
Genetic Characterization and Variation of African Swine Fever Virus China/GD/2019 Strain in Domestic Pigs


Abstract
:1. Introduction
2. Results
2.1. Complete Genome Sequence of ASFV China/GD/2019 Strain
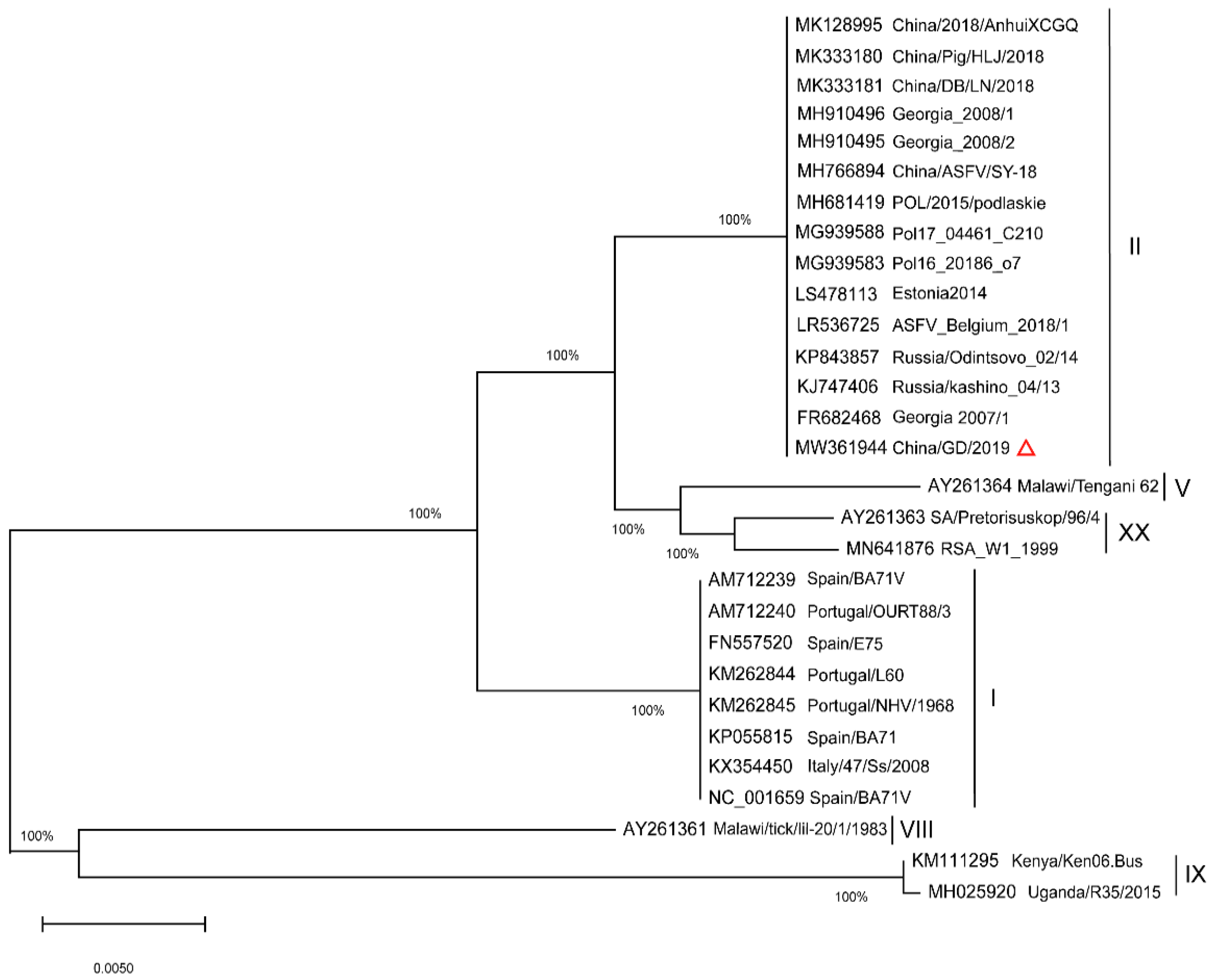
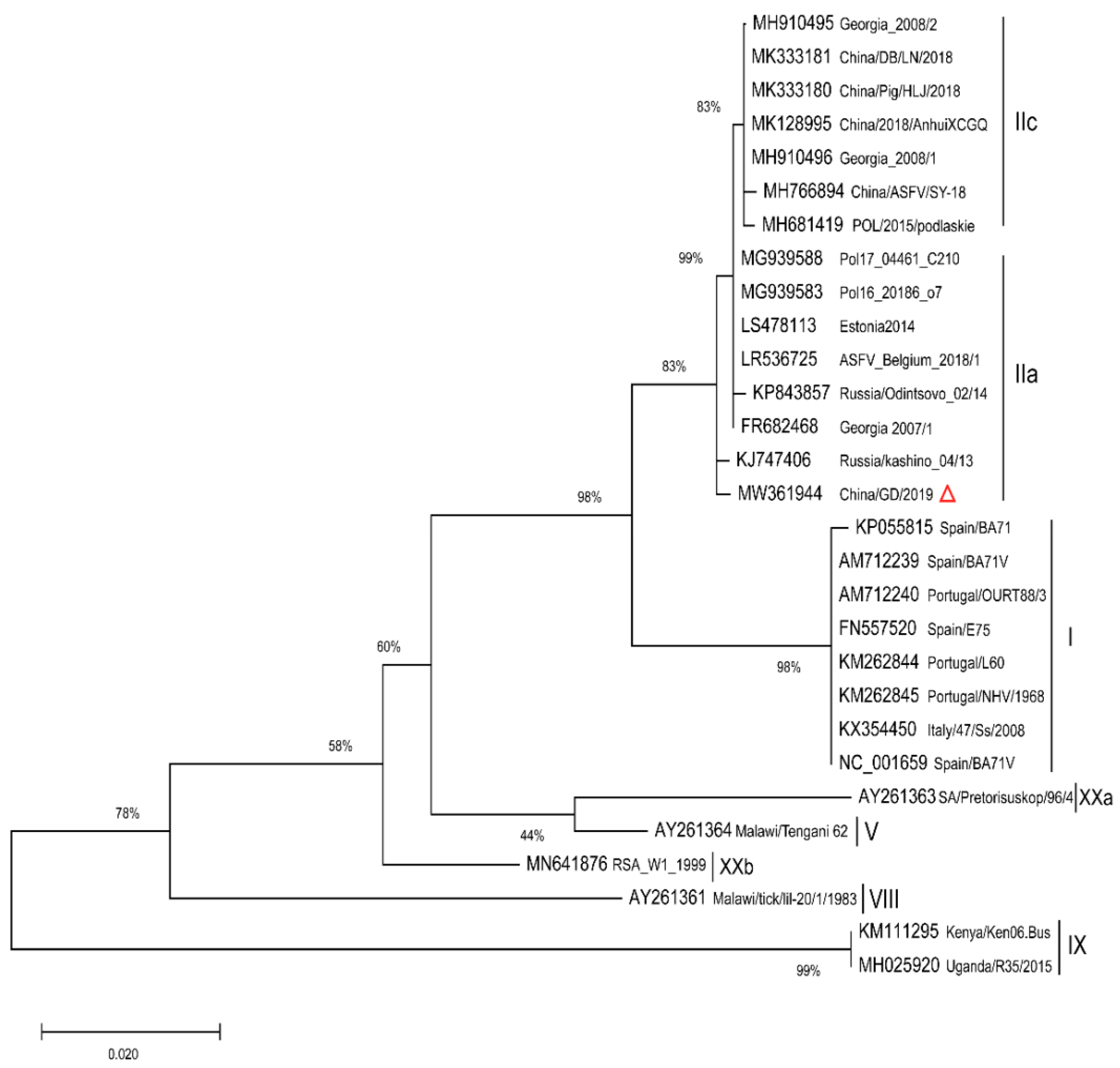
2.2. Phylogenetic Analysis of Full Length p72 (B646L) and p54 (E183L) Genes
2.3. Core Genes and Specific Genes Analysis
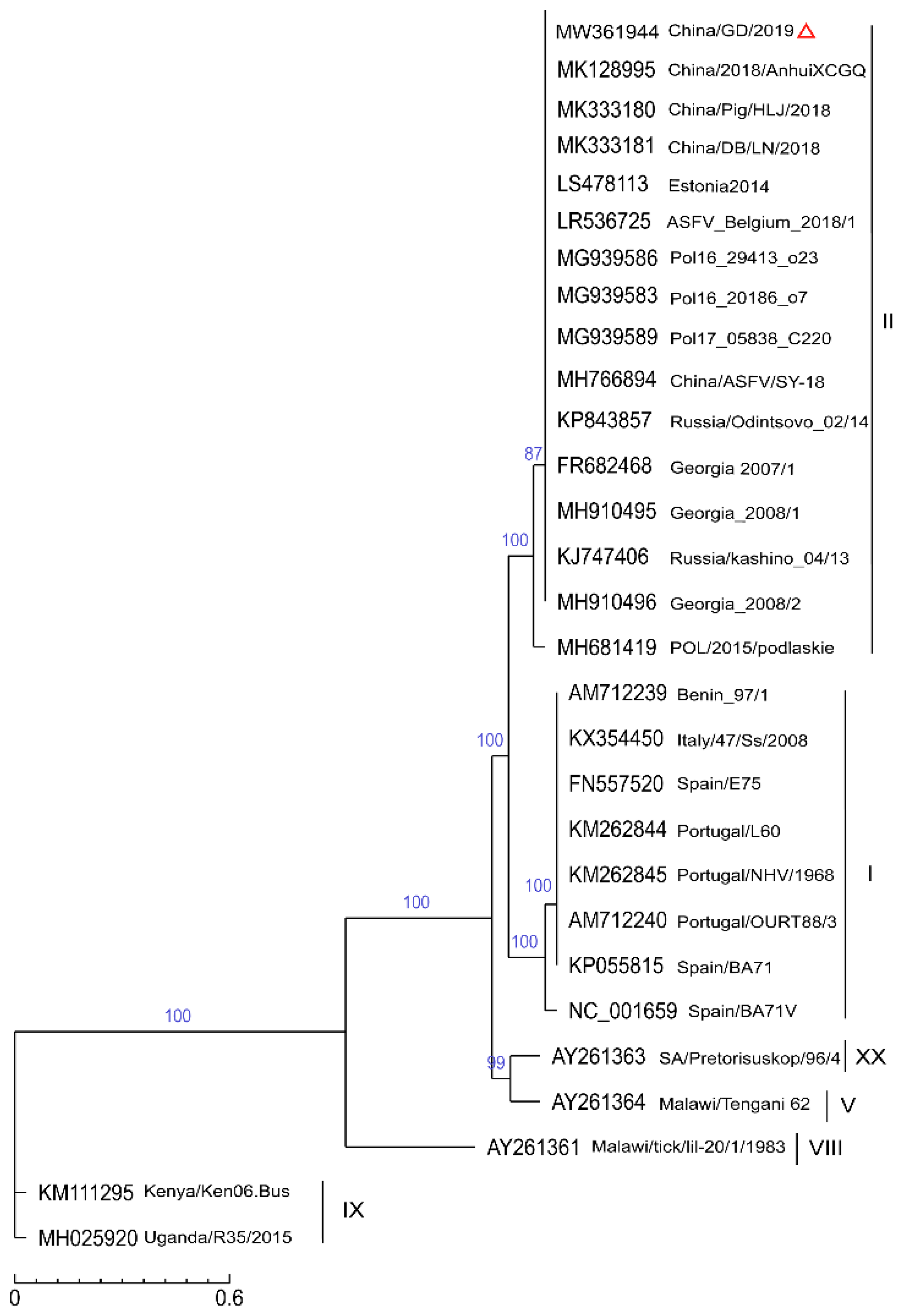
2.4. Phylogenetic Analysis of the SNP
2.5. SNP/InDel Analysis of China/GD/2019 Strain
3. Discussion
4. Materials and Methods
4.1. Ethics Approval and Consent to Participate
4.2. Field Samples
4.3. DNA Extraction
4.4. Genome Sequencing and Assembly Analysis
4.5. SNP/InDel Analysis
4.6. Core Genes and Pan Genes Analysis
4.7. Phylogenetic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Enjuanes, L.; Carrascosa, A.L.; Viñuela, E. Isolation and properties of the DNA of African swine fever (ASF) virus. J. Gen. Virol. 1976, 32, 479–492. [Google Scholar] [CrossRef]
- Dixon, L.K. Molecular cloning and restriction enzyme mapping of an African swine fever virus isolate from Malawi. J. Gen. Virol. 1988, 69, 1683–1694. [Google Scholar] [CrossRef]
- Galindo, I.; Alonso, C. African Swine Fever Virus: A Review. Viruses 2017, 9, 103. [Google Scholar] [CrossRef] [Green Version]
- Martins, C.L.; Leitão, A.C. Porcine immune responses to African swine fever virus (ASFV) infection. Vet. Immunol. Immunopathol. 1994, 43, 99–106. [Google Scholar] [CrossRef]
- Ortín, J.; Enjuanes, L.; Viñuela, E. Cross-links in African swine fever virus DNA. J. Virol. 1979, 31, 579–583. [Google Scholar] [CrossRef] [Green Version]
- Costard, S.; Wieland, B.; de Glanville, W.; Jori, F.; Rowlands, R.; Vosloo, W.; Roger, F.; Pfeiffer, D.U.; Dixon, L.K. African swine fever: How can global spread be prevented? Philos. Trans. R. Soc. Lond. Ser. Biol. Sci. 2009, 364, 2683–2696. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Ren, Z.; Wang, Q.; Ge, S.; Liu, Y.; Liu, C.; Liu, F.; Hu, Y.; Li, J.; Bao, J.; et al. Infection of African swine fever in wild boar, China, 2018. Transbound. Emerg. Dis. 2019, 66, 1395–1398. [Google Scholar] [CrossRef]
- Bao, J.; Wang, Q.; Lin, P.; Liu, C.; Li, L.; Wu, X.; Chi, T.; Xu, T.; Ge, S.; Liu, Y.; et al. Genome comparison of African swine fever virus China/2018/AnhuiXCGQ strain and related European p72 Genotype II strains. Transbound. Emerg. Dis. 2019, 66, 1167–1176. [Google Scholar] [CrossRef]
- Zhao, D.; Liu, R.; Zhang, X.; Li, F.; Wang, J.; Zhang, J.; Liu, X.; Wang, L.; Zhang, J.; Wu, X.; et al. Replication and virulence in pigs of the first African swine fever virus isolated in China. Emerg. Microbes. Infect. 2019, 8, 438–447. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Li, N.; Luo, Y.; Liu, Y.; Miao, F.; Chen, T.; Zhang, S.; Cao, P.; Li, X.; Tian, K.; et al. Emergence of African Swine Fever in China, 2018. Transbound. Emerg. Dis. 2018, 65, 1482–1484. [Google Scholar] [CrossRef] [Green Version]
- Yáñez, R.J.; Rodríguez, J.M.; Nogal, M.L.; Yuste, L.; Enríquez, C.; Rodriguez, J.F.; Viñuela, E. Analysis of the complete nucleotide sequence of African swine fever virus. Virology 1995, 208, 249–278. [Google Scholar] [CrossRef] [Green Version]
- Blasco, R.; de la Vega, I.; Almazán, F.; Agüero, M.; Viñuela, E. Genetic variation of African swine fever virus: Variable regions near the ends of the viral DNA. Virology 1989, 173, 251–257. [Google Scholar] [CrossRef]
- Dixon, L.K.; Twigg, S.R.; Baylis, S.A.; Vydelingum, S.; Bristow, C.; Hammond, J.M.; Smith, G.L. Nucleotide sequence of a 55 kbp region from the right end of the genome of a pathogenic African swine fever virus isolate (Malawi LIL20/1). J. Gen. Virol. 1994, 75, 1655–1684. [Google Scholar] [CrossRef]
- Blasco, R.; Agüero, M.; Almendral, J.M.; Viñuela, E. Variable and constant regions in African swine fever virus DNA. Virology 1989, 168, 330–338. [Google Scholar] [CrossRef]
- de la Vega, I.; Viñuela, E.; Blasco, R. Genetic variation and multigene families in African swine fever virus. Virology 1990, 179, 234–246. [Google Scholar] [CrossRef]
- Vydelingum, S.; Baylis, S.A.; Bristow, C.; Smith, G.L.; Dixon, L.K. Duplicated genes within the variable right end of the genome of a pathogenic isolate of African swine fever virus. J. Gen. Virol. 1993, 74, 2125–2130. [Google Scholar] [CrossRef]
- Zhu, Z.; Chen, H.; Liu, L.; Cao, Y.; Jiang, T.; Zou, Y.; Peng, Y. Classification and characterization of multigene family proteins of African swine fever viruses. Brief. Bioinform. 2020, 22, 4. [Google Scholar] [CrossRef]
- Gallardo, C.; Fernández-Pinero, J.; Pelayo, V.; Gazaev, I.; Markowska-Daniel, I.; Pridotkas, G.; Nieto, R.; Fernández-Pacheco, P.; Bokhan, S.; Nevolko, O.; et al. Genetic variation among African swine fever genotype II viruses, eastern and central Europe. Emerg. Infect. 2014, 20, 1544–1547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simulundu, E.; Sinkala, Y.; Chambaro, H.M.; Chinyemba, A.; Banda, F.; Mooya, L.E.; Ndebe, J.; Chitanga, S.; Makungu, C.; Munthali, G.; et al. Genetic characterisation of African swine fever virus from 2017 outbreaks in Zambia: Identification of p72 genotype II variants in domestic pigs. Onderstepoort J. Vet. Res. 2018, 85, e1–e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, X.; He, X.; Zhang, X.; Zhang, X.; Liu, L.; Guan, Y.; Zhang, Y.; Bu, Z. Genome sequences derived from pig and dried blood pig feed samples provide important insights into the transmission of African swine fever virus in China in 2018. Emerg. Microbes Infect. 2019, 8, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Tao, D.; Sun, D.; Liu, Y.; Wei, S.; Yang, Z.; An, T.; Shan, F.; Chen, Z.; Liu, J. One year of African swine fever outbreak in China. Acta Trop. 2020, 211, 105602. [Google Scholar] [CrossRef]
- Bastos, A.D.; Penrith, M.L.; Crucière, C.; Edrich, J.L.; Hutchings, G.; Roger, F.; Couacy-Hymann, E.G.; Thomson, G.R. Genotyping field strains of African swine fever virus by partial p72 gene characterisation. Arch. Virol. 2003, 148, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.M.; Balaji, S.; Koonin, E.V.; Aravind, L. Evolutionary genomics of nucleo-cytoplasmic large DNA viruses. Virus Res. 2006, 117, 156–184. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Morrissy, C.J.; Westbury, H.A. Strong sequence conservation of African swine fever virus p72 protein provides the molecular basis for its antigenic stability. Arch. Virol. 1996, 141, 1795–1802. [Google Scholar] [CrossRef] [PubMed]
- Irusta, P.M.; Borca, M.V.; Kutish, G.F.; Lu, Z.; Caler, E.; Carrillo, C.; Rock, D.L. Amino acid tandem repeats within a late viral gene define the central variable region of African swine fever virus. Virology 1996, 220, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Boinas, F.S.; Hutchings, G.H.; Dixon, L.K.; Wilkinson, P.J. Characterization of pathogenic and non-pathogenic African swine fever virus isolates from Ornithodoros erraticus inhabiting pig premises in Portugal. J. Gen. Virol. 2004, 85, 2177–2187. [Google Scholar] [CrossRef] [PubMed]
- Chapman, D.A.G.; Tcherepanov, V.; Upton, C.; Dixon, L.K. Comparison of the genome sequences of non-pathogenic and pathogenic African swine fever virus isolates. J. Gen. Virol. 2008, 89, 397–408. [Google Scholar] [CrossRef]
- Kipanyula, M.J.; Nong′ona, S.W. Variations in clinical presentation and anatomical distribution of gross lesions of African swine fever in domestic pigs in the southern highlands of Tanzania: A field experience. Trop. Anim. Health Prod. 2017, 49, 303–310. [Google Scholar] [CrossRef]
- de Villiers, E.P.; Gallardo, C.; Arias, M.; da Silva, M.; Upton, C.; Martin, R.; Bishop, R.P. Phylogenomic analysis of 11 complete African swine fever virus genome sequences. Virology 2010, 400, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Farlow, J.; Donduashvili, M.; Kokhreidze, M.; Kotorashvili, A.; Vepkhvadze, N.G.; Kotaria, N.; Gulbani, A. Intra-epidemic genome variation in highly pathogenic African swine fever virus (ASFV) from the country of Georgia. Virol. J. 2018, 15, 190. [Google Scholar] [CrossRef] [Green Version]
- Owolodun, O.A.; Obishakin, E.T.; Ekong, P.S.; Yakubu, B. Investigation of African swine fever in slaughtered pigs, Plateau state, Nigeria, 2004-2006. Trop. Anim. Health Prod. 2010, 42, 1605–1610. [Google Scholar] [CrossRef]
- Phologane, S.B.; Bastos, A.D.; Penrith, M.L. Intra- and inter-genotypic size variation in the central variable region of the 9RL open reading frame of diverse African swine fever viruses. Virus. Genes 2005, 31, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Pan, I.C. African swine fever virus: Generation of subpopulations with altered immunogenicity and virulence following passage in cell cultures. J. Vet. Med. Sci. 1992, 54, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Afonso, C.L.; Piccone, M.E.; Zaffuto, K.M.; Neilan, J.; Kutish, G.F.; Lu, Z.; Balinsky, C.A.; Gibb, T.R.; Bean, T.J.; Zsak, L.; et al. African swine fever virus multigene family 360 and 530 genes affect host interferon response. J. Virol. 2004, 78, 1858–1864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Luo, Y.; Zhao, Y.; Gao, G.F.; Bi, Y.; Qiu, H.J. Comparative genomic analysis reveals an ’open’ pan-genome of African swine fever virus. Transbound. Emerg. Dis. 2020, 67, 1553–1562. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Name | GenBank No. | Origin | Year | p72 gt | Reference |
|---|---|---|---|---|---|
| China/GD/2019 | MW361944 | China | 2019 | II | This study |
| China/2018/AnhuiXCGQ | MK128995 | China | 2018 | II | Bao, J. et al. (2019) |
| POL/2015/Podlaskie | MH681419 | Poland | 2015 | II | Olesen et al. (2018) |
| Georgia 2007/1 | FR682468 | Georgia | 2007 | II | Chapman et al. (2011) |
| Estonia 2014 | LS478113 | Estonia | 2014 | II | Zani et al. (2018) |
| Russia/Odintsovo_02/14 | KP843857 | Russia | 2014 | II | Unpublished |
| Spain/E75 | FN557520 | Spain | 1975 | I | de Villiers et al. (2010) |
| Malawi/Tengani 62 | AY261364 | Malawi | 1962 | V/I | Pan (1992) |
| SA/Pretorisuskop/96/4 | AY261363 | South Africa | 1996 | XX/I | Zsak et al. (2005) |
| Malawi/tick/Lil-20/1/1983 | AY261361 | Malawi | 1983 | VIII | Haresnape and Wilkinson (1989) |
| Portugal/OURT88/3 | AM712240 | Portugal | 1988 | I | Chapman et al. (2008) |
| Benin 97/1 | AM712239 | Benin | 1997 | I | Chapman et al. (2008) |
| Kenya/Ken06.Bus | KM111295 | Kenya | 2006 | IX | Bishop et al. (2015) |
| Portugal/L60 | KM262844 | Portugal | 1960 | I | Portugal et al. (2015) |
| Portugal/NHV/1968 | KM262845 | Portugal | 1968 | I | Portugal et al. (2015) |
| Italy/47/Ss/2008 | KX354450 | Italy | 2008 | I | Granberg et al. (2016) |
| Spain/BA71 | KP055815 | Spain | 1971 | I | Rodriguez et al. (2015) |
| Uganda/R35/2015 | MH025920 | Uganda | 2015 | IX | Unpublished |
| China/Pig/HLJ/2018 | MK333180 | China | 2018 | II | Wen, X. et al. (2019) |
| China/DB/LN/2018 | MK333181 | China | 2018 | II | Wen, X. et al. (2019) |
| Georgia_2008/1 | MH910495 | Georgia | 2008 | II | Farlow et al. (2018) |
| Georgia_2008/2 | MH910496 | Georgia | 2008 | II | Farlow et al. (2018) |
| China/2018/SY-18 | MH766894 | China | 2018 | II | Miao, F. et al. (2018) |
| Pol16_20186_o7 | MG939583 | Poland | 2016 | II | Wozniakowski et al. (2018) |
| Pol16_29413_o23 | MG939586 | Poland | 2016 | II | Wozniakowski et al. (2018) |
| Pol17_04461_C210 | MG939588 | Poland | 2017 | II | Wozniakowski et al. (2018) |
| Pol17_05838_C220 | MG939589 | Poland | 2017 | II | Wozniakowski et al. (2018) |
| Russia/Kashino_04/13 | KJ747406 | Russia | 2014 | II | Vlasova et al. (2014) |
| ASFV_Belgium_2018/1 | LR536725 | Belgium | 2018 | II | Forth et al. (2019) |
| Pol16_20186_o7 | MG939583 | Poland | 2018 | II | Wozniakowski et al. (2018) |
| Spain/BA71V | NC_00165 | Spain | 1971 | I | Yanez et al. (1995) |
| Variation Type | China/2018/AnhuiXCGQ | China/Pig/HLJ/2018 | China/DB/LN/2018 | Pol16_20186_o7 | Pol16_29413_o23 | Pol17_05838_C220 | Estonia 2014 | Belgium_2018/1 |
|---|---|---|---|---|---|---|---|---|
| Synonymous | 1 | 1 | 1 | 1 | 1 | 1 | 63 | 1 |
| Nonsynonymous | 1 | 1 | 1 | 4 | 4 | 3 | 102 | 2 |
| Intergenic | 6 | 4 | 4 | 4 | 4 | 4 | 4 | 4 |
| Deletion | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
| Premature-stop | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 |
| Frameshift mutation | 0 | 1 | 1 | 0 | 0 | 0 | 0 | 0 |
| Type | Name | China/2018/ AnhuiXCGQ | China/Pig/ HLJ/2018 | China/DB/ LN/2018 | Pol16_20186 _o7 | Pol16_29413 _o23 | Pol17_05838 _C220 | Belgium_2018 /1 | China/GD/2019 |
|---|---|---|---|---|---|---|---|---|---|
| Nonsyn | MGF_360 -6L | T | T | T | T | T | T | T | G |
| D117L | C | C | C | C | C | C | T | C | |
| MGF_505 -4R | G | G | G | A | A | A | G | G | |
| K145R | C | C | C | A | A | A | C | C | |
| E199L | C | C | C | T | C | C | C | C | |
| E184L | C | C | C | C | T | C | C | C | |
| Syn | F317L | A | A | A | A | A | A | A | T |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Wang, X.; Zhang, X.; He, S.; Chen, Y.; Liu, X.; Guo, C. Genetic Characterization and Variation of African Swine Fever Virus China/GD/2019 Strain in Domestic Pigs. Pathogens 2022, 11, 97. https://doi.org/10.3390/pathogens11010097
Wang X, Wang X, Zhang X, He S, Chen Y, Liu X, Guo C. Genetic Characterization and Variation of African Swine Fever Virus China/GD/2019 Strain in Domestic Pigs. Pathogens. 2022; 11(1):97. https://doi.org/10.3390/pathogens11010097
Chicago/Turabian StyleWang, Xun, Xiaoying Wang, Xiaoxiao Zhang, Sheng He, Yaosheng Chen, Xiaohong Liu, and Chunhe Guo. 2022. "Genetic Characterization and Variation of African Swine Fever Virus China/GD/2019 Strain in Domestic Pigs" Pathogens 11, no. 1: 97. https://doi.org/10.3390/pathogens11010097