Green Version]
- Mehrotra, P.; Jamwal, S.V.; Saquib, N.; Sinha, N.; Siddiqui, Z.; Manivel, V.; Chatterjee, S.; Rao, K.V. Pathogenicity of Mycobacterium tuberculosis is expressed by regulating metabolic thresholds of the host macrophage. PLoS Pathog. 2014, 10, e1004265. [Google Scholar] [CrossRef] [Green Version]
- Sorgi, C.A.; Soares, E.M.; Rosada, R.S.; Bitencourt, C.S.; Zoccal, K.F.; Pereira, P.A.T.; Fontanari, C.; Brandao, I.; Masson, A.P.; Ramos, S.G.; et al. Eicosanoid pathway on host resistance and inflammation during Mycobacterium tuberculosis infection is comprised by LTB4 reduction but not PGE2 increment. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165574. [Google Scholar] [CrossRef]
- Lee, J.Y.; Jung, Y.W.; Jeong, I.; Joh, J.S.; Sim, S.Y.; Choi, B.; Jee, H.G.; Lim, D.G. Immune parameters differentiating active from latent tuberculosis infection in humans. Tuberculosis 2015, 95, 758–763. [Google Scholar] [CrossRef] [PubMed]
- Rosas, I.O.; Diaz, G.; Gottlieb, R.L.; Lobo, S.M.; Robinson, P.; Hunter, B.D.; Cavalcante, A.W.; Overcash, J.S.; Hanania, N.A.; Skarbnik, A.; et al. Tocilizumab and remdesivir in hospitalized patients with severe COVID-19 pneumonia: A randomized clinical trial. Intensive Care Med. 2021, 47, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Esposito, F.; Matthes, H.; Schad, F. Seven COVID-19 Patients Treated with C-Reactive Protein (CRP) Apheresis. J. Clin. Med. 2022, 11, 1956. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, A.R.; Nishiguchi, T.; Grimm, S.L.; Schlesinger, L.S.; Graviss, E.A.; Cirillo, J.D.; Coarfa, C.; Mandalakas, A.M.; Heyckendorf, J.; Kaufmann, S.H.E.; et al. Tuberculosis endotypes to guide stratified host-directed therapy. Med 2021, 2, 217–232. [Google Scholar] [CrossRef]
- Tobin, D.M.; Vary, J.C., Jr.; Ray, J.P.; Walsh, G.S.; Dunstan, S.J.; Bang, N.D.; Hagge, D.A.; Khadge, S.; King, M.C.; Hawn, T.R.; et al. The lta4h locus modulates susceptibility to mycobacterial infection in zebrafish and humans. Cell 2010, 140, 717–730. [Google Scholar] [CrossRef] [Green Version]
- Roca, F.J.; Whitworth, L.J.; Prag, H.A.; Murphy, M.P.; Ramakrishnan, L. Tumor necrosis factor induces pathogenic mitochondrial ROS in tuberculosis through reverse electron transport. Science 2022, 376, eabh2841. [Google Scholar] [CrossRef]
- Lee, Y.J.; Han, S.K.; Park, J.H.; Lee, J.K.; Kim, D.K.; Chung, H.S.; Heo, E.Y. The effect of metformin on culture conversion in tuberculosis patients with diabetes mellitus. Korean J. Intern. Med. 2018, 33, 933–940. [Google Scholar] [CrossRef]
- Anand, K.; Sahu, G.; Burns, E.; Ensor, A.; Ensor, J.; Pingali, S.R.; Subbiah, V.; Iyer, S.P. Mycobacterial infections due to PD-1 and PD-L1 checkpoint inhibitors. ESMO Open 2020, 5, e000866. [Google Scholar] [CrossRef]
- Barber, D.L.; Sakai, S.; Kudchadkar, R.R.; Fling, S.P.; Day, T.A.; Vergara, J.A.; Ashkin, D.; Cheng, J.H.; Lundgren, L.M.; Raabe, V.N.; et al. Tuberculosis following PD-1 blockade for cancer immunotherapy. Sci. Transl. Med. 2019, 11, eaat2702. [Google Scholar] [CrossRef]
- Lee, J.J.; Chan, A.; Tang, T. Tuberculosis reactivation in a patient receiving anti-programmed death-1 (PD-1) inhibitor for relapsed Hodgkin’s lymphoma. Acta. Oncol. 2016, 55, 519–520. [Google Scholar] [CrossRef] [PubMed]
- Van Eeden, R.; Rapoport, B.L.; Smit, T.; Anderson, R. Tuberculosis Infection in a Patient Treated With Nivolumab for Non-small Cell Lung Cancer: Case Report and Literature Review. Front. Oncol. 2019, 9, 659. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Major Target and Drugs | Mode of Action | Effect | Developmental Stage as HDT for TB | Licensed | Ref. |
|---|---|---|---|---|---|
| Phagosome maturation and autophagy | |||||
| Imatinib | induces v-ATPase pump subunit expression and recruitment | promoting phagosomal acidification | preclinical, randomized clinical trial | Leukemia (licensed) | [83] |
| Imiquimod | stimulates TLR7 | activating autophagy and increasing NO production | preclinical | Superficial basal cell carcinoma (licensed) | [84,85] |
| Isoniazid | activates AMPK through intracellular Ca2+ influx, inducing Ca2+-dependent autophagy | activating autophagy, increasing NOX2-induced mtROS | preclinical | Anti-TB drug (licensed) | [86] |
| Pyrazinamide | activates AMPK through intracellular Ca2+ influx, inducing Ca2+-dependent autophagy | activating autophagy, increasing NOX2-induced mtROS | preclinical | Anti-TB drug (licensed) | [86] |
| Carbamazepine | induces mTOR-independent autophagy through AMPK activation | activating autophagy | preclinical | Anti-convulsant (licensed) | [87] |
| Pasakbumin A | ERK1/2-mediated signaling | activating autophagy, inducing phagosome maturation | preclinical | Natural compound (preclinical) | [88] |
| Resveratrol | activates SIRT1 | activating autophagy, inducing phagosome-lysosome fusion | preclinical | Nutritional supplement (licensed) | [89] |
| Honokiol | activates SIRT3 | activating autophagy, enhancing antimicrobial response, reducing mitochondrial damage and oxidative stress | preclinical | Nutritional supplement (licensed) | [90] |
| Inflammation | |||||
| CC-11050 | inhibits PDE-4 and downregulates TNF-α | resolving inflammation, improving therapeutic effect of isoniazid | preclinical | Leprosy, cancer (licensed) | [91,92] |
| Prednisone, Dexamethasone | Anti-inflammation and immunosuppression | resolving inflammation, attenuating pathology | preclinical | Anti-inflammatory (licensed) | [93] |
| Adaptive immunity | |||||
| G1-4A | stimulates TLR4 | upregulating expression of MHC class II and CD86, increasing secretion of proinflammatory cytokine and NO | preclinical | Natural compound (preclinical) | [94] |
| Nivolumab, Pembrolizumab | anti-PD-1 mAb | enhancing degranulation of CD8+ T cell, increasing ratio of IFN-γ producing lymphocytes | preclinical | Cancer (licensed) | [95] |
| Cell death | |||||
| Alisporivir | inhibits ROS-induced necroptosis | attenuating tissue damage induced by excessive TNF-α through synergistic effect with Desipramine | preclinical | Hepatitis C (clinical trial) | [96] |
| Desipramine | inhibits ROS-induced necroptosis | attenuating tissue damage induced by excessive TNF-α through synergistic effect with Alisporivir | preclinical | Antidepressant (licensed) | [96] |
| Ferrostatin-1 | antioxidant that eliminates lipid ROS | inhibiting lipid peroxidation | preclinical | Huntington’s disease, periventricular leukomalacia, kidney dysfunction(preclinical) | [58,97] |
| Metabolism | |||||
| Metformin | activates AMPK and inhibits mitochondrial respiratory chain | promoting phagosome-lysosome fusion, increasing mtROS | randomized clinical trial | Diabetes (licensed) | [98,99] |
| Statins | inhibits HMG-CoA reductase | promoting phagosome maturation and autophagy | preclinical | Hypercholesterolemia (licensed) | [100] |
| Zileuton | inhibits 5-LOX | inhibiting leukotriene synthesis | preclinical | Asthma (licensed) | [101] |
| Ibuprofen, acetylsalicylic acid | inhibits COX | inhibiting PGE2 synthesis, improving therapeutic effect of pyrazinamide | preclinical | Pain, fever (licensed) | [102] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, E.-K.; Lee, H.-J.; Jung, Y.-J. Host-Directed Therapies for Tuberculosis. Pathogens 2022, 11, 1291. https://doi.org/10.3390/pathogens11111291
Jeong E-K, Lee H-J, Jung Y-J. Host-Directed Therapies for Tuberculosis. Pathogens. 2022; 11(11):1291. https://doi.org/10.3390/pathogens11111291
Chicago/Turabian StyleJeong, Eui-Kwon, Hyo-Ji Lee, and Yu-Jin Jung. 2022. "Host-Directed Therapies for Tuberculosis" Pathogens 11, no. 11: 1291. https://doi.org/10.3390/pathogens11111291