
Staphylococcus ratti sp. nov. Isolated from a Lab Rat

 , , , , , , , and
, , , , , , , and
Abstract
:1. Introduction
2. Results and Discussion
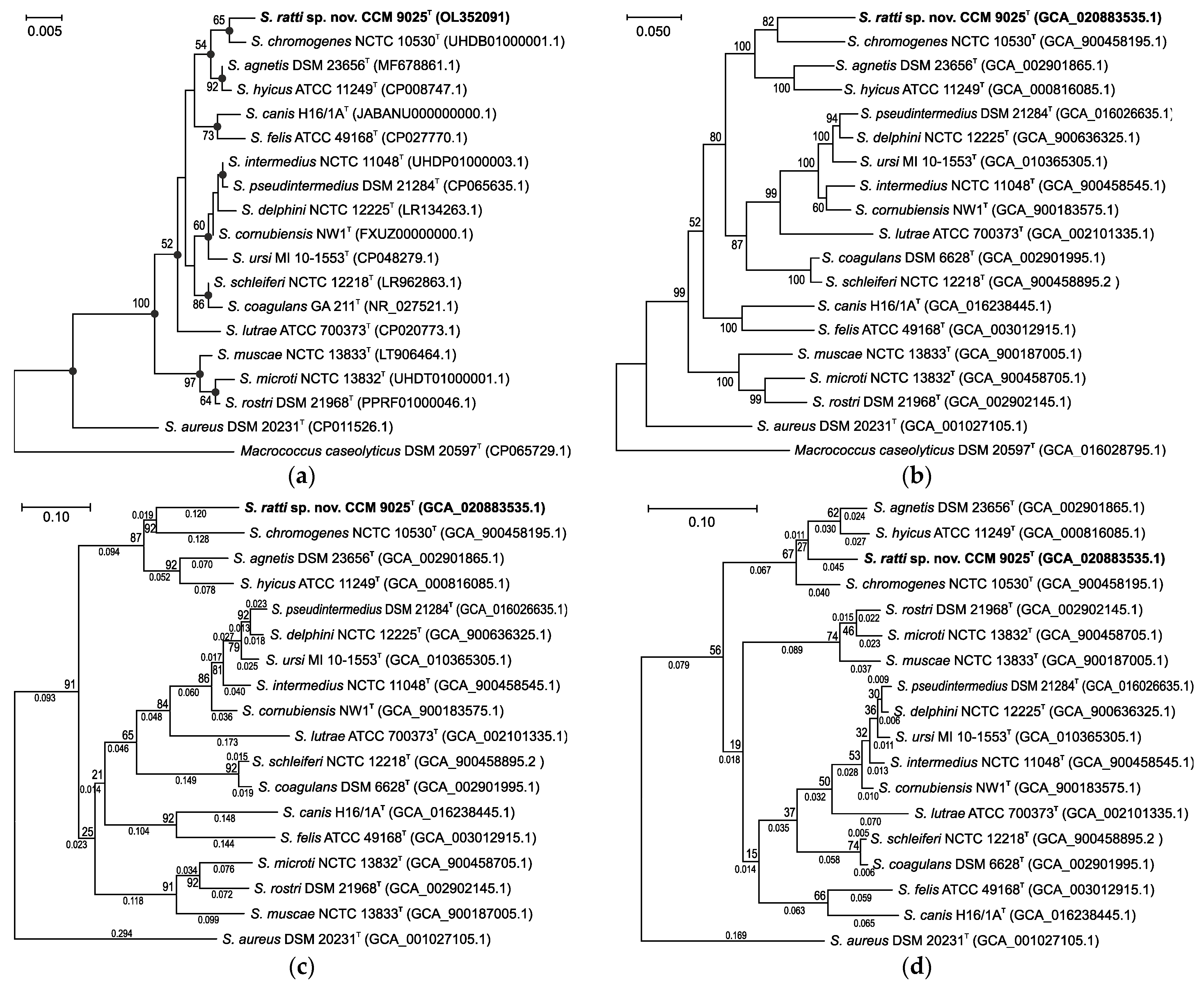
2.1. Phylogenetic Analyses
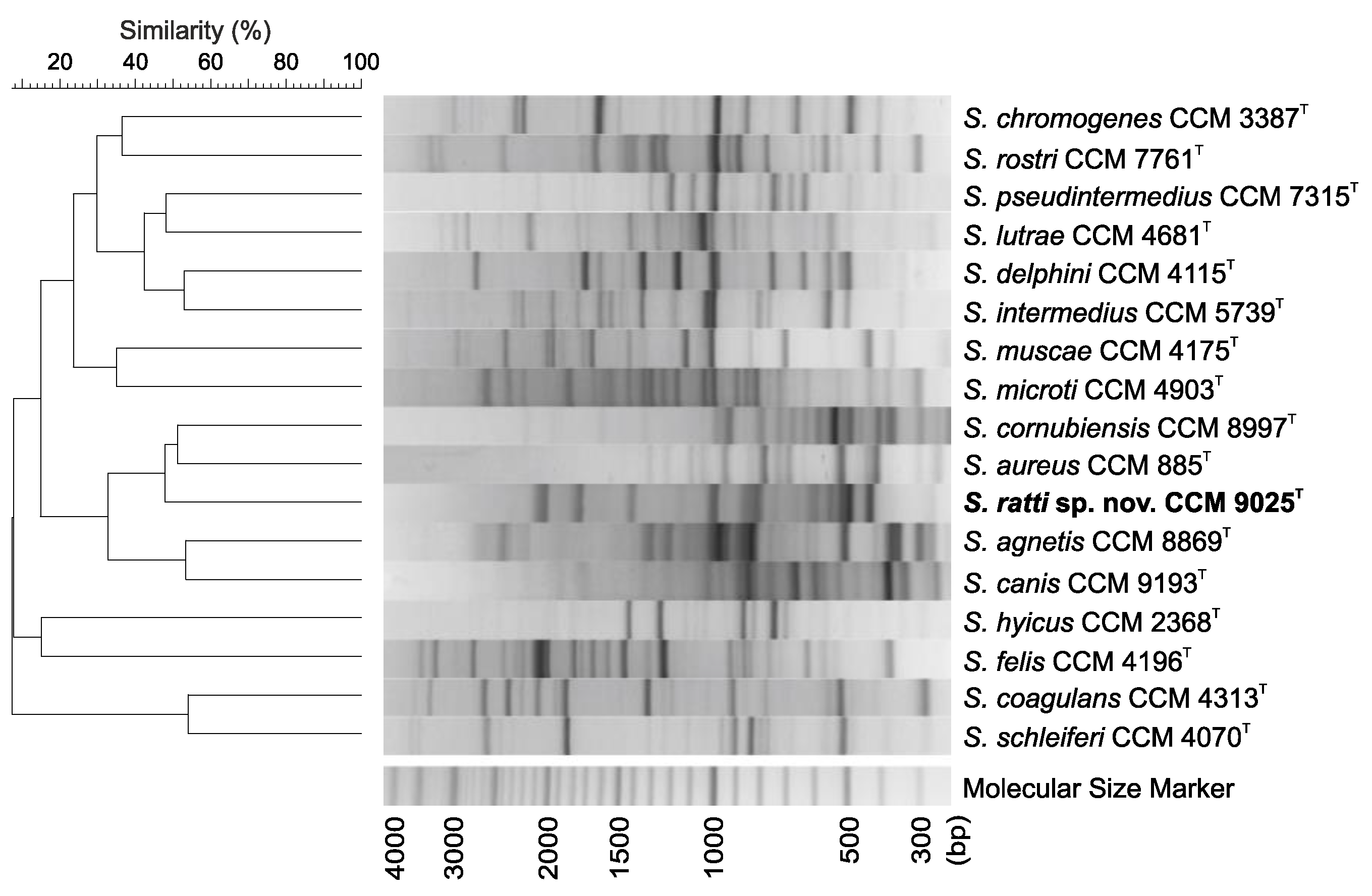
2.2. Phenotypic, Genotypic and Chemotaxonomic Characteristics
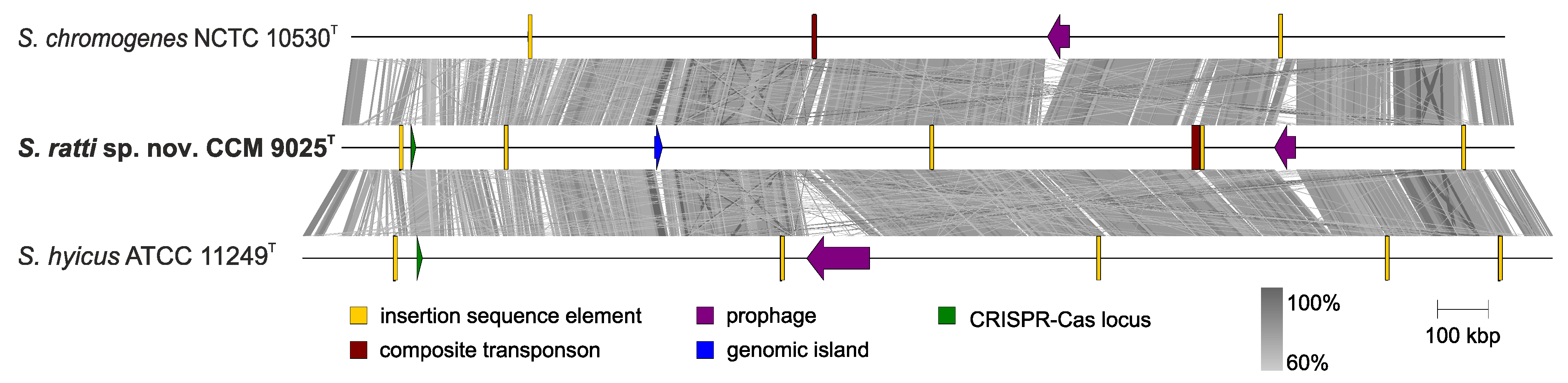
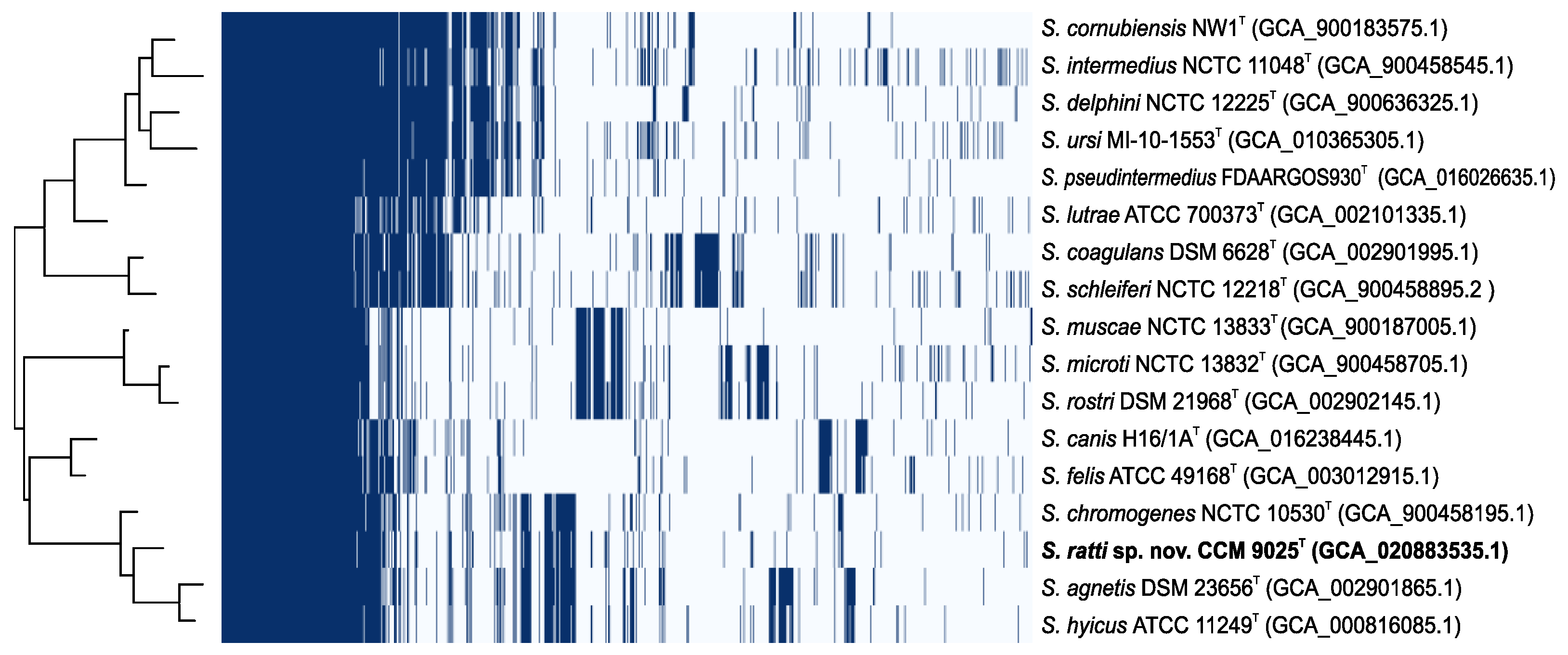
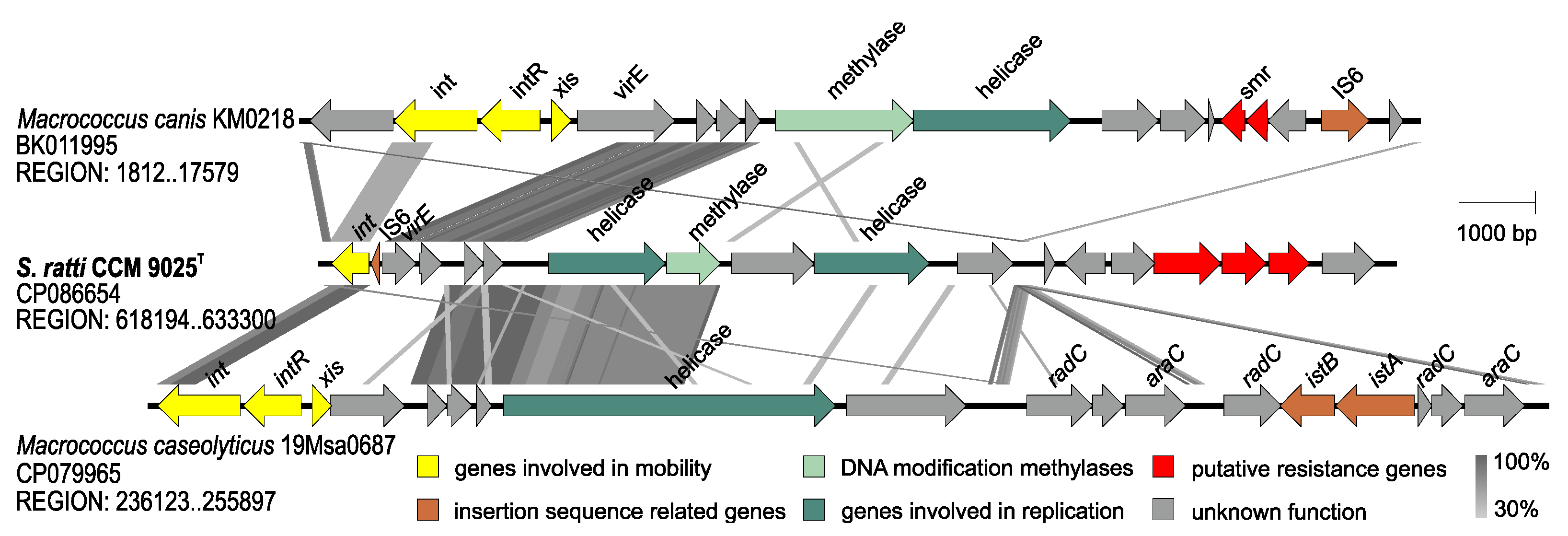
2.3. Whole Genome Characterization of Staphylococcus ratti sp. nov.
2.4. Description of Staphylococcus ratti sp. nov.
3. Materials and Methods
3.1. Bacterial Strains and Cultivation
3.2. Phenotypic Characterization
3.3. Transmission Electron Microscopy
3.4. Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry (MALDI-TOF MS) Analysis
3.5. Chemotaxonomic Analyses
3.6. Genotypic Analysis by (GTG)5-PCR Fingerprinting
3.7. Phylogenetic Analysis Based on 16S rRNA, Housekeeping Genes and Core Genome
3.8. Genome Sequencing and Bioinformatics Analyses
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Götz, F.; Bannerman, T.; Schleifer, K.-H. The Genera Staphylococcus and Macrococcus. In The Prokaryotes; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H., Stackebrandt, E., Eds.; Springer: New York, NY, USA, 2006; pp. 5–75. [Google Scholar] [CrossRef]
- Nováková, D.; Pantůček, R.; Hubálek, Z.; Falsen, E.; Busse, H.-J.; Schumann, P.; Sedláček, I. Staphylococcus microti sp. nov., isolated from the common vole (Microtus arvalis). Int. J. Syst. Evol. Microbiol. 2010, 60, 566–573. [Google Scholar] [CrossRef] [Green Version]
- Hauschild, T. Phenotypic and genotypic identification of staphylococci isolated from wild small mammals. Syst. Appl. Microbiol. 2001, 24, 411–416. [Google Scholar] [CrossRef]
- Hauschild, T.; Slizewski, P.; Masiewicz, P. Species distribution of staphylococci from small wild mammals. Syst. Appl. Microbiol. 2010, 33, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, A.; Ozaki, J.; Kawano, J.; Saitoh, Y.; Kimura, S. Distribution of Staphylococcus species on animal skin. J. Vet. Med. Sci. 1992, 54, 355–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raafat, D.; Mrochen, D.M.; Al’Sholui, F.; Heuser, E.; Ryll, R.; Pritchett-Corning, K.R.; Jacob, J.; Walther, B.; Matuschka, F.R.; Richter, D.; et al. Molecular epidemiology of methicillin-susceptible and methicillin-resistant Staphylococcus aureus in wild, captive and laboratory rats: Effect of habitat on the nasal S. aureus population. Toxins 2020, 12, 80. [Google Scholar] [CrossRef] [Green Version]
- Mrochen, D.M.; Grumann, D.; Schulz, D.; Gumz, J.; Trube, P.; Pritchett-Corning, K.; Johnson, S.; Nicklas, W.; Kirsch, P.; Martelet, K.; et al. Global spread of mouse-adapted Staphylococcus aureus lineages CC1, CC15, and CC88 among mouse breeding facilities. Int. J. Med. Microbiol. 2018, 308, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Hanses, F.; Roux, C.; Dunman, P.M.; Salzberger, B.; Lee, J.C. Staphylococcus aureus gene expression in a rat model of infective endocarditis. Genome Med. 2014, 6, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Power, M.E.; Olson, M.E.; Domingue, P.A.; Costerton, J.W. A rat model of Staphylococcus aureus chronic osteomyelitis that provides a suitable system for studying the human infection. J. Med. Microbiol. 1990, 33, 189–198. [Google Scholar] [CrossRef]
- Donnelly, T.M.; Stark, D.M. Susceptibility of laboratory rats, hamsters, and mice to wound infection with Staphylococcus aureus. Am. J. Vet. Res. 1985, 46, 2634–2638. [Google Scholar]
- Devriese, L.A. Staphylococci in healthy and diseased animals. J. Appl. Microbiol. 1990, 69, 71S–80S. [Google Scholar] [CrossRef]
- Bannoehr, J.; Guardabassi, L. Staphylococcus pseudintermedius in the dog: Taxonomy, diagnostics, ecology, epidemiology and pathogenicity. Vet. Dermatol. 2012, 23, 253-e52. [Google Scholar] [CrossRef] [PubMed]
- Devriese, L.A.; Vancanneyt, M.; Baele, M.; Vaneechoutte, M.; De Graef, E.; Snauwaert, C.; Cleenwerck, I.; Dawyndt, P.; Swings, J.; Decostere, A.; et al. Staphylococcus pseudintermedius sp. nov., a coagulase-positive species from animals. Int. J. Syst. Evol. Microbiol. 2005, 55, 1569–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igimi, S.; Takahashi, E.; Mitsuoka, T. Staphylococcus schleiferi subsp. coagulans subsp. nov., isolated from the external auditory meatus of dogs with external ear otitis. Int. J. Syst. Bacteriol. 1990, 40, 409–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newstead, L.L.; Harris, J.; Goodbrand, S.; Varjonen, K.; Nuttall, T.; Paterson, G.K. Staphylococcus caledonicus sp. nov. and Staphylococcus canis sp. nov. isolated from healthy domestic dogs. Int. J. Syst. Evol. Microbiol. 2021, 71, 004878. [Google Scholar] [CrossRef] [PubMed]
- Vrbovská, V.; Sedláček, I.; Zeman, M.; Švec, P.; Kovařovic, V.; Šedo, O.; Laichmanová, M.; Doskař, J.; Pantůček, R. Characterization of Staphylococcus intermedius group isolates associated with animals from Antarctica and emended description of Staphylococcus delphini. Microorganisms 2020, 8, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perreten, V.; Kania, S.A.; Bemis, D. Staphylococcus ursi sp. nov., a new member of the ‘Staphylococcus intermedius group’ isolated from healthy black bears. Int. J. Syst. Evol. Microbiol. 2020, 70, 4637–4645. [Google Scholar] [CrossRef]
- Worthing, K.; Pang, S.; Trott, D.J.; Abraham, S.; Coombs, G.W.; Jordan, D.; McIntyre, L.; Davies, M.R.; Norris, J. Characterisation of Staphylococcus felis isolated from cats using whole genome sequencing. Vet. Microbiol. 2018, 222, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Guardabassi, L.; Loeber, M.E.; Jacobson, A. Transmission of multiple antimicrobial-resistant Staphylococcus intermedius between dogs affected by deep pyoderma and their owners. Vet. Microbiol. 2004, 98, 23–27. [Google Scholar] [CrossRef]
- Boerlin, P.; Eugster, S.; Gaschen, F.; Straub, R.; Schawalder, P. Transmission of opportunistic pathogens in a veterinary teaching hospital. Vet. Microbiol. 2001, 82, 347–359. [Google Scholar] [CrossRef]
- Fudaba, Y.; Nishifuji, K.; Andresen, L.O.; Yamaguchi, T.; Komatsuzawa, H.; Amagai, M.; Sugai, M. Staphylococcus hyicus exfoliative toxins selectively digest porcine desmoglein 1. Microb. Pathog. 2005, 39, 171–176. [Google Scholar] [CrossRef]
- Casanova, C.; Iselin, L.; von Steiger, N.; Droz, S.; Sendi, P. Staphylococcus hyicus bacteremia in a farmer. J. Clin. Microbiol. 2011, 49, 4377–4378. [Google Scholar] [CrossRef] [Green Version]
- Foissac, M.; Lekaditi, M.; Loutfi, B.; Ehrhart, A.; Dauchy, F.A. Spondylodiscitis and bacteremia due to Staphylococcus hyicus in an immunocompetent man. Germs 2016, 6, 106–110. [Google Scholar] [CrossRef] [Green Version]
- Taponen, S.; Supre, K.; Piessens, V.; Van Coillie, E.; De Vliegher, S.; Koort, J.M.K. Staphylococcus agnetis sp. nov., a coagulase-variable species from bovine subclinical and mild clinical mastitis. Int. J. Syst. Evol. Microbiol. 2012, 62, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, L.L.; Thofner, I.; Bisgaard, M.; Olsen, R.H.; Christensen, J.P.; Christensen, H. Staphylococcus agnetis, a potential pathogen in broiler breeders. Vet. Microbiol. 2017, 212, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Al-Rubaye, A.A.; Couger, M.B.; Ojha, S.; Pummill, J.F.; Koon, J.A., 2nd; Wideman, R.F., Jr.; Rhoads, D.D. Genome analysis of Staphylococcus agnetis, an agent of lameness in broiler chickens. PLoS ONE 2015, 10, e0143336. [Google Scholar] [CrossRef] [PubMed]
- Devriese, L.A.; Baele, M.; Vaneechoutte, M.; Martel, A.; Haesebrouck, F. Identification and antimicrobial susceptibility of Staphylococcus chromogenes isolates from intramammary infections of dairy cows. Vet. Microbiol. 2002, 87, 175–182. [Google Scholar] [CrossRef]
- Andresen, L.O.; Ahrens, P.; Daugaard, L.; Bille-Hansen, V. Exudative epidermitis in pigs caused by toxigenic Staphylococcus chromogenes. Vet. Microbiol. 2005, 105, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Andrews, A.H.; Lamport, A. Isolation of Staphylococcus chromogenes from an unusual case of impetigo in a goat. Vet. Rec. 1997, 140, 584. [Google Scholar] [CrossRef]
- Schmidt, T.; Kock, M.M.; Ehlers, M.M. Diversity and antimicrobial susceptibility profiling of staphylococci isolated from bovine mastitis cases and close human contacts. J. Dairy Sci. 2015, 98, 6256–6269. [Google Scholar] [CrossRef]
- Lamers, R.P.; Muthukrishnan, G.; Castoe, T.A.; Tafur, S.; Cole, A.M.; Parkinson, C.L. Phylogenetic relationships among Staphylococcus species and refinement of cluster groups based on multilocus data. BMC Evol. Biol. 2012, 12, 171. [Google Scholar] [CrossRef] [Green Version]
- Goris, J.; Konstantinidis, K.T.; Klappenbach, J.A.; Coenye, T.; Vandamme, P.; Tiedje, J.M. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 2007, 57, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Schleifer, K.-H.; Bell, J.A. Staphylococcus. In Bergey’s Manual of Systematics of Archaea and Bacteria, Online ed.; Whitman, W.B., Ed.; Wiley & Sons and Bergey’s Manual Trust: Hoboken, NJ, USA, 2015. [Google Scholar] [CrossRef]
- Nahaie, M.R.; Goodfellow, M.; Minnikin, D.E.; Hájek, V. Polar lipid and isoprenoid quinone composition in the classification of Staphylococcus. J. Gen. Microbiol. 1984, 130, 2427–2437. [Google Scholar] [CrossRef] [Green Version]
- Schumann, P. Peptidoglycan Structure. Methods Microbiol. 2011, 38, 101–129. [Google Scholar] [CrossRef]
- Devriese, L.A.; Hajek, V.; Oeding, P.; Meyer, S.A.; Schleifer, K.H. Staphylococcus hyicus (Sompolinsky 1953) comb. nov. and Staphylococcus hyicus subsp. chromogenes subsp. nov. Int. J. Syst. Bacteriol. 1978, 28, 482–490. [Google Scholar] [CrossRef] [Green Version]
- Shwani, A.; Adkins, P.R.F.; Ekesi, N.S.; Alrubaye, A.; Calcutt, M.J.; Middleton, J.R.; Rhoads, D.D. Whole-genome comparisons of Staphylococcus agnetis isolates from cattle and chickens. Appl. Environ. Microbiol. 2020, 86, e00484-20. [Google Scholar] [CrossRef] [PubMed]
- Luthje, P.; von Kockritz-Blickwede, M.; Schwarz, S. Identification and characterization of nine novel types of small staphylococcal plasmids carrying the lincosamide nucleotidyltransferase gene lnu(A). J. Antimicrob. Chemother. 2007, 59, 600–606. [Google Scholar] [CrossRef] [Green Version]
- Burkhart, B.J.; Schwalen, C.J.; Mann, G.; Naismith, J.H.; Mitchell, D.A. YcaO-dependent posttranslational amide activation: Biosynthesis, structure, and function. Chem. Rev. 2017, 117, 5389–5456. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.G.; Frankel, M.B.; Missiakas, D.; Schneewind, O. SagB glucosaminidase is a determinant of Staphylococcus aureus glycan chain length, antibiotic susceptibility, and protein secretion. J. Bacteriol. 2016, 198, 1123–1136. [Google Scholar] [CrossRef] [Green Version]
- Dean, B.A.; Williams, R.E.O.; Hall, F.; Corse, J. Phage typing of coagulase-negative staphylococci and micrococci. Epidemiol. Infect. (J. Hyg.) 1973, 71, 261–270. [Google Scholar] [CrossRef] [Green Version]
- Kwan, T.; Liu, J.; DuBow, M.; Gros, P.; Pelletier, J. The complete genomes and proteomes of 27 Staphylococcus aureus bacteriophages. Proc. Natl. Acad. Sci. USA 2005, 102, 5174–5179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, D.; Adriaenssens, E.M.; Martinez, B.; Rodriguez, A.; Lavigne, R.; Kropinski, A.M.; Garcia, P. Three proposed new bacteriophage genera of staphylococcal phages: “3alikevirus”, “77likevirus” and “Phietalikevirus”. Arch. Virol. 2014, 159, 389–398. [Google Scholar] [CrossRef] [Green Version]
- Tetens, J.; Sprotte, S.; Thimm, G.; Wagner, N.; Brinks, E.; Neve, H.; Holzel, C.S.; Franz, C.M.A.P. First molecular characterization of Siphoviridae-like bacteriophages infecting Staphylococcus hyicus in a case of exudative epidermitis. Front. Microbiol. 2021, 12, 653501. [Google Scholar] [CrossRef]
- Schwendener, S.; Dona, V.; Perreten, V. The novel macrolide resistance genes mef(D), msr(F), and msr(H) are present on resistance islands in Macrococcus canis, Macrococcus caseolyticus, and Staphylococcus aureus. Antimicrob. Agents Chemother. 2020, 64, e00160-20. [Google Scholar] [CrossRef]
- Rosey, E.L.; Oskouian, B.; Stewart, G.C. Lactose metabolism by Staphylococcus aureus: Characterization of lacABCD, the structural genes of the tagatose 6-phosphate pathway. J. Bacteriol. 1991, 173, 5992–5998. [Google Scholar] [CrossRef] [Green Version]
- Naushad, S.; Barkema, H.W.; Luby, C.; Condas, L.A.; Nobrega, D.B.; Carson, D.A.; De Buck, J. Comprehensive phylogenetic analysis of bovine non-aureus staphylococci species based on whole-genome sequencing. Front. Microbiol. 2016, 7, 1990. [Google Scholar] [CrossRef] [Green Version]
- Haft, D.H.; Selengut, J.; Mongodin, E.F.; Nelson, K.E. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput. Biol. 2005, 1, e60. [Google Scholar] [CrossRef]
- Pantůček, R.; Švec, P.; Dajcs, J.J.; Machová, I.; Černohlavková, J.; Šedo, O.; Gelbíčová, T.; Mašlaňová, I.; Doškař, J.; Zdráhal, Z.; et al. Staphylococcus petrasii sp. nov. including S. petrasii subsp. petrasii subsp. nov. and S. petrasii subsp. croceilyticus subsp. nov., isolated from human clinical specimens and human ear infections. Syst. Appl. Microbiol. 2013, 36, 90–95. [Google Scholar] [CrossRef] [PubMed]
- Mannerová, S.; Pantůček, R.; Doškař, J.; Švec, P.; Snauwaert, C.; Vancanneyt, M.; Swings, J.; Sedláček, I. Macrococcus brunensis sp. nov., Macrococcus hajekii sp. nov. and Macrococcus lamae sp. nov., from the skin of llamas. Int. J. Syst. Evol. Microbiol. 2003, 53, 1647–1654. [Google Scholar] [CrossRef] [PubMed]
- Mašlaňová, I.; Wertheimer, Z.; Sedláček, I.; Švec, P.; Indráková, A.; Kovařovic, V.; Schumann, P.; Spröer, C.; Králová, S.; Šedo, O.; et al. Description and comparative genomics of Macrococcus caseolyticus subsp. hominis subsp. nov., Macrococcus goetzii sp. nov., Macrococcus epidermidis sp. nov., and Macrococcus bohemicus sp. nov., novel macrococci from human clinical material with virulence potential and suspected uptake of foreign DNA by natural transformation. Front. Microbiol. 2018, 9, 1178. [Google Scholar] [CrossRef] [PubMed]
- EUCAST. Breakpoint Tables for Interpretation of MICs and Zone Diameters. The European Committee on Antimicrobial Susceptibility Testing: Version 6.0. 2016. Available online: https://www.eucast.org (accessed on 10 June 2019).
- Pantůček, R.; Sedláček, I.; Indraková, A.; Vrbovská, V.; Mašlaňová, I.; Kovařovic, V.; Švec, P.; Králová, S.; Krištofová, L.; Kekláková, J.; et al. Staphylococcus edaphicus sp. nov., isolated in Antarctica, harbors the mecC gene and genomic islands with a suspected role in adaptation to extreme environments. Appl. Env. Microbiol. 2018, 84, e01746-17. [Google Scholar] [CrossRef] [Green Version]
- Sasser, M. Identification of Bacteria by Gas Chromatography of Cellular Fatty Acids, MIDI Technical Note 101, Revision July 2006 ed.; MIDI Inc.: Newark, DE, USA, 1990. [Google Scholar]
- Schumann, P.; Kalensee, F.; Cao, J.; Criscuolo, A.; Clermont, D.; Kohler, J.M.; Meier-Kolthoff, J.P.; Neumann-Schaal, M.; Tindall, B.J.; Pukall, R. Reclassification of Haloactinobacterium glacieicola as Occultella glacieicola gen. nov., comb. nov., of Haloactinobacterium album as Ruania alba comb. nov, with an emended description of the genus Ruania, recognition that the genus names Haloactinobacterium and Ruania are heterotypic synonyms and description of Occultella aeris sp. nov., a halotolerant isolate from surface soil sampled at an ancient copper smelter. Int. J. Syst. Evol. Microbiol. 2021, 71, 004769. [Google Scholar] [CrossRef]
- Kämpfer, P.; McInroy, J.A.; Clermont, D.; Neumann-Schaal, M.; Criscuolo, A.; Busse, H.-J.; Glaeser, S.P. Leucobacter soli sp. nov., from soil amended with humic acid. Int. J. Syst. Evol. Microbiol. 2021, 71, 005156. [Google Scholar] [CrossRef] [PubMed]
- Vieira, S.; Huber, K.J.; Neumann-Schaal, M.; Geppert, A.; Luckner, M.; Wanner, G.; Overmann, J. Usitatibacter rugosus gen. nov., sp. nov. and Usitatibacter palustris sp. nov., novel members of Usitatibacteraceae fam. nov. within the order Nitrosomonadales isolated from soil. Int. J. Syst. Evol. Microbiol. 2021, 71, 004631. [Google Scholar] [CrossRef] [PubMed]
- Švec, P.; Pantůček, R.; Petráš, P.; Sedláček, I.; Nováková, D. Identification of Staphylococcus spp. using (GTG)5-PCR fingerprinting. Syst. Appl. Microbiol. 2010, 33, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Pantůček, R.; Sedláček, I.; Petráš, P.; Koukalová, D.; Švec, P.; Štětina, V.; Vancanneyt, M.; Chrastinová, L.; Vokurková, J.; Růžičková, V.; et al. Staphylococcus simiae sp. nov., isolated from South American squirrel monkeys. Int. J. Syst. Evol. Microbiol. 2005, 55, 1953–1958. [Google Scholar] [CrossRef]
- Mellmann, A.; Becker, K.; von Eiff, C.; Keckevoet, U.; Schumann, P.; Harmsen, D. Sequencing and staphylococci identification. Emerg. Infect. Dis. 2006, 12, 333–336. [Google Scholar] [CrossRef]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef]
- Lagesen, K.; Hallin, P.; Rodland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Kitts, P.A.; Church, D.M.; Thibaud-Nissen, F.; Choi, J.; Hem, V.; Sapojnikov, V.; Smith, R.G.; Tatusova, T.; Xiang, C.; Zherikov, A.; et al. Assembly: A resource for assembled genomes at NCBI. Nucleic Acids Res. 2016, 44, D73–D80. [Google Scholar] [CrossRef]
- Sichtig, H.; Minogue, T.; Yan, Y.; Stefan, C.; Hall, A.; Tallon, L.; Sadzewicz, L.; Nadendla, S.; Klimke, W.; Hatcher, E.; et al. FDA-ARGOS is a database with public quality-controlled reference genomes for diagnostic use and regulatory science. Nat. Commun. 2019, 10, 3313. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Na, S.I.; Kim, Y.O.; Yoon, S.H.; Ha, S.M.; Baek, I.; Chun, J. UBCG: Up-to-date bacterial core gene set and pipeline for phylogenomic tree reconstruction. J. Microbiol. 2018, 56, 280–285. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 2021, 49, gkab902. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez, R.L.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef] [Green Version]
- Fišarová, L.; Botka, T.; Du, X.; Mašlaňová, I.; Bárdy, P.; Pantůček, R.; Benešík, M.; Roudnický, P.; Winstel, V.; Larsen, J.; et al. Staphylococcus epidermidis phages transduce antimicrobial resistance plasmids and mobilize chromosomal islands. mSphere 2021, 6, e00223-21. [Google Scholar] [CrossRef]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; O’Neill, K.R.; Haft, D.H.; DiCuccio, M.; Chetvernin, V.; Badretdin, A.; Coulouris, G.; Chitsaz, F.; Derbyshire, M.K.; Durkin, A.S.; et al. RefSeq: Expanding the Prokaryotic Genome Annotation Pipeline reach with protein family model curation. Nucleic Acids Res. 2021, 49, D1020–D1028. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; The UGENE Team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [Green Version]
- Edwards, R.; Decewicz, P.; Katelyn; Daniel, S.; Laurasisk. linsalrob/PhiSpy: Dropped prophages (v.4.2.19). In Zenodo; CERN: Geneva, Switzerland, 2021. [Google Scholar] [CrossRef]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Simon Fraser University Research Computing Group; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34, D32–D36. [Google Scholar] [CrossRef] [Green Version]
- Russel, J.; Pinilla-Redondo, R.; Mayo-Munoz, D.; Shah, S.A.; Sorensen, S.J. CRISPRCasTyper: Automated identification, annotation, and classification of CRISPR-Cas loci. CRISPR J. 2020, 3, 462–469. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | S. ratti sp. nov. CCM 9025T | S. agnetis CCM 8869T | S. hyicus CCM 2368T | S. chromogenes CCM 3387T |
|---|---|---|---|---|
| Pigment | white | white | white | orange |
| Growth at 15 °C | − | − | + | + |
| Growth in 15% NaCl | + | + | + | − |
| Hemolysis | + | − | − | − |
| Voges–Proskauer test 1 | − | + | − | − |
| Acid from trehalose | + | − | + | + |
| Tween 80 hydrolysis | − | + | + | − |
| Alkaline phosphatase | + | − | + | + |
| β-glucuronidase 2 | + | w | + | − |
| Urease | + | − | − | + |
| Fatty Acid | S. ratti sp. nov. CCM 9025T | S. chromogenes CCM 3387T | S. agnetis CCM 8869T | S. hyicus CCM 2368T |
|---|---|---|---|---|
| iso-C14:0 | TR | 1.0 | TR | TR |
| C14:0 | TR | TR | 1.0 | 1.0 |
| iso-C15:0 | 35.0 | 16.5 | 43.9 | 33.7 |
| anteiso-C15:0 | 24.1 | 36.4 | 25.1 | 32.5 |
| iso-C16:0 | 1.5 | 1.5 | TR | 1.2 |
| C16:0 | 1.5 | 1.5 | 2.4 | 2.7 |
| iso-C17:0 | 10.8 | 6.0 | 9.2 | 7.4 |
| anteiso-C17:0 | 10.2 | 13.4 | 4.8 | 6.9 |
| C18:0 | 2.6 | 5.5 | 2.9 | 4.0 |
| iso-C19:0 | 4.4 | 3.2 | 2.1 | 2.1 |
| anteiso-C19:0 | 2.3 | 2.9 | TR | 1.0 |
| C20:0 | 5.8 | 10.4 | 6.1 | 6.2 |
| Function and Role | Virulence Factors | Related Genes | Prediction in CCM 9025T Genome |
|---|---|---|---|
| Adherence | Clumping factor B | clfB | LN051_01230 |
| Fibronectin binding proteins | fnbA | LN051_04265 | |
| Ser-Asp rich fibrinogen-binding proteins | sdrC | LN051_00305 | |
| Enzymes | Cysteine protease | sspB | LN051_01195 |
| Hyaluronate lyase | hysA | LN051_02175 | |
| Lipase | geh | LN051_01425 | |
| Thermonuclease | nuc | LN051_06665 | |
| Secretion system | Type VII secretion system | esaA | LN051_10885; LN051_10890 |
| esaB | LN051_10875 | ||
| essC | LN051_10865 | ||
| esxA | LN051_10895 | ||
| Surface protein anchoring | Lipoprotein diacylglyceryl transferase | lgt | LN051_09220 |
| Lipoprotein-specific signal peptidase II | lspA | LN051_07315 | |
| Immune evasion | Capsule | Undetermined | LN051_02355; LN051_03230; LN051_07970 |
| Toxin | β-hemolysin | hlb | LN051_01075 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovařovic, V.; Sedláček, I.; Petráš, P.; Králová, S.; Mašlaňová, I.; Švec, P.; Neumann-Schaal, M.; Botka, T.; Gelbíčová, T.; Staňková, E.; et al. Staphylococcus ratti sp. nov. Isolated from a Lab Rat. Pathogens 2022, 11, 51. https://doi.org/10.3390/pathogens11010051
Kovařovic V, Sedláček I, Petráš P, Králová S, Mašlaňová I, Švec P, Neumann-Schaal M, Botka T, Gelbíčová T, Staňková E, et al. Staphylococcus ratti sp. nov. Isolated from a Lab Rat. Pathogens. 2022; 11(1):51. https://doi.org/10.3390/pathogens11010051
Chicago/Turabian StyleKovařovic, Vojtěch, Ivo Sedláček, Petr Petráš, Stanislava Králová, Ivana Mašlaňová, Pavel Švec, Meina Neumann-Schaal, Tibor Botka, Tereza Gelbíčová, Eva Staňková, and et al. 2022. "Staphylococcus ratti sp. nov. Isolated from a Lab Rat" Pathogens 11, no. 1: 51. https://doi.org/10.3390/pathogens11010051