
Characterization of the Rhipicephalus (Boophilus) microplus Sialotranscriptome Profile in Response to Theileria equi Infection

 ,
,  , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
2.1. Raw RNA-seq Data
2.2. Transcriptome de novo Assembly
2.3. Annotation of Sialotranscriptome Profiles
2.4. Functional Analysis of Sialotranscriptome Profiles
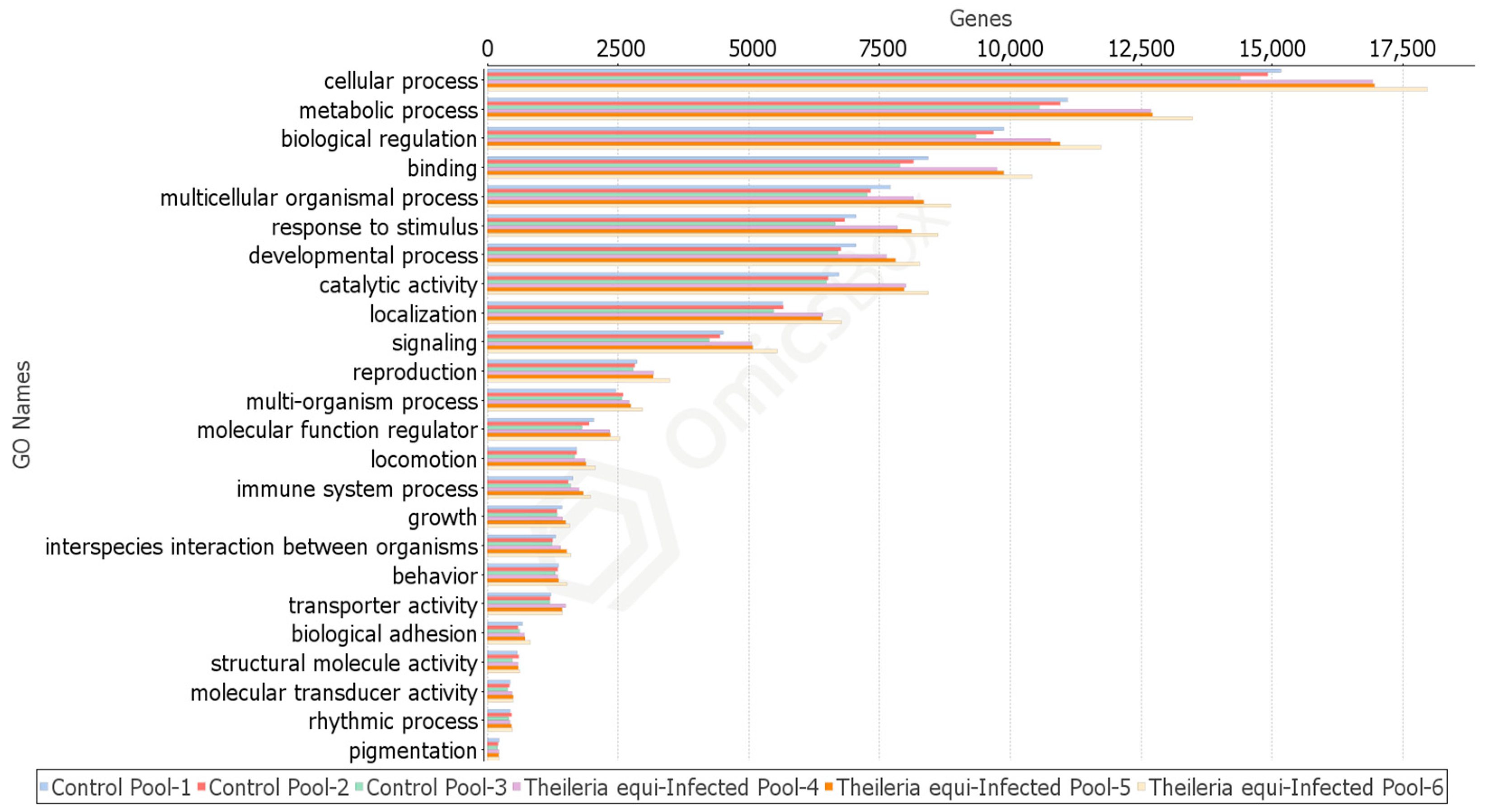
2.4.1. Rhipicephalus microplus Sialotranscriptome Profiles
2.4.2. Theileria equi Transcriptomic Profile during Sporogony
2.4.3. GO Terms in Theileria equi Profile Highlights Parasite–Host Interactions
2.5. Gene Expression in Response to Theileria equi Infection
2.5.1. Redox Metabolism
2.5.2. Carbohydrate-Related Proteins
2.5.3. Antimicrobial Activity
2.5.4. Signaling Pathways
2.5.5. Blood Feeding/Mitigation of Host Defenses
2.6. Transcriptome Validation through qPCR
3. Discussion
3.1. Theileria equi’ Invasion Process
3.2. An Insight into Theileria Intracellular Maintenance
3.3. Theileria equi: Drivers of Cell Changes
3.4. Theileria Endeavors Manipulation of Host Signaling Routes and the Ticks’ Response
3.5. Rhipicephalus Microplus Response to Theileria equi: Co-Evolutional Status
3.6. Recognition of Theileria and Antimicrobial Activity
3.7. Ticks’ Oxidative Response to Theileria equi Infection and Its Defensive Reaction
4. Material and Methods
4.1. Study Design and Rationale
4.2. Rhipicephalus microplus Colony
4.2.1. Production of Uninfected and Theileria equi Infected Rhipicephalus microplus Ticks
4.2.2. Tick Dissection
4.3. RNA Extraction, Quantification, and Parasitic Status Confirmation
4.4. RNA-seq
4.5. RNA-seq Data Analysis
4.6. Validation of RNA-seq Data
4.6.1. Target Transcripts
4.6.2. Gene Expression Measured by qPCR Assays
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- OIE-Listed Diseases 2020: OIE—World Organisation for Animal Health. Available online: https://www.oie.int/animal-health-in-the-world/oie-listed-diseases-2020/ (accessed on 23 September 2020).
- Onyiche, T.E.; Suganuma, K.; Igarashi, I.; Yokoyama, N.; Xuan, X.; Thekisoe, O. A Review on Equine Piroplasmosis: Epidemiology, Vector Ecology, Risk Factors, Host Immunity, Diagnosis and Control. Int. J. Environ. Res. Public Health 2019, 16, 1736. [Google Scholar] [CrossRef] [Green Version]
- Tirosh-Levy, S.; Gottlieb, Y.; Fry, L.M.; Knowles, D.P.; Steinman, A. Twenty Years of Equine Piroplasmosis Research: Global Distribution, Molecular Diagnosis, and Phylogeny. Pathogens 2020, 9, 926. [Google Scholar] [CrossRef]
- Friedhoff, K.T.; Tenter, A.M.; Müller, I. Haemoparasites of equines: impact on international trade of horses. Rev. Sci. Tech. 1990, 9, 1187–1194. [Google Scholar]
- Ueti, M.W.; Palmer, G.H.; Kappmeyer, L.S.; Statdfield, M.; Scoles, G.A.; Knowles, D.P. Ability of the Vector Tick Boophilus microplus To Acquire and Transmit Babesia equi following Feeding on Chronically Infected Horses with Low-Level Parasitemia. J. Clin. Microbiol. 2005, 43, 3755–3759. [Google Scholar] [CrossRef] [Green Version]
- Scoles, G.A.; Ueti, M.W. Vector Ecology of Equine Piroplasmosis. Annu. Rev. Èntomol. 2015, 60, 561–580. [Google Scholar] [CrossRef]
- Ikadai, H.; Matsuu, A.; Sasaki, M.; Ishida, H.; Fujisaki, K.; Oyamada, T.; Igarashi, I. Molecular evidence of babesia equi transmission in haemaphysalis longicornis. Am. J. Trop. Med. Hyg. 2007, 76, 694–697. [Google Scholar] [CrossRef]
- Guimarães, A.M.; Lima, J.D.; Ribeiro, M.F.B.; Camargos, E.R.S.; Bozzi, I.A. Ultrastructure of sporogony in Babesia equi in salivary glands of adult female Boophilus microplus ticks. Parasitol. Res. 1997, 84, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Lima, J.D.; Ribeiro, M.F.B.; Guimarães, A.M. Sporogony and experimental transmission of Babesia equi by Boophilus microplus. Parasitol. Res. 1998, 84, 323–327. [Google Scholar] [CrossRef]
- Ueti, M.W.; Palmer, G.H.; Scoles, G.A.; Kappmeyer, L.S.; Knowles, D.P. Persistently Infected Horses Are Reservoirs for Intrastadial Tick-Borne Transmission of the Apicomplexan Parasite Babesia equi. Infect. Immun. 2008, 76, 3525–3529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peckle, M.; Pires, M.S.; Dos Santos, T.M.; Roier, E.C.R.; Da Silva, C.B.; Vilela, J.A.R.; Santos, H.A.; Massard, C.L. Molecular epidemiology of Theileria equi in horses and their association with possible tick vectors in the state of Rio de Janeiro, Brazil. Parasitol. Res. 2013, 112, 2017–2025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ndawula, J.C.; Tabor, A.E. Cocktail Anti-Tick Vaccines: The Unforeseen Constraints and Approaches toward Enhanced Efficacies. Vaccines 2020, 8, 457. [Google Scholar] [CrossRef]
- De La Fuente, J.; Kocan, K.M. Strategies for development of vaccines for control of ixodid tick species. Parasite Immunol. 2006, 28, 275–283. [Google Scholar] [CrossRef]
- Šimo, L.; Kazimirova, M.; Richardson, J.; Bonnet, S.I. The Essential Role of Tick Salivary Glands and Saliva in Tick Feeding and Pathogen Transmission. Front. Cell. Infect. Microbiol. 2017, 7, 281. [Google Scholar] [CrossRef]
- Antunes, S.; Couto, J.; Ferrolho, J.; Rodrigues, F.; Nobre, J.; Santos, A.S.; Santos-Silva, M.M.; De La Fuente, J.; Domingos, A. Rhipicephalus bursa Sialotranscriptomic Response to Blood Feeding and Babesia ovis Infection: Identification of Candidate Protective Antigens. Front. Cell. Infect. Microbiol. 2018, 8, 116. [Google Scholar] [CrossRef]
- Zivkovic, Z.; Esteves, E.; Almazán, C.; Daffre, S.; Nijhof, A.M.; Kocan, K.M.; Jongejan, F.; De La Fuente, J. Differential expression of genes in salivary glands of male Rhipicephalus (Boophilus)microplus in response to infection with Anaplasma marginale. BMC Genom. 2010, 11, 186. [Google Scholar] [CrossRef] [Green Version]
- De La Fuente, J.; Antunes, S.; Bonnet, S.; Cabezas-Cruz, A.; Domingos, A.G.; Estrada-Peña, A.; Johnson, N.; Kocan, K.M.; Mansfield, K.L.; Nijhof, A.M.; et al. Tick-Pathogen Interactions and Vector Competence: Identification of Molecular Drivers for Tick-Borne Diseases. Front. Cell. Infect. Microbiol. 2017, 7, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramamoorthi, N.; Narasimhan, S.; Pal, U.; Bao, F.; Yang, X.F.; Fish, D.; Anguita, J.; Norgard, M.V.; Kantor, F.S.; Anderson, J.F.; et al. The Lyme disease agent exploits a tick protein to infect the mammalian host. Nat. Cell Biol. 2005, 436, 573–577. [Google Scholar] [CrossRef] [Green Version]
- Merino, O.; Antunes, S.; Mosqueda, J.; Moreno-Cid, J.A.; De La Lastra, J.M.P.; Rosario-Cruz, R.; Rodríguez, S.; Domingos, A.; De La Fuente, J.; Rosario-Cruz, R. Vaccination with proteins involved in tick–pathogen interactions reduces vector infestations and pathogen infection. Vaccine 2013, 31, 5889–5896. [Google Scholar] [CrossRef] [PubMed]
- Couto, J.; Villar, M.; Mateos-Hernández, L.; Ferrolho, J.; Sanches, G.S.; Santos, A.S.; Santos, A.S.; Nobre, J.; Moreira, O.; Antunes, S.; et al. Quantitative Proteomics Identifies Metabolic Pathways Affected by Babesia Infection and Blood Feeding in the Sialoproteome of the Vector Rhipicephalus bursa. Vaccines 2020, 8, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Narasimhan, S.; Zhang, L.; Liu, L.; Wang, P.; Fikrig, E. Tick Histamine Release Factor Is Critical for Ixodes scapularis Engorgement and Transmission of the Lyme Disease Agent. PLOS Pathog. 2010, 6, e1001205. [Google Scholar] [CrossRef]
- Villar, M.; Ayllón, N.; Busby, A.T.; Galindo, R.C.; Blouin, E.F.; Kocan, K.M.; Bonzón-Kulichenko, E.; Zivkovic, Z.; Almazán, C.; Torina, A.; et al. Expression of Heat Shock and Other Stress Response Proteins in Ticks and Cultured Tick Cells in Response to Anaplasma spp. Infection and Heat Shock. Int. J. Proteom. 2010, 2010, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Shiels, B.R.; McKellar, S.; Katzer, F.; Lyons, K.; Kinnaird, J.; Ward, C.; Wastling, J.M.; Swan, D. A Theileria annulata DNA Binding Protein Localized to the Host Cell Nucleus Alters the Phenotype of a Bovine Macrophage Cell Line. Eukaryot. Cell 2004, 3, 495–505. [Google Scholar] [CrossRef] [Green Version]
- Kalil, S.P.; Rosa, R.; Capelli-Peixoto, J.; Pohl, P.C.; Oliveira, P.L.; Fogaça, A.C.; Daffre, S. Immune-related redox metabolism of embryonic cells of the tick Rhipicephalus microplus (BME26) in response to infection with Anaplasma marginale. Parasites Vectors 2017, 10, 613. [Google Scholar] [CrossRef] [Green Version]
- Tonui, T.; Corredor-Moreno, P.; Kanduma, E.; Njuguna, J.; Njahira, M.N.; Nyanjom, S.G.; Silva, J.C.; Djikeng, A.; Pelle, R. Transcriptomics reveal potential vaccine antigens and a drastic increase of up-regulated genes during Theileria parva development from arthropod to bovine infective stages. PLoS ONE 2018, 13, e0204047. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, S.; Roy, S.; Mistry, H.U.; Murthy, S.; George, N.; Bhandari, V.; Sharma, P. Potential Sabotage of Host Cell Physiology by Apicomplexan Parasites for Their Survival Benefits. Front. Immunol. 2017, 8, 8. [Google Scholar] [CrossRef] [Green Version]
- Shaw, M.K. Theileria parvaSporozoite Entry into Bovine Lymphocytes Involves both Parasite and Host Cell Signal Transduction Processes. Exp. Parasitol. 1996, 84, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Chin, D.; Means, A.R. Calmodulin: A prototypical calcium sensor. Trends Cell Biol. 2000, 10, 322–328. [Google Scholar] [CrossRef]
- Villares, M.; Berthelet, J.; Weitzman, J.B. The clever strategies used by intracellular parasites to hijack host gene expression. Semin. Immunopathol. 2020, 42, 215–226. [Google Scholar] [CrossRef]
- Shaw, M.K. Cell invasion by Theileria sporozoites. Trends Parasitol. 2003, 19, 2–6. [Google Scholar] [CrossRef]
- Hayashida, K.; Hara, Y.; Abe, T.; Yamasaki, C.; Toyoda, A.; Kosuge, T.; Suzuki, Y.; Sato, Y.; Kawashima, S.; Katayama, T.; et al. Comparative Genome Analysis of Three Eukaryotic Parasites with Differing Abilities To Transform Leukocytes Reveals Key Mediators of Theileria-Induced Leukocyte Transformation. mBio 2012, 3, e00204-12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsolier, J.; Weitzman, J.B. Comment le parasite Apicomplexe Theileria manipule-t-il l’identité cellulaire de son hôte bovin? Biol. Aujourd’hui 2014, 208, 311–323. [Google Scholar] [CrossRef]
- Pain, A. Genome of the Host-Cell Transforming Parasite Theileria annulata Compared with T. parva. Science 2005, 309, 131–133. [Google Scholar] [CrossRef] [Green Version]
- Swan, D.G.; Phillips, K.; Tait, A.; Shiels, B.R. Evidence for localisation of a Theileria parasite AT hook DNA-binding protein to the nucleus of immortalised bovine host cells. Mol. Biochem. Parasitol. 1999, 101, 117–129. [Google Scholar] [CrossRef]
- Shiels, B.R.; Langsley, G.; Weir, W.; Pain, A.; McKellar, S.; Dobbelaere, D. Alteration of host cell phenotype by Theileria annulata and Theileria parva: mining for manipulators in the parasite genomes. Int. J. Parasitol. 2006, 36, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, J.D.; Ueti, M.W.; Johnson, W.C.; Scoles, G.A.; Knowles, D.P.; Mealey, R.H. Lymphocytes and Macrophages Are Infected by Theileria equi, but T Cells and B Cells Are Not Required to Establish Infection In Vivo. PLOS ONE 2013, 8, e76996. [Google Scholar] [CrossRef] [PubMed]
- Cock-Rada, A.M.; Medjkane, S.; Janski, N.; Yousfi, N.; Perichon, M.; Chaussepied, M.; Chluba, J.; Langsley, G.; Weitzman, J.B. SMYD3 Promotes Cancer Invasion by Epigenetic Upregulation of the Metalloproteinase MMP-9. Cancer Res. 2012, 72, 810–820. [Google Scholar] [CrossRef] [Green Version]
- Cheeseman, K.; Weitzman, J.B. Host-parasite interactions; an intimate epigenetic relationship. Cell. Microbiol. 2015, 17, 1121–1132. [Google Scholar] [CrossRef]
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [Green Version]
- Jo, Y.H.; Kim, Y.J.; Park, K.B.; Seong, J.H.; Kim, S.G.; Park, S.; Noh, M.Y.; Lee, Y.S.; Han, Y.S. TmCactin plays an important role in Gram-negative and -positive bacterial infection by regulating expression of 7 AMP genes in Tenebrio molitor. Sci. Rep. 2017, 7, srep46459. [Google Scholar] [CrossRef]
- Schuster, M.; Annemann, M.; Plaza-Sirvent, C.; Schmitz, I. Atypical IκB proteins—Nuclear modulators of NF-κB signaling. Cell Commun. Signal. 2013, 11, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Fenwick, C. A Subclass of Ras Proteins That Regulate the Degradation of IB. Science 2000, 287, 869–873. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Sun, M.; Zheng, X.; Li, L.; Sun, L.; Chen, D.; Sun, Q. Cell-surface localization of Pellino antagonizes Toll-mediated innate immune signalling by controlling MyD88 turnover in Drosophila. Nat. Commun. 2014, 5, 3458. [Google Scholar] [CrossRef] [Green Version]
- Haller, D.; Mackiewicz, M.; Gerber, S.; Beyer, D.; Kullmann, B.; Schneider, I.; Ahmed, J.S.; Seitzer, U. Cytoplasmic sequestration of p53 promotes survival in leukocytes transformed by Theileria. Oncogene 2010, 29, 3079–3086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antunes, S.; Couto, J.; Ferrolho, J.; Sanches, G.S.; Charrez, J.O.M.; Hernández, N.D.L.C.; Mazuz, M.; Villar, M.; Shkap, V.; De La Fuente, J.; et al. Transcriptome and Proteome Response of Rhipicephalus annulatus Tick Vector to Babesia bigemina Infection. Front. Physiol. 2019, 10, 318. [Google Scholar] [CrossRef] [PubMed]
- Mondekova, H.H.; Sima, R.; Urbanova, V.; Kovar, V.; Rego, R.O.M.; Grubhoffer, L.; Kopacek, P.; Hajdusek, O. Characterization of Ixodes ricinus Fibrinogen-Related Proteins (Ixoderins) Discloses Their Function in the Tick Innate Immunity. Front. Cell. Infect. Microbiol. 2017, 7, 509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Fujisaki, K. A cysteine protease inhibitor (cystatin) from the tick Haemaphysalis longicornis is involved in tick innate immunity. In Proceedings of the Trends in Acarology; Springer: Berlin/Heidelberg, Germany, 2010; pp. 227–231. [Google Scholar]
- Williams, M.; Baxter, R.H.G. The structure and function of thioester-containing proteins in arthropods. Biophys. Rev. 2014, 6, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.-Y.; Xue, J.; Wu, W.-J.; Wang, Y.; Lv, Z.-Y.; Zhang, C.-X. An immune-induced Reeler protein is involved in the Bombyx mori melanization cascade. Insect Biochem. Mol. Biol. 2011, 41, 696–706. [Google Scholar] [CrossRef]
- Giachetto, P.F.; Cunha, R.C.; Nhani, A.J.; Garcia, M.V.; Ferro, J.A.; Andreotti, R. Gene Expression in the Salivary Gland of Rhipicephalus (Boophilus) microplus Fed on Tick-Susceptible and Tick-Resistant Hosts. Front. Cell. Infect. Microbiol. 2020, 9, 477. [Google Scholar] [CrossRef]
- Narasimhan, S.; Schuijt, T.J.; Abraham, N.M.; Rajeevan, N.; Coumou, J.; Graham, M.; Robson, A.; Wu, M.-J.; Daffre, S.; Hovius, J.W.; et al. Modulation of the tick gut milieu by a secreted tick protein favors Borrelia burgdorferi colonization. Nat. Commun. 2017, 8, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, W.; Rajendran, R.; Zhao, Y.; Tan, B.; Wu, G.; Bazer, F.W.; Zhu, G.; Peng, Y.; Huang, X.; Deng, J.; et al. Amino Acids As Mediators of Metabolic Cross Talk between Host and Pathogen. Front. Immunol. 2018, 9, 319. [Google Scholar] [CrossRef]
- Radakovic, M.; Davitkov, D.; Borozan, S.; Stojanovic, S.; Stevanovic, J.; Krstic, V.; Stanimirovic, Z. Oxidative stress and DNA damage in horses naturally infected with Theileria equi. Veter. J. 2016, 217, 112–118. [Google Scholar] [CrossRef]
- Ahantarig, A.; Trinachartvanit, W.; Baimai, V.; Grubhoffer, L. Hard ticks and their bacterial endosymbionts (or would be pathogens). Folia Microbiol. 2013, 58, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Durrani, Z.; Weir, W.; Pillai, S.; Kinnaird, J.; Shiels, B.R. Modulation of activation-associated host cell gene expression by the apicomplexan parasite T heileria annulata. Cell. Microbiol. 2012, 14, 1434–1454. [Google Scholar] [CrossRef] [Green Version]
- Tschoeke, D.A.; Nunes, G.L.; Jardim, R.; Lima, J.; Dumaresq, A.S.; Gomes, M.R.; de Mattos Pereira, L.; Loureiro, D.R.; Stoco, P.H.; de Matos Guedes, H.L.; et al. The Comparative Genomics and Phylogenomics of Leishmania Amazonensis parasite. Evol. Bioinform. 2014, 10, 131–153. [Google Scholar] [CrossRef]
- Mano, J.; Belles-Boix, E.; Babiychuk, E.; Inzé, D.; Torii, Y.; Hiraoka, E.; Takimoto, K.; Slooten, L.; Asada, K.; Kushnir, S. Protection against Photooxidative Injury of Tobacco Leaves by 2-Alkenal Reductase. Detoxication of Lipid Peroxide-Derived Reactive Carbonyls. Plant Physiol. 2005, 139, 1773–1783. [Google Scholar] [CrossRef] [Green Version]
- Battesti, D.M.B.; Arzua, M.; Bechara, G.H. Carrapatos de importância médico-veterinária da região neotropical: Um guia ilustrado para identificação de espécies. Available online: http://bases.bireme.br/cgi-bin/wxislind.exe/iah/online/?IsisScript=iah/iah.xis&src=google&base=LILACS&lang=p&nextAction=lnk&exprSearch=443314&indexSearch=ID (accessed on 23 September 2020).
- Bhoora, R.; Quan, M.; Franssen, L.; Butler, C.M.; Van Der Kolk, J.H.; Guthrie, A.J.; Zweygarth, E.; Jongejan, F.; Collins, N.E. Development and evaluation of real-time PCR assays for the quantitative detection of Babesia caballi and Theileria equi infections in horses from South Africa. Veter. Parasitol. 2010, 168, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.R.; Brown, G.K.; Dunstan, R.H.; Roberts, T.K. Anaplasma platys: An improved PCR for its detection in dogs. Exp. Parasitol. 2005, 109, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.-M.; Blanco, L.B.C.; Alhassan, A.; Iseki, H.; Yokoyama, N.; Xuan, X.; Igarashi, I. Diagnostic real-time PCR assay for the quantitative detection of Theileria equi from equine blood samples. Veter. Parasitol. 2008, 151, 158–163. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- TransDecoder/TransDecoder. Available online: https://github.com/TransDecoder/TransDecoder (accessed on 23 September 2020).
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S. Blast2GO: A Comprehensive Suite for Functional Analysis in Plant Genomics. Int. J. Plant Genom. 2008, 2008, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kriventseva, E.V.; Kuznetsov, D.; Tegenfeldt, F.; Manni, M.; Dias, R.; Simão, F.A.; Zdobnov, E.M. OrthoDB v10: Sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 2019, 47, D807–D811. [Google Scholar] [CrossRef] [Green Version]
- Seppey, M.; Manni, M.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness. In Gene Prediction; Kollmar, M., Ed.; Methods in Molecular Biology; Humana: New York, NY, USA, 2019; Volume 1962, pp. 227–245. [Google Scholar]
- The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; Von Mering, C.; Bork, P. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, 34. [Google Scholar] [CrossRef] [Green Version]
- Nijhof, A.M.; Balk, J.A.; Postigo, M.; Jongejan, F. Selection of reference genes for quantitative RT-PCR studies in Rhipicephalus (Boophilus) microplus and Rhipicephalus appendiculatus ticks and determination of the expression profile of Bm86. BMC Mol. Biol. 2009, 10, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Total Trinity transcripts | 295,406 |
| % GC | 46.21 |
| Contig ExN10 | 8044 |
| Contig ExN20 | 5968 |
| Contig ExN30 | 4649 |
| Contig ExN50 | 2721 |
| Median contig length | 340 |
| Average contig | 961.12 |
| Total assembled bases | 283,930,859 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paulino, P.; Vitari, G.; Rezende, A.; Couto, J.; Antunes, S.; Domingos, A.; Peckle, M.; Massard, C.; Araújo, F.; Santos, H. Characterization of the Rhipicephalus (Boophilus) microplus Sialotranscriptome Profile in Response to Theileria equi Infection. Pathogens 2021, 10, 167. https://doi.org/10.3390/pathogens10020167
Paulino P, Vitari G, Rezende A, Couto J, Antunes S, Domingos A, Peckle M, Massard C, Araújo F, Santos H. Characterization of the Rhipicephalus (Boophilus) microplus Sialotranscriptome Profile in Response to Theileria equi Infection. Pathogens. 2021; 10(2):167. https://doi.org/10.3390/pathogens10020167
Chicago/Turabian StylePaulino, Patrícia, Gabriela Vitari, Antonio Rezende, Joana Couto, Sandra Antunes, Ana Domingos, Maristela Peckle, Carlos Massard, Flávio Araújo, and Huarrisson Santos. 2021. "Characterization of the Rhipicephalus (Boophilus) microplus Sialotranscriptome Profile in Response to Theileria equi Infection" Pathogens 10, no. 2: 167. https://doi.org/10.3390/pathogens10020167