
Role of Apoptotic Cell Clearance in Pneumonia and Inflammatory Lung Disease


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Overview of Apoptosis
3. Mechanisms of Efferocytosis
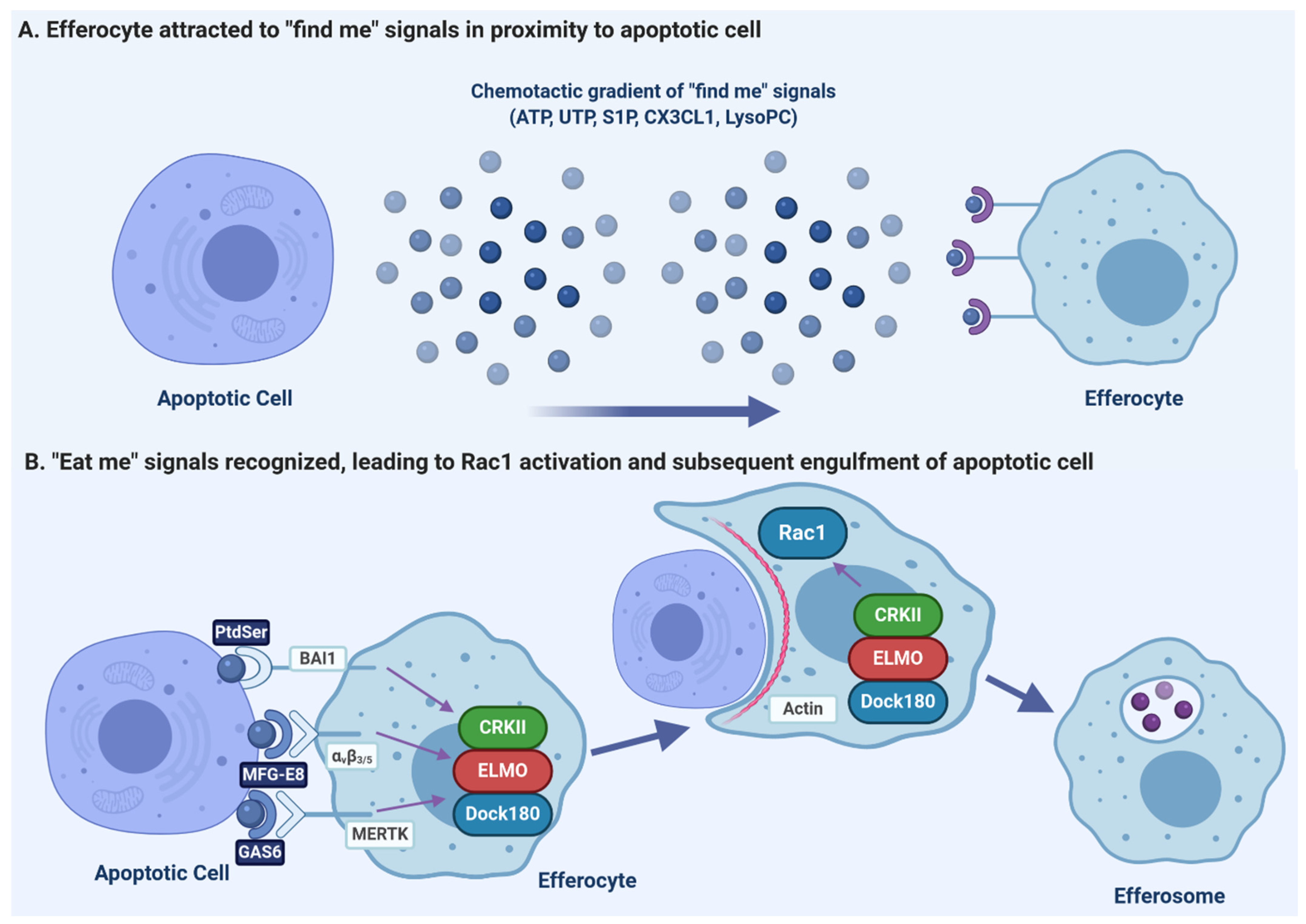
3.1. Release of “Find-Me” Signals by Apoptotic Cells
3.2. Recognition of Apoptotic Cells via “Eat-Me” Signals
3.3. Receptor Signaling and Internalization
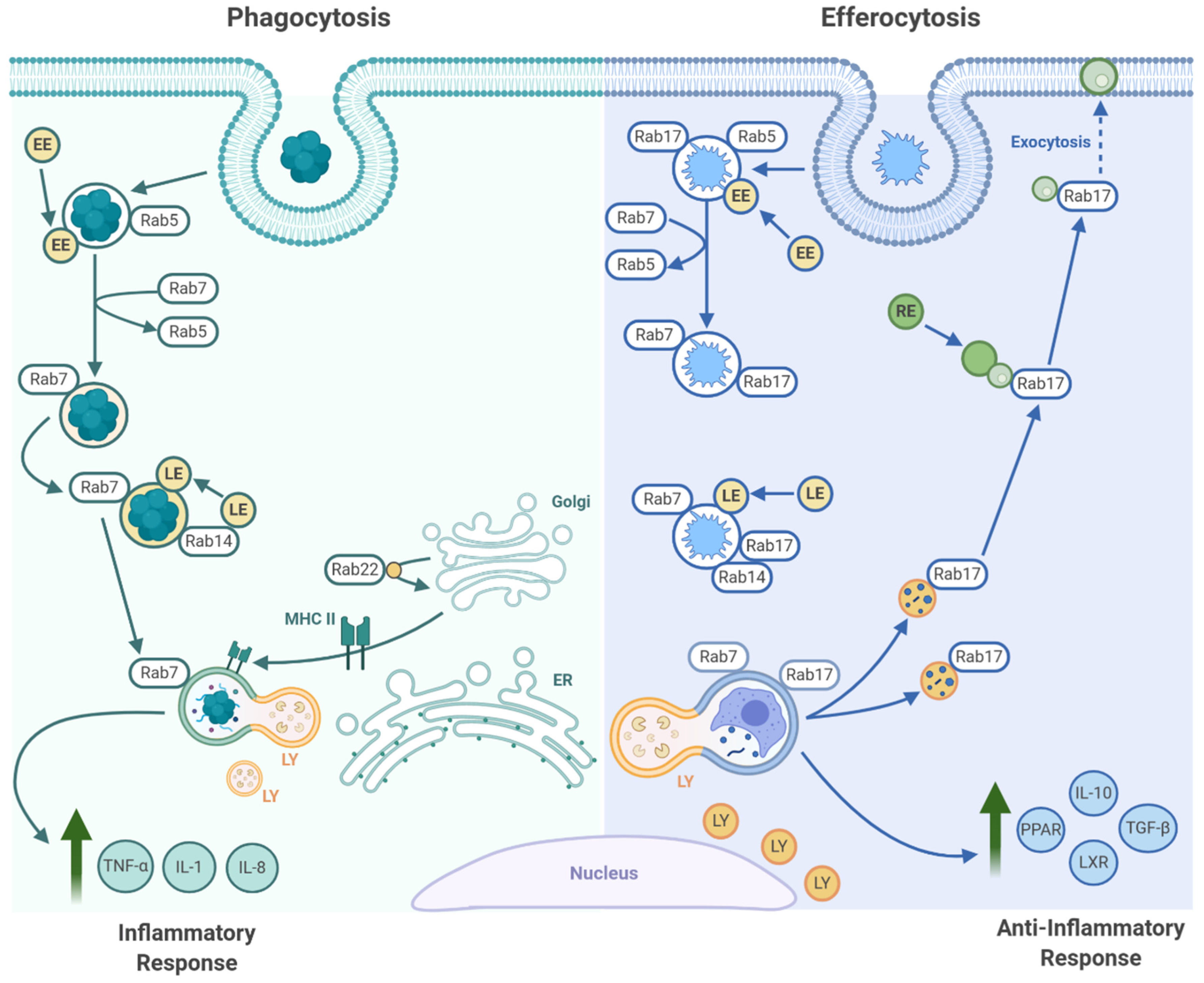
3.4. Efferosome Maturation
4. Efferocyte Metabolism, Polarization, and Inflammation
4.1. Efferocyte Metabolism and Polarization
4.2. Specialized Pro-Resolving Mediators
5. Alveolar Macrophages and Efferocytosis in the Lung
6. Impaired Efferocytosis in Lung Disease
6.1. Chronic Obstrructive Pulmonary Disorder
6.2. Asthma
6.3. Cystic Fibrosis
6.4. Lung Cancer
6.5. Community-Aquired Pneumonia
7. Pathogen Manipulation of Efferocytosis
7.1. Subverting Efferocytosis
7.1.1. Streptococcus pneumoniae: Inducing Apoptosis to Limit Microbicidal Activity
7.1.2. Legionella, Salmonella and Tuberculosis: Inhibiting Apoptosis
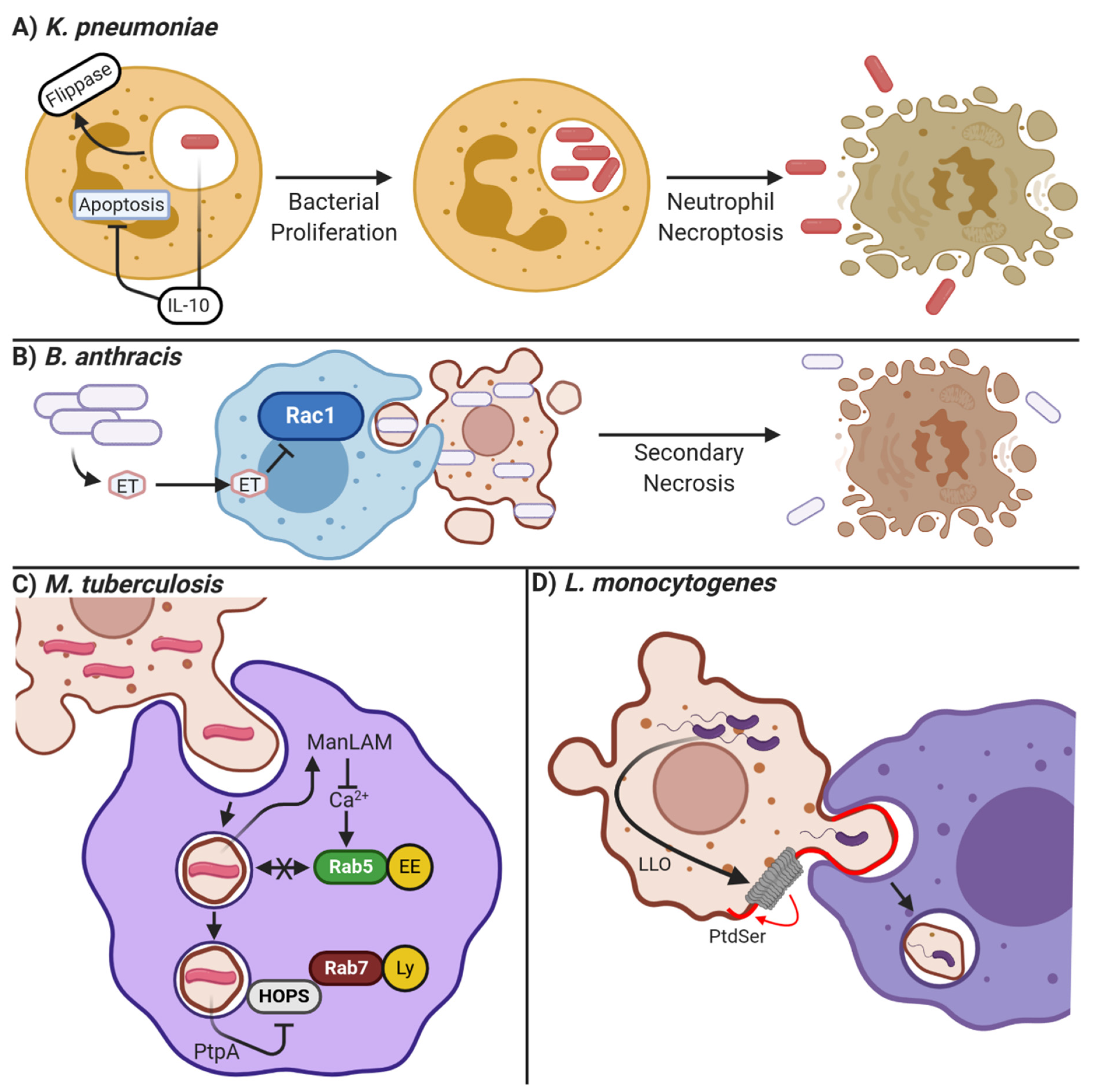
7.1.3. Klebsiella pneumoniae: Manipulating Cell Death Pathways
7.1.4. Staphylococcus aureus: Cell- Specific Manipulation of Apoptosis and Efferocytosis
7.1.5. Francisella novicida and Bacillus anthracis: Inhibiting Efferocytic Receptors
7.1.6. Altered Efferosome Maturation
7.2. Manipulating Efferocytosis
8. Therapeutic Interventions that Manipulate Efferocytosis
8.1. Existing Therapeutics
8.2. Novel Therapeutic Targets
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- D’Arcy, M.S. Cell Death: A Review of the Major Forms of Apoptosis, Necrosis and Autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Hochreiter-Hufford, A.; Ravichandran, K.S. Clearing the Dead: Apoptotic Cell Sensing, Recognition, Engulfment, and Digestion. Cold Spring Harb. Perspect. Biol. 2013, 5, a008748. [Google Scholar] [CrossRef] [Green Version]
- McCubbrey, A.L.; Curtis, J.L. Efferocytosis and Lung Disease. Chest 2013, 143, 1750–1757. [Google Scholar] [CrossRef] [Green Version]
- Korns, D.; Frasch, S.C.; Fernandez-Boyanapalli, R.; Henson, P.M.; Bratton, D.L. Modulation of Macrophage Efferocytosis in Inflammation. Front. Immunol. 2011, 2, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Flannagan, R.S.; Jaumouillé, V.; Grinstein, S. The Cell Biology of Phagocytosis. Annu. Rev. Pathol. 2011, 7, 61–98. [Google Scholar] [CrossRef]
- Taefehshokr, N.; Yin, C.; Heit, B. Rab GTPases in the Differential Processing of Phagocytosed Pathogens versus Efferocytosed Apoptotic Cells. Histol. Histopathol. 2020, 18252. [Google Scholar] [CrossRef]
- Kawano, M.; Nagata, S. Efferocytosis and Autoimmune Disease. Int. Immunol. 2018, 30, 551–558. [Google Scholar] [CrossRef] [Green Version]
- Vandivier, R.W.; Henson, P.M.; Douglas, I.S. Burying the Dead: The Impact of Failed Apoptotic Cell Removal (Efferocytosis) on Chronic Inflammatory Lung Disease. Chest 2006, 129, 1673–1682. [Google Scholar] [CrossRef]
- Demedts, I.K.; Demoor, T.; Bracke, K.R.; Joos, G.F.; Brusselle, G.G. Role of Apoptosis in the Pathogenesis of COPD and Pulmonary Emphysema. Respir. Res. 2006, 7, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandivier, R.W.; Fadok, V.A.; Hoffmann, P.R.; Bratton, D.L.; Penvari, C.; Brown, K.K.; Brain, J.D.; Accurso, F.J.; Henson, P.M. Elastase-Mediated Phosphatidylserine Receptor Cleavage Impairs Apoptotic Cell Clearance in Cystic Fibrosis and Bronchiectasis. J. Clin. Investig. 2002, 109, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Wootton, D.G.; Diggle, P.J.; Court, J.; Eneje, O.; Keogan, L.; Macfarlane, L.; Wilks, S.; Woodhead, M.; Gordon, S.B. Recovery from Pneumonia Requires Efferocytosis Which Is Impaired in Smokers and Those with Low Body Mass Index and Enhanced by Statins. Thorax 2016, 71, 1052–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bates, M.; Stolberg, V.R.; Freeman, C.M.; Kady, M.R.; Alikaj, H.; McCloskey, L.; Curtis, J.L. Glucocorticoid-Augmented Efferocytosis Reduces Pneumococcal Killing by Human Alveolar Macrophages by Blocking Phagosome Acidification. In C31. Mechanistic Insights into Lung Infection, Proceedings of the American Thoracic Society International Conference Abstracts, San Diego, CA, USA, 18–23 May 2018; American Thoracic Society: New York, NY, USA, 2018; p. A4701. [Google Scholar]
- Stolberg, V.R.; McCubbrey, A.L.; Freeman, C.M.; Brown, J.P.; Crudgington, S.W.; Taitano, S.H.; Saxton, B.L.; Mancuso, P.; Curtis, J.L. Glucocorticoid-Augmented Efferocytosis Inhibits Pulmonary Pneumococcal Clearance in Mice by Reducing Alveolar Macrophage Bactericidal Function. J. Immunol. 2015, 195, 174–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wang, R.; Wang, H.; Yang, X.; Yang, J.; Xiong, W.; Wen, Q.; Ma, L. Glucocorticoids Suppress Antimicrobial Autophagy and Nitric Oxide Production and Facilitate Mycobacterial Survival in Macrophages. Sci. Rep. 2017, 7, 982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Green, D.R.; Llambi, F. Cell Death Signaling. Cold Spring Harb. Perspect. Biol. 2015, 7. [Google Scholar] [CrossRef]
- Kiraz, Y.; Adan, A.; Kartal Yandim, M.; Baran, Y. Major Apoptotic Mechanisms and Genes Involved in Apoptosis. Tumor Biol. 2016, 37, 8471–8486. [Google Scholar] [CrossRef] [Green Version]
- Julien, O.; Wells, J.A. Caspases and Their Substrates. Cell Death Differ. 2017, 24, 1380–1389. [Google Scholar] [CrossRef]
- Yang, J.; Liu, X.; Bhalla, K.; Kim, C.N.; Ibrado, A.M.; Cai, J.; Peng, T.I.; Jones, D.P.; Wang, X. Prevention of Apoptosis by Bcl-2: Release of Cytochrome c from Mitochondria Blocked. Science 1997, 275, 1129–1132. [Google Scholar] [CrossRef]
- Kluck, R.M.; Bossy-Wetzel, E.; Green, D.R.; Newmeyer, D.D. The Release of Cytochrome c from Mitochondria: A Primary Site for Bcl-2 Regulation of Apoptosis. Science 1997, 275, 1132–1136. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.H.; Zhao, Y.; Barber, M.J.; Kuharsky, D.K.; Yin, X.M. Bid-Induced Cytochrome c Release Is Mediated by a Pathway Independent of Mitochondrial Permeability Transition Pore and Bax. J. Biol. Chem. 2000, 275, 39474–39481. [Google Scholar] [CrossRef] [Green Version]
- Morin, D.; Pires, F.; Plin, C.; Tillement, J.-P. Role of the Permeability Transition Pore in Cytochrome C Release from Mitochondria during Ischemia-Reperfusion in Rat Liver. Biochem. Pharmacol. 2004, 68, 2065–2073. [Google Scholar] [CrossRef] [PubMed]
- Hill, M.M.; Adrain, C.; Duriez, P.J.; Creagh, E.M.; Martin, S.J. Analysis of the Composition, Assembly Kinetics and Activity of Native Apaf-1 Apoptosomes. EMBO J. 2004, 23, 2134–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, H.; Li, Y.; Liu, X.; Wang, X. An APAF-1. Cytochrome c Multimeric Complex Is a Functional Apoptosome That Activates Procaspase-9. J. Biol. Chem. 1999, 274, 11549–11556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, N.; Yonehara, S.; Ishii, A.; Yonehara, M.; Mizushima, S.; Sameshima, M.; Hase, A.; Seto, Y.; Nagata, S. The Polypeptide Encoded by the CDNA for Human Cell Surface Antigen Fas Can Mediate Apoptosis. Cell 1991, 66, 233–243. [Google Scholar] [CrossRef]
- Pan, G.; O’Rourke, K.; Chinnaiyan, A.M.; Gentz, R.; Ebner, R.; Ni, J.; Dixit, V.M. The Receptor for the Cytotoxic Ligand TRAIL. Science 1997, 276, 111–113. [Google Scholar] [CrossRef]
- Chaudhary, P.M.; Eby, M.; Jasmin, A.; Bookwalter, A.; Murray, J.; Hood, L. Death Receptor 5, a New Member of the TNFR Family, and DR4 Induce FADD-Dependent Apoptosis and Activate the NF-KappaB Pathway. Immunity 1997, 7, 821–830. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, T.; Strasser, A.; Jost, P.J. Fas Death Receptor Signalling: Roles of Bid and XIAP. Cell Death Differ. 2012, 19, 42–50. [Google Scholar] [CrossRef] [Green Version]
- Schug, Z.T.; Gonzalvez, F.; Houtkooper, R.H.; Vaz, F.M.; Gottlieb, E. BID Is Cleaved by Caspase-8 within a Native Complex on the Mitochondrial Membrane. Cell Death Differ. 2011, 18, 538–548. [Google Scholar] [CrossRef]
- Heit, B.; Yeung, T.; Grinstein, S. Changes in Mitochondrial Surface Charge Mediate Recruitment of Signaling Molecules during Apoptosis. Am. J. Physiol. 2011, 300, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Ozören, N.; El-Deiry, W.S. Defining Characteristics of Types I and II Apoptotic Cells in Response to TRAIL. Neoplasia 2002, 4, 551–557. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, T.; Katayama, T.; Kohama, K.; Endo, Y.; Sawasaki, T. Myosin Phosphatase Is Inactivated by Caspase-3 Cleavage and Phosphorylation of Myosin Phosphatase Targeting Subunit 1 during Apoptosis. Mol. Biol. Cell 2013, 24, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Enari, M.; Sakahira, H.; Yokoyama, H.; Okawa, K.; Iwamatsu, A.; Nagata, S. A Caspase-Activated DNase That Degrades DNA during Apoptosis, and Its Inhibitor ICAD. Nature 1998, 391, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Sandilos, J.K.; Chiu, Y.-H.; Chekeni, F.B.; Armstrong, A.J.; Walk, S.F.; Ravichandran, K.S.; Bayliss, D.A. Pannexin 1, an ATP Release Channel, Is Activated by Caspase Cleavage of Its Pore-Associated C-Terminal Autoinhibitory Region. J. Biol. Chem. 2012, 287, 11303–11311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Jiang, H.; Luo, S.; Zhang, M.; Zhang, Y.; Sun, F.; Huang, S.; Li, H. Caspase-Mediated Cleavage of C53/LZAP Protein Causes Abnormal Microtubule Bundling and Rupture of the Nuclear Envelope. Cell Res. 2013, 23, 691–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeCathelineau, A.M.; Henson, P.M. The Final Step in Programmed Cell Death: Phagocytes Carry Apoptotic Cells to the Grave. Essays Biochem. 2003, 39, 105–117. [Google Scholar] [CrossRef]
- Kimani, S.G.; Geng, K.; Kasikara, C.; Kumar, S.; Sriram, G.; Wu, Y.; Birge, R.B. Contribution of Defective PS Recognition and Efferocytosis to Chronic Inflammation and Autoimmunity. Front. Immunol. 2014, 5, 566. [Google Scholar] [CrossRef]
- Thorp, E.B. Mechanisms of Failed Apoptotic Cell Clearance by Phagocyte Subsets in Cardiovascular Disease. Apoptosis 2010, 15, 1124–1136. [Google Scholar] [CrossRef] [Green Version]
- Silva, M.T. Secondary Necrosis: The Natural Outcome of the Complete Apoptotic Program. FEBS Lett. 2010, 584, 4491–4499. [Google Scholar] [CrossRef] [Green Version]
- Silva, M.T.; do Vale, A.; dos Santos, N.M.N. Secondary Necrosis in Multicellular Animals: An Outcome of Apoptosis with Pathogenic Implications. Apoptosis 2008, 13, 463–482. [Google Scholar] [CrossRef]
- Vanden Berghe, T.; Vanlangenakker, N.; Parthoens, E.; Deckers, W.; Devos, M.; Festjens, N.; Guerin, C.J.; Brunk, U.T.; Declercq, W.; Vandenabeele, P. Necroptosis, Necrosis and Secondary Necrosis Converge on Similar Cellular Disintegration Features. Cell Death Differ. 2010, 17, 922–930. [Google Scholar] [CrossRef] [Green Version]
- Bruce, J.I. Plasma Membrane Calcium Pump Regulation by Metabolic Stress. World J. Biol. Chem. 2010, 1, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Steenbergen, C. Mechanisms Underlying Acute Protection from Cardiac Ischemia-Reperfusion Injury. Physiol. Rev. 2008, 88, 581–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, B.S.; Belenghi, B.; Levine, A. Oxidative Stress Increased Respiration and Generation of Reactive Oxygen Species, Resulting in ATP Depletion, Opening of Mitochondrial Permeability Transition, and Programmed Cell Death. Plant. Physiol. 2002, 128, 1271–1281. [Google Scholar] [CrossRef] [Green Version]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.-S. Calcium, ATP, and ROS: A Mitochondrial Love-Hate Triangle. Am. J. Physiol. Cell Physiol. 2004, 287, 817–833. [Google Scholar] [CrossRef] [PubMed]
- Sachet, M.; Liang, Y.Y.; Oehler, R. The Immune Response to Secondary Necrotic Cells. Apoptosis 2017, 22, 1189–1204. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Molinaro, C.; Johnson, N.; Casiano, C.A. Secondary Necrosis Is a Source of Proteolytically Modified Forms of Specific Intracellular Autoantigens: Implications for Systemic Autoimmunity. Arthritis Rheum. 2001, 44, 2642–2652. [Google Scholar] [CrossRef]
- Martin, C.J.; Booty, M.G.; Rosebrock, T.R.; Nunes-Alves, C.; Desjardins, D.M.; Keren, I.; Fortune, S.M.; Remold, H.G.; Behar, S.M. Efferocytosis Is an Innate Antibacterial Mechanism. Cell Host Microbe 2012, 12, 289–300. [Google Scholar] [CrossRef] [Green Version]
- Karaji, N.; Sattentau, Q.J. Efferocytosis of Pathogen-Infected Cells. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Rosales, C.; Uribe-Querol, E. Phagocytosis: A Fundamental Process in Immunity. Biomed. Res. Int. 2017, 2017, 9042851. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.; Plüddemann, A. Macrophage Clearance of Apoptotic Cells: A Critical Assessment. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Elliott, M.R.; Koster, K.M.; Murphy, P.S. Efferocytosis Signaling in the Regulation of Macrophage Inflammatory Responses. J. Immunol. 2017, 198, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, K.S. Find-Me and Eat-Me Signals in Apoptotic Cell Clearance: Progress and Conundrums. J. Exp. Med. 2010, 207, 1807–1817. [Google Scholar] [CrossRef] [PubMed]
- Lauber, K.; Bohn, E.; Kröber, S.M.; Xiao, Y.; Blumenthal, S.G.; Lindemann, R.K.; Marini, P.; Wiedig, C.; Zobywalski, A.; Baksh, S.; et al. Apoptotic Cells Induce Migration of Phagocytes via Caspase-3-Mediated Release of a Lipid Attraction Signal. Cell 2003, 113, 717–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peter, C.; Waibel, M.; Keppeler, H.; Lehmann, R.; Xu, G.; Halama, A.; Adamski, J.; Schulze-Osthoff, K.; Wesselborg, S.; Lauber, K. Release of Lysophospholipid “find-Me” Signals during Apoptosis Requires the ATP-Binding Cassette Transporter A1. Autoimmunity 2012, 45, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovich, M.; Sharma, P.; et al. Nucleotides Released by Apoptotic Cells Act as a Find-Me Signal to Promote Phagocytic Clearance. Nature 2009, 461, 282–286. [Google Scholar] [CrossRef] [Green Version]
- Heit, B.; Robbins, S.M.; Downey, C.M.; Guan, Z.; Colarusso, P.; Miller, B.J.; Jirik, F.R.; Kubes, P. PTEN Functions to “prioritize” Chemotactic Cues and Prevent “Distraction” in Migrating Neutrophils. Nat. Immunol. 2008, 9, 743–752. [Google Scholar] [CrossRef]
- Truman, L.A.; Ford, C.A.; Pasikowska, M.; Pound, J.D.; Wilkinson, S.J.; Dumitriu, I.E.; Melville, L.; Melrose, L.A.; Ogden, C.A.; Nibbs, R.; et al. CX3CL1/Fractalkine Is Released from Apoptotic Lymphocytes to Stimulate Macrophage Chemotaxis. Blood 2008, 112, 5026–5036. [Google Scholar] [CrossRef] [Green Version]
- Gude, D.R.; Alvarez, S.E.; Paugh, S.W.; Mitra, P.; Yu, J.; Griffiths, R.; Barbour, S.E.; Milstien, S.; Spiegel, S. Apoptosis Induces Expression of Sphingosine Kinase 1 to Release Sphingosine-1-Phosphate as a “Come-and-Get-Me” Signal. FASEB J. 2008, 22, 2629–2638. [Google Scholar] [CrossRef] [Green Version]
- Wong, H.S.; Jaumouillé, V.; Heit, B.; Doodnauth, S.A.; Patel, S.; Huang, Y.-W.; Grinstein, S.; Robinson, L.A. Cytoskeletal Confinement of CX3CL1 Limits Its Susceptibility to Proteolytic Cleavage by ADAM10. Mol. Biol. Cell 2014, 25, 3884–3899. [Google Scholar] [CrossRef]
- Elliott, M.R.; Ravichandran, K.S. The Dynamics of Apoptotic Cell Clearance. Dev. Cell 2016, 38, 147–160. [Google Scholar] [CrossRef] [Green Version]
- Mitra, P.; Oskeritzian, C.A.; Payne, S.G.; Beaven, M.A.; Milstien, S.; Spiegel, S. Role of ABCC1 in Export of Sphingosine-1-Phosphate from Mast Cells. Proc. Natl. Acad. Sci. USA 2006, 103, 16394–16399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, B.; Gan, W.; Liu, Z.; Shen, Z.; Wang, J.; Shi, R.; Liu, Y.; Liu, Y.; Jiang, M.; Zhang, Z.; et al. Erythropoeitin Signaling in Macrophages Promotes Dying Cell Clearance and Immune Tolerance. Immunity 2016, 44, 287–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, Y.-S.; Kim, S.-Y.; Kim, M.-J.; Lim, J.-H.; Cho, M.-S.; Kang, J.L. PPARγ Activation Following Apoptotic Cell Instillation Promotes Resolution of Lung Inflammation and Fibrosis via Regulation of Efferocytosis and Proresolving Cytokines. Mucosal Immunol. 2015, 8, 1031–1046. [Google Scholar] [CrossRef] [PubMed]
- Segawa, K.; Nagata, S. An Apoptotic “Eat Me” Signal: Phosphatidylserine Exposure. Trends Cell Biol. 2015, 25, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Bian, Z.; Shi, L.; Niu, S.; Ha, B.; Tremblay, A.; Li, L.; Zhang, X.; Paluszynski, J.; Liu, M.; et al. Loss of Cell Surface CD47 Clustering Formation and Binding Avidity to SIRPα Facilitate Apoptotic Cell Clearance by Macrophages. J. Immunol. 2015, 195, 661–671. [Google Scholar] [CrossRef] [Green Version]
- Tsai, R.K.; Discher, D.E. Inhibition of “Self” Engulfment through Deactivation of Myosin-II at the Phagocytic Synapse between Human Cells. J. Cell Biol. 2008, 180, 989–1003. [Google Scholar] [CrossRef] [Green Version]
- Fadok, V.A.; Warner, M.L.; Bratton, D.L.; Henson, P.M. CD36 Is Required for Phagocytosis of Apoptotic Cells by Human Macrophages That Use Either a Phosphatidylserine Receptor or the Vitronectin Receptor (Alpha v Beta 3). J. Immunol. 1998, 161, 6250–6257. [Google Scholar] [PubMed]
- Fadok, V.A.; Voelker, D.R.; Campbell, P.A.; Cohen, J.J.; Bratton, D.L.; Henson, P.M. Exposure of Phosphatidylserine on the Surface of Apoptotic Lymphocytes Triggers Specific Recognition and Removal by Macrophages. J. Immunol. 1992, 148, 2207–2216. [Google Scholar] [PubMed]
- Païdassi, H.; Tacnet-Delorme, P.; Garlatti, V.; Darnault, C.; Ghebrehiwet, B.; Gaboriaud, C.; Arlaud, G.J.; Frachet, P. C1q Binds Phosphatidylserine and Likely Acts as a Multiligand-Bridging Molecule in Apoptotic Cell Recognition. J. Immunol. 2008, 180, 2329–2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Liu, J.; Piao, C.; Shao, J.; Du, J. ICAM-1 Suppresses Tumor Metastasis by Inhibiting Macrophage M2 Polarization through Blockade of Efferocytosis. Cell Death Dis. 2015, 6, e1780. [Google Scholar] [CrossRef] [Green Version]
- Greenberg, M.E.; Sun, M.; Zhang, R.; Febbraio, M.; Silverstein, R.; Hazen, S.L. Oxidized Phosphatidylserine-CD36 Interactions Play an Essential Role in Macrophage-Dependent Phagocytosis of Apoptotic Cells. J. Exp. Med. 2006, 203, 2613–2625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardai, S.J.; McPhillips, K.A.; Frasch, S.C.; Janssen, W.J.; Starefeldt, A.; Murphy-Ullrich, J.E.; Bratton, D.L.; Oldenborg, P.-A.; Michalak, M.; Henson, P.M. Cell-Surface Calreticulin Initiates Clearance of Viable or Apoptotic Cells through Trans-Activation of LRP on the Phagocyte. Cell 2005, 123, 321–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segawa, K.; Suzuki, J.; Nagata, S. Flippases and Scramblases in the Plasma Membrane. Cell Cycle 2014, 13, 2990–2991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagata, S.; Suzuki, J.; Segawa, K.; Fujii, T. Exposure of Phosphatidylserine on the Cell Surface. Cell Death Differ. 2016, 23, 952–961. [Google Scholar] [CrossRef] [Green Version]
- Kay, J.G.; Koivusalo, M.; Ma, X.; Wohland, T.; Grinstein, S. Phosphatidylserine Dynamics in Cellular Membranes. Mol. Biol. Cell 2012, 23, 2198–2212. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Imanishi, E.; Nagata, S. Xkr8 Phospholipid Scrambling Complex in Apoptotic Phosphatidylserine Exposure. Proc. Natl. Acad. Sci. USA 2016, 113, 9509–9514. [Google Scholar] [CrossRef] [Green Version]
- Segawa, K.; Kurata, S.; Yanagihashi, Y.; Brummelkamp, T.R.; Matsuda, F.; Nagata, S. Caspase-Mediated Cleavage of Phospholipid Flippase for Apoptotic Phosphatidylserine Exposure. Science 2014, 344, 1164–1168. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, J.; Denning, D.P.; Imanishi, E.; Horvitz, H.R.; Nagata, S. Xk-Related Protein 8 and CED-8 Promote Phosphatidylserine Exposure in Apoptotic Cells. Science 2013, 341, 403–406. [Google Scholar] [CrossRef] [Green Version]
- Flannagan, R.S.; Canton, J.; Furuya, W.; Glogauer, M.; Grinstein, S. The Phosphatidylserine Receptor TIM4 Utilizes Integrins as Coreceptors to Effect Phagocytosis. Mol. Biol. Cell 2014, 25, 1511–1522. [Google Scholar] [CrossRef]
- Park, D.; Tosello-Trampont, A.-C.; Elliott, M.R.; Lu, M.; Haney, L.B.; Ma, Z.; Klibanov, A.L.; Mandell, J.W.; Ravichandran, K.S. BAI1 Is an Engulfment Receptor for Apoptotic Cells Upstream of the ELMO/Dock180/Rac Module. Nature 2007, 450, 430–434. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-Y.; Kim, I.-S. Stabilin Receptors: Role as Phosphatidylserine Receptors. Biomolecules 2019, 9, 387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, Y.; Tian, L.; Voss, O.H.; Margulies, D.H.; Krzewski, K.; Coligan, J.E. CD300b Regulates the Phagocytosis of Apoptotic Cells via Phosphatidylserine Recognition. Cell Death Differ. 2014, 21, 1746–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friggeri, A.; Banerjee, S.; Biswas, S.; de Freitas, A.; Liu, G.; Bierhaus, A.; Abraham, E. Participation of the Receptor for Advanced Glycation End Products in Efferocytosis. J. Immunol. 2011, 186, 6191–6198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackburn, J.W.D.; Lau, D.H.C.; Liu, E.Y.; Ellins, J.; Vrieze, A.M.; Pawlak, E.N.; Dikeakos, J.D.; Heit, B. Soluble CD93 Is an Apoptotic Cell Opsonin Recognized by Ax Β2. Eur. J. Immunol. 2019, 49, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Nandrot, E.F.; Anand, M.; Almeida, D.; Atabai, K.; Sheppard, D.; Finnemann, S.C. Essential Role for MFG-E8 as Ligand for Alphavbeta5 Integrin in Diurnal Retinal Phagocytosis. Proc. Natl. Acad. Sci. USA 2007, 104, 12005–12010. [Google Scholar] [CrossRef] [Green Version]
- Jun, J.-I.; Kim, K.-H.; Lau, L.F. The Matricellular Protein CCN1 Mediates Neutrophil Efferocytosis in Cutaneous Wound Healing. Nat. Commun. 2015, 6, 7386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, M.O.; Obin, M.S.; Heeb, M.J.; Burgess, B.L.; Abrams, T.A. Both Protein S and Gas6 Stimulate Outer Segment Phagocytosis by Cultured Rat Retinal Pigment Epithelial Cells. Exp. Eye Res. 2005, 81, 581–591. [Google Scholar] [CrossRef]
- Hurtado, B.; Muñoz, X.; Recarte-Pelz, P.; García, N.; Luque, A.; Krupinski, J.; Sala, N.; de Frutos, P.G. Expression of the Vitamin K-Dependent Proteins GAS6 and Protein S and the TAM Receptor Tyrosine Kinases in Human Atherosclerotic Carotid Plaques. Thromb. Haemost. 2011, 105, 873–882. [Google Scholar] [CrossRef] [Green Version]
- Morizono, K.; Xie, Y.; Olafsen, T.; Lee, B.; Dasgupta, A.; Wu, A.M.; Chen, I.S.Y. The Soluble Serum Protein Gas6 Bridges Virion Envelope Phosphatidylserine to the TAM Receptor Tyrosine Kinase Axl to Mediate Viral Entry. Cell Host Microbe 2011, 9, 286–298. [Google Scholar] [CrossRef] [Green Version]
- Fadok, V.A.; Bratton, D.L.; Konowal, A.; Freed, P.W.; Westcott, J.Y.; Henson, P.M. Macrophages That Have Ingested Apoptotic Cells in vitro Inhibit Proinflammatory Cytokine Production through Autocrine/Paracrine Mechanisms Involving TGF-Beta, PGE2, and PAF. J. Clin. Investig. 1998, 101, 890–898. [Google Scholar] [CrossRef] [Green Version]
- Freire-de-Lima, C.G.; Xiao, Y.Q.; Gardai, S.J.; Bratton, D.L.; Schiemann, W.P.; Henson, P.M. Apoptotic Cells, through Transforming Growth Factor-Beta, Coordinately Induce Anti-Inflammatory and Suppress pro-Inflammatory Eicosanoid and NO Synthesis in Murine Macrophages. J. Biol. Chem. 2006, 281, 38376–38384. [Google Scholar] [CrossRef] [Green Version]
- Bevers, E.M.; Comfurius, P.; van Rijn, J.L.; Hemker, H.C.; Zwaal, R.F. Generation of Prothrombin-Converting Activity and the Exposure of Phosphatidylserine at the Outer Surface of Platelets. Eur. J. Biochem. 1982, 122, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Segawa, K.; Suzuki, J.; Nagata, S. Constitutive Exposure of Phosphatidylserine on Viable Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 19246–19251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, S.; Heinisch, I.; Ross, E.; Shaw, K.; Buckley, C.D.; Savill, J. Apoptosis Disables CD31-Mediated Cell Detachment from Phagocytes Promoting Binding and Engulfment. Nature 2002, 418, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Bradley, C.A. CD24—A Novel ‘Don’t Eat Me’ Signal. Nat. Rev. Cancer 2019, 19, 541. [Google Scholar] [CrossRef] [PubMed]
- Azuma, Y.; Nakagawa, H.; Dote, K.; Higai, K.; Matsumoto, K. Decreases in CD31 and CD47 Levels on the Cell Surface during Etoposide-Induced Jurkat Cell Apoptosis. Biol. Pharm. Bull. 2011, 34, 1828–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigotti, A.; Acton, S.L.; Krieger, M. The Class B Scavenger Receptors SR-BI and CD36 Are Receptors for Anionic Phospholipids. J. Biol. Chem. 1995, 270, 16221–16224. [Google Scholar] [CrossRef] [Green Version]
- Wiesolek, H.L.; Bui, T.M.; Lee, J.J.; Dalal, P.; Finkielsztein, A.; Batra, A.; Thorp, E.B.; Sumagin, R. Intercellular Adhesion Molecule 1 Functions as an Efferocytosis Receptor in Inflammatory Macrophages. Am. J. Pathol. 2020, 190, 874–885. [Google Scholar] [CrossRef]
- Osman, R.; Tacnet-Delorme, P.; Kleman, J.-P.P.; Millet, A.; Frachet, P. Calreticulin Release at an Early Stage of Death Modulates the Clearance by Macrophages of Apoptotic Cells. Front. Immunol. 2017, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Heit, B.; Kim, H.; Cosío, G.; Castaño, D.; Collins, R.; Lowell, C.A.; Kain, K.C.; Trimble, W.S.; Grinstein, S. Multimolecular Signaling Complexes Enable Syk-Mediated Signaling of CD36 Internalization. Dev. Cell 2013, 24, 372–383. [Google Scholar] [CrossRef] [Green Version]
- Torres-Gomez, A.; Cabañas, C.; Lafuente, E.M. Phagocytic Integrins: Activation and Signaling. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Yancey, P.G.; Babaev, V.R.; Blakemore, J.L.; Zhang, Y.; Ding, L.; Fazio, S.; Linton, M.F. Macrophage SR-BI Mediates Efferocytosis via Src/PI3K/Rac1 Signaling and Reduces Atherosclerotic Lesion Necrosis. J. Lipid Res. 2015, 56, 1449–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eom, H.; Kaushik, N.; Yoo, K.; Shim, J.; Kwon, M.; Choi, M.-Y.; Yoon, T.; Kang, S.; Lee, S. MerTK Mediates STAT3-KRAS/SRC-Signaling Axis for Glioma Stem Cell Maintenance. Artif. Cells Nanomed. Biotechnol. 2018, 46, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Shelby, S.J.; Colwill, K.; Dhe-Paganon, S.; Pawson, T.; Thompson, D.A. MERTK Interactions with SH2-Domain Proteins in the Retinal Pigment Epithelium. PLoS ONE 2013, 8, e53964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brugnera, E.; Haney, L.; Grimsley, C.; Lu, M.; Walk, S.F.; Tosello-Trampont, A.-C.; Macara, I.G.; Madhani, H.; Fink, G.R.; Ravichandran, K.S. Unconventional Rac-GEF Activity Is Mediated through the Dock180-ELMO Complex. Nat. Cell Biol. 2002, 4, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Ravichandran, K.S. Dock180-ELMO Cooperation in Rac Activation. Methods Enzymol. 2006, 406, 388–402. [Google Scholar] [CrossRef]
- Wu, Y.; Singh, S.; Georgescu, M.-M.; Birge, R.B. A Role for Mer Tyrosine Kinase in Alphavbeta5 Integrin-Mediated Phagocytosis of Apoptotic Cells. J. Cell Sci. 2005, 118, 539–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, M.L.; Kim, J.I.; Birge, R.B. Alphavbeta5 Integrin Recruits the CrkII-Dock180-Rac1 Complex for Phagocytosis of Apoptotic Cells. Nat. Cell Biol. 2000, 2, 899–905. [Google Scholar] [CrossRef]
- Kitano, M.; Nakaya, M.; Nakamura, T.; Nagata, S.; Matsuda, M. Imaging of Rab5 Activity Identifies Essential Regulators for Phagosome Maturation. Nature 2008, 453, 241–245. [Google Scholar] [CrossRef] [Green Version]
- Vieira, O.V.; Botelho, R.J.; Rameh, L.; Brachmann, S.M.; Matsuo, T.; Davidson, H.W.; Schreiber, A.; Backer, J.M.; Cantley, L.C.; Grinstein, S. Distinct Roles of Class I and Class III Phosphatidylinositol 3-Kinases in Phagosome Formation and Maturation. J. Cell Biol. 2001, 155, 19–25. [Google Scholar] [CrossRef]
- Christoforidis, S.; McBride, H.M.; Burgoyne, R.D.; Zerial, M. The Rab5 Effector EEA1 Is a Core Component of Endosome Docking. Nature 1999, 397, 621–625. [Google Scholar] [CrossRef] [PubMed]
- Eathiraj, S.; Pan, X.; Ritacco, C.; Lambright, D.G. Structural Basis of Family-Wide Rab GTPase Recognition by Rabenosyn-5. Nature 2005, 436, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Kleine Balderhaar, H.J.; Ungermann, C. CORVET and HOPS Tethering Complexes—Coordinators of Endosome and Lysosome Fusion. J. Cell Sci. 2013, 126, 1307–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becken, U.; Jeschke, A.; Veltman, K.; Haas, A. Cell-Free Fusion of Bacteria-Containing Phagosomes with Endocytic Compartments. Proc. Natl. Acad. Sci. USA 2010, 107, 20726–20731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonsen, A.; Gaullier, J.M.; D’Arrigo, A.; Stenmark, H. The Rab5 Effector EEA1 Interacts Directly with Syntaxin-6. J. Biol. Chem. 1999, 274, 28857–28860. [Google Scholar] [CrossRef] [Green Version]
- Kinchen, J.M.; Ravichandran, K.S. Identification of Two Evolutionarily Conserved Genes Regulating Processing of Engulfed Apoptotic Cells. Nature 2010, 464, 778–782. [Google Scholar] [CrossRef] [Green Version]
- Johansson, M.; Rocha, N.; Zwart, W.; Jordens, I.; Janssen, L.; Kuijl, C.; Olkkonen, V.M.; Neefjes, J. Activation of Endosomal Dynein Motors by Stepwise Assembly of Rab7-RILP-P150Glued, ORP1L, and the Receptor βIII Spectrin. J. Biol. 2007, 176, 459–471. [Google Scholar] [CrossRef] [Green Version]
- Langemeyer, L.; Borchers, A.-C.; Herrmann, E.; Füllbrunn, N.; Han, Y.; Perz, A.; Auffarth, K.; Kümmel, D.; Ungermann, C. A Conserved and Regulated Mechanism Drives Endosomal Rab Transition. eLife 2020, 9, e56090. [Google Scholar] [CrossRef]
- Cantalupo, G.; Alifano, P.; Roberti, V.; Bruni, C.B.; Bucci, C. Rab-Interacting Lysosomal Protein (RILP): The Rab7 Effector Required for Transport to Lysosomes. EMBO J. 2001, 20, 683–693. [Google Scholar] [CrossRef]
- Harrison, R.E.; Bucci, C.; Vieira, O.V.; Schroer, T.A.; Grinstein, S. Phagosomes Fuse with Late Endosomes and/or Lysosomes by Extension of Membrane Protrusions along Microtubules: Role of Rab7 and RILP. Mol. Cell. Biol. 2003, 23, 6494–6506. [Google Scholar] [CrossRef]
- Downey, G.P.; Botelho, R.J.; Butler, J.R.; Moltyaner, Y.; Chien, P.; Schreiber, A.D.; Grinstein, S. Phagosomal Maturation, Acidification, and Inhibition of Bacterial Growth in Nonphagocytic Cells Transfected with FcgammaRIIA Receptors. J. Biol. Chem. 1999, 274, 28436–28444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, S.; Faulhaber, A.; Sieber, C.; Pfeifer, D.; Hochberg, T.; Gansz, M.; Deshmukh, S.D.; Dauth, S.; Brix, K.; Saftig, P.; et al. The Endolysosomal Cysteine Cathepsins L and K Are Involved in Macrophage-Mediated Clearance of Staphylococcus Aureus and the Concomitant Cytokine Induction. FASEB J. 2014, 28, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Savina, A.; Jancic, C.; Hugues, S.; Guermonprez, P.; Vargas, P.; Moura, I.C.; Lennon-Duménil, A.-M.; Seabra, M.C.; Raposo, G.; Amigorena, S. NOX2 Controls Phagosomal PH to Regulate Antigen Processing during Crosspresentation by Dendritic Cells. Cell 2006, 126, 205–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes, P.; Demaurex, N.; Dinauer, M.C. Regulation of the NADPH Oxidase and Associated Ion Fluxes during Phagocytosis. Traffic 2013, 14, 1118–1131. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, M.; Huber, L.A.; Parton, R.G.; Griffiths, G. Biogenesis of Phagolysosomes Proceeds through a Sequential Series of Interactions with the Endocytic Apparatus. J. Cell Biol. 1994, 124, 677–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fairn, G.D.; Grinstein, S. How Nascent Phagosomes Mature to Become Phagolysosomes. Trends Immunol. 2012, 1–9. [Google Scholar] [CrossRef]
- Mantegazza, A.R.; Zajac, A.L.; Twelvetrees, A.; Holzbaur, E.L.F.; Amigorena, S.; Marks, M.S. TLR-Dependent Phagosome Tubulation in Dendritic Cells Promotes Phagosome Cross-Talk to Optimize MHC-II Antigen Presentation. Proc. Natl. Acad. Sci. USA 2014, 111, 15508–15513. [Google Scholar] [CrossRef] [Green Version]
- Mantegazza, A.R.; Magalhaes, J.G.; Amigorena, S.; Marks, M.S. Presentation of Phagocytosed Antigens by MHC Class I and II. Traffic 2013, 14, 135–152. [Google Scholar] [CrossRef] [Green Version]
- Moghaddami, M.; Mayrhofer, G.; Cleland, L.G. MHC Class II Compartment, Endocytosis and Phagocytic Activity of Macrophages and Putative Dendritic Cells Isolated from Normal Tissues Rich in Synovium. Int. Immunol. 2005, 17, 1117–1130. [Google Scholar] [CrossRef]
- Mori, Y.; Matsui, T.; Fukuda, M. Rabex-5 Protein Regulates Dendritic Localization of Small GTPase Rab17 and Neurite Morphogenesis in Hippocampal Neurons. J. Biol. Chem. 2013, 288, 9835–9847. [Google Scholar] [CrossRef] [Green Version]
- Yin, C.; Kim, Y.; Argintaru, D.; Heit, B. Rab17 Mediates Differential Antigen Sorting Following Efferocytosis and Phagocytosis. Cell Death Dis. 2016, 7, e2529. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Argintaru, D.; Heit, B. Rab17 Mediates Intermixing of Phagocytosed Apoptotic Cells with Recycling Endosomes. Small GTPases 2019, 10, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Blander, J.M.; Medzhitov, R. Toll-Dependent Selection of Microbial Antigens for Presentation by Dendritic Cells. Nature 2006, 440, 808–812. [Google Scholar] [CrossRef] [PubMed]
- López-Haber, C.; Levin-Konigsberg, R.; Zhu, Y.; Bi-Karchin, J.; Balla, T.; Grinstein, S.; Marks, M.S.; Mantegazza, A.R. Phosphatidylinositol-4-Kinase IIα Licenses Phagosomes for TLR4 Signaling and MHC-II Presentation in Dendritic Cells. Proc. Natl. Acad. Sci. USA 2020, 117, 28251–28262. [Google Scholar] [CrossRef]
- Voll, R.E.; Herrmann, M.; Roth, E.A.; Stach, C.; Kalden, J.R.; Girkontaite, I. Immunosuppressive Effects of Apoptotic Cells. Nature 1997, 390, 350–351. [Google Scholar] [CrossRef]
- Tibrewal, N.; Wu, Y.; D’mello, V.; Akakura, R.; George, T.C.; Varnum, B.; Birge, R.B. Autophosphorylation Docking Site Tyr-867 in Mer Receptor Tyrosine Kinase Allows for Dissociation of Multiple Signaling Pathways for Phagocytosis of Apoptotic Cells and down-Modulation of Lipopolysaccharide-Inducible NF-KappaB Transcriptional Activation. J. Biol. Chem. 2008, 283, 3618–3627. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.-J.; Han, J.-Y.; Byun, J.; Park, H.-J.; Park, E.-M.; Chong, Y.H.; Cho, M.-S.; Kang, J.L. Inhibiting Mer Receptor Tyrosine Kinase Suppresses STAT1, SOCS1/3, and NF-ΚB Activation and Enhances Inflammatory Responses in Lipopolysaccharide-Induced Acute Lung Injury. J. Leukoc. Biol. 2012, 91, 921–932. [Google Scholar] [CrossRef]
- Zhang, B.; Fang, L.; Wu, H.-M.; Ding, P.-S.; Xu, K.; Liu, R.-Y. Mer Receptor Tyrosine Kinase Negatively Regulates Lipoteichoic Acid-Induced Inflammatory Response via PI3K/Akt and SOCS3. Mol. Immunol. 2016, 76, 98–107. [Google Scholar] [CrossRef]
- Rébé, C.; Raveneau, M.; Chevriaux, A.; Lakomy, D.; Sberna, A.-L.; Costa, A.; Bessède, G.; Athias, A.; Steinmetz, E.; Lobaccaro, J.M.; et al. Induction of Transglutaminase 2 by a Liver X Receptor/Retinoic Acid Receptor Alpha Pathway Increases the Clearance of Apoptotic Cells by Human Macrophages. Circ. Res. 2009, 105, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Mukundan, L.; Odegaard, J.I.; Morel, C.R.; Heredia, J.E.; Mwangi, J.W.; Ricardo-Gonzalez, R.R.; Goh, Y.P.S.; Eagle, A.R.; Dunn, S.E.; Awakuni, J.U.H.; et al. PPAR-Delta Senses and Orchestrates Clearance of Apoptotic Cells to Promote Tolerance. Nat. Med. 2009, 15, 1266–1272. [Google Scholar] [CrossRef]
- Gonzalez, N.A.; Bensinger, S.J.; Hong, C.; Beceiro, S.; Bradley, M.N.; Zelcer, N.; Deniz, J.; Ramirez, C.; Díaz, M.; Gallardo, G.; et al. Apoptotic Cells Promote Their Own Clearance and Immune Tolerance through Activation of the Nuclear Receptor LXR. Immunity 2009, 31, 245–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chawla, A.; Boisvert, W.A.; Lee, C.-H.; Laffitte, B.A.; Barak, Y.; Joseph, S.B.; Liao, D.; Nagy, L.; Edwards, P.A.; Curtiss, L.K.; et al. A PPARγ-LXR-ABCA1 Pathway in Macrophages Is Involved in Cholesterol Efflux and Atherogenesis. Mol. Cell 2001, 7, 161–171. [Google Scholar] [CrossRef]
- Beyer, M.; Mallmann, M.R.; Xue, J.; Staratschek-Jox, A.; Vorholt, D.; Krebs, W.; Sommer, D.; Sander, J.; Mertens, C.; Nino-Castro, A.; et al. High-Resolution Transcriptome of Human Macrophages. PLoS ONE 2012, 7, e45466. [Google Scholar] [CrossRef]
- Boergesen, M.; Pedersen, T.Å.; Gross, B.; van Heeringen, S.J.; Hagenbeek, D.; Bindesbøll, C.; Caron, S.; Lalloyer, F.; Steffensen, K.R.; Nebb, H.I.; et al. Genome-Wide Profiling of Liver X Receptor, Retinoid X Receptor, and Peroxisome Proliferator-Activated Receptor α in Mouse Liver Reveals Extensive Sharing of Binding Sites. Mol. Cell Biol. 2012, 32, 852–867. [Google Scholar] [CrossRef] [Green Version]
- Croasdell, A.; Duffney, P.F.; Kim, N.; Lacy, S.H.; Sime, P.J.; Phipps, R.P. PPARγ and the Innate Immune System Mediate the Resolution of Inflammation. PPAR Res. 2015, 2015, 549691. [Google Scholar] [CrossRef] [Green Version]
- Johann, A.M.; von Knethen, A.; Lindemann, D.; Brüne, B. Recognition of Apoptotic Cells by Macrophages Activates the Peroxisome Proliferator-Activated Receptor-Gamma and Attenuates the Oxidative Burst. Cell Death Differ. 2006, 13, 1533–1540. [Google Scholar] [CrossRef] [Green Version]
- Majai, G.; Sarang, Z.; Csomós, K.; Zahuczky, G.; Fésüs, L. PPARgamma-Dependent Regulation of Human Macrophages in Phagocytosis of Apoptotic Cells. Eur. J. Immunol. 2007, 37, 1343–1354. [Google Scholar] [CrossRef]
- Wang, L.H.; Yang, X.Y.; Zhang, X.; Huang, J.; Hou, J.; Li, J.; Xiong, H.; Mihalic, K.; Zhu, H.; Xiao, W.; et al. Transcriptional Inactivation of STAT3 by PPARgamma Suppresses IL-6-Responsive Multiple Myeloma Cells. Immunity 2004, 20, 205–218. [Google Scholar] [CrossRef] [Green Version]
- Ide, T.; Shimano, H.; Yoshikawa, T.; Yahagi, N.; Amemiya-Kudo, M.; Matsuzaka, T.; Nakakuki, M.; Yatoh, S.; Iizuka, Y.; Tomita, S.; et al. Cross-Talk between Peroxisome Proliferator-Activated Receptor (PPAR) Alpha and Liver X Receptor (LXR) in Nutritional Regulation of Fatty Acid Metabolism. II. LXRs Suppress Lipid Degradation Gene Promoters through Inhibition of PPAR Signaling. Mol. Endocrinol. 2003, 17, 1255–1267. [Google Scholar] [CrossRef] [Green Version]
- Teissier, E.; Nohara, A.; Chinetti, G.; Paumelle, R.; Cariou, B.; Fruchart, J.-C.; Brandes, R.P.; Shah, A.; Staels, B. Peroxisome Proliferator–Activated Receptor α Induces NADPH Oxidase Activity in Macrophages, Leading to the Generation of LDL with PPAR-α Activation Properties. Circ. Res. 2004, 95, 1174–1182. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, W.F.; Kumar, A.P.; Piedrafita, F.J. The Human Myeloperoxidase Gene Is Regulated by LXR and PPARα Ligands. Biochem. Biophys. Res. Commun. 2006, 349, 846–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serghides, L.; Kain, K.C. Peroxisome Proliferator-Activated Receptor Gamma-Retinoid X Receptor Agonists Increase CD36-Dependent Phagocytosis of Plasmodium Falciparum-Parasitized Erythrocytes and Decrease Malaria-Induced TNF-Alpha Secretion by Monocytes/Macrophages. J. Immunol. 2001, 166, 6742–6748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walcher, D.; Kümmel, A.; Kehrle, B.; Bach, H.; Grüb, M.; Durst, R.; Hombach, V.; Marx, N. LXR Activation Reduces Proinflammatory Cytokine Expression in Human CD4-Positive Lymphocytes. Arter. Thromb. Vasc. Biol. 2006, 26, 1022–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontaine, C.; Rigamonti, E.; Nohara, A.; Gervois, P.; Teissier, E.; Fruchart, J.-C.; Staels, B.; Chinetti-Gbaguidi, G. Liver X Receptor Activation Potentiates the Lipopolysaccharide Response in Human Macrophages. Circ. Res. 2007, 101, 40–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.; Weinberg, S.; DeBerge, M.; Gainullina, A.; Schipma, M.; Kinchen, J.M.; Ben-Sahra, I.; Gius, D.R.; Yvan-Charvet, L.; Chandel, N.S.; et al. Efferocytosis Fuels Requirements of Fatty Acid Oxidation and the Electron Transport Chain to Polarize Macrophages for Tissue Repair. Cell Metab. 2019, 29, 443–456.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costet, P.; Lalanne, F.; Gerbod-Giannone, M.C.; Molina, J.R.; Fu, X.; Lund, E.G.; Gudas, L.J.; Tall, A.R. Retinoic Acid Receptor-Mediated Induction of ABCA1 in Macrophages. Mol. Cell Biol. 2003, 23, 7756–7766. [Google Scholar] [CrossRef] [Green Version]
- Yvan-Charvet, L.; Pagler, T.A.; Seimon, T.A.; Thorp, E.; Welch, C.L.; Witztum, J.L.; Tabas, I.; Tall, A.R. ABCA1 and ABCG1 Protect against Oxidative Stress-Induced Macrophage Apoptosis during Efferocytosis. Circ. Res. 2010, 106, 1861–1869. [Google Scholar] [CrossRef] [Green Version]
- Viaud, M.; Ivanov, S.; Vujic, N.; Duta-Mare, M.; Aira, L.-E.; Barouillet, T.; Garcia, E.; Orange, F.; Dugail, I.; Hainault, I.; et al. Lysosomal Cholesterol Hydrolysis Couples Efferocytosis to Anti-Inflammatory Oxysterol Production. Circ. Res. 2018, 122, 1369–1384. [Google Scholar] [CrossRef]
- Yurdagul, A.; Subramanian, M.; Wang, X.; Crown, S.B.; Ilkayeva, O.R.; Darville, L.; Kolluru, G.K.; Rymond, C.C.; Gerlach, B.D.; Zheng, Z.; et al. Macrophage Metabolism of Apoptotic Cell-Derived Arginine Promotes Continual Efferocytosis and Resolution of Injury. Cell Metab. 2020, 31, 518–533.e10. [Google Scholar] [CrossRef]
- Yin, C.; Vrieze, A.M.; Rosoga, M.; Akingbasote, J.; Pawlak, E.N.; Jacob, R.A.; Hu, J.; Sharma, N.; Dikeakos, J.D.; Barra, L.; et al. Efferocytic Defects in Early Atherosclerosis Are Driven by GATA2 Overexpression in Macrophages. Front. Immunol. 2020, 11, 594136. [Google Scholar] [CrossRef]
- Gonzalez, N.A.; Quintana, J.A.; García-Silva, S.; Mazariegos, M.; González de la Aleja, A.; Nicolás-Ávila, J.A.; Walter, W.; Adrover, J.M.; Crainiciuc, G.; Kuchroo, V.K.; et al. Phagocytosis Imprints Heterogeneity in Tissue-Resident Macrophages. J. Exp. Med. 2017, 214, 1281–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basil, M.C.; Levy, B.D. Specialized Pro-Resolving Mediators: Endogenous Regulators of Infection and Inflammation. Nat. Rev. Immunol. 2016, 16, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Dalli, J.; Serhan, C.N. Pro-Resolving Mediators in Regulating and Conferring Macrophage Function. Front. Immunol. 2017, 8, 1400. [Google Scholar] [CrossRef] [Green Version]
- Levy, B.D.; Clish, C.B.; Schmidt, B.; Gronert, K.; Serhan, C.N. Lipid Mediator Class Switching during Acute Inflammation: Signals in Resolution. Nat. Immunol. 2001, 2, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Mancini, A.D.; Di Battista, J.A. The Cardinal Role of the Phospholipase A(2)/Cyclooxygenase-2/Prostaglandin E Synthase/Prostaglandin E(2) (PCPP) Axis in Inflammostasis. Inflamm. Res. 2011, 60, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- Levy, B.D.; Romano, M.; Chapman, H.A.; Reilly, J.J.; Drazen, J.; Serhan, C.N. Human Alveolar Macrophages Have 15-Lipoxygenase and Generate 15(S)-Hydroxy-5,8,11-Cis-13-Trans-Eicosatetraenoic Acid and Lipoxins. J. Clin. Investig. 1993, 92, 1572–1579. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Hamberg, M.; Samuelsson, B. Lipoxins: Novel Series of Biologically Active Compounds Formed from Arachidonic Acid in Human Leukocytes. Proc. Natl. Acad. Sci. USA 1984, 81, 5335–5339. [Google Scholar] [CrossRef] [Green Version]
- Arita, M.; Bianchini, F.; Aliberti, J.; Sher, A.; Chiang, N.; Hong, S.; Yang, R.; Petasis, N.A.; Serhan, C.N. Stereochemical Assignment, Antiinflammatory Properties, and Receptor for the Omega-3 Lipid Mediator Resolvin E1. J. Exp. Med. 2005, 201, 713–722. [Google Scholar] [CrossRef]
- Serhan, C.N.; Clish, C.B.; Brannon, J.; Colgan, S.P.; Chiang, N.; Gronert, K. Novel Functional Sets of Lipid-Derived Mediators with Antiinflammatory Actions Generated from Omega-3 Fatty Acids via Cyclooxygenase 2-Nonsteroidal Antiinflammatory Drugs and Transcellular Processing. J. Exp. Med. 2000, 192, 1197–1204. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Yang, R.; Martinod, K.; Kasuga, K.; Pillai, P.S.; Porter, T.F.; Oh, S.F.; Spite, M. Maresins: Novel Macrophage Mediators with Potent Antiinflammatory and Proresolving Actions. J. Exp. Med. 2009, 206, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Langmead, C.J.; Riddy, D.M. New Advances in Targeting the Resolution of Inflammation: Implications for Specialized Pro-Resolving Mediator GPCR Drug Discovery. ACS Pharmacol. Transl. Sci. 2020, 3, 88–106. [Google Scholar] [CrossRef] [PubMed]
- Codagnone, M.; Cianci, E.; Lamolinara, A.; Mari, V.C.; Nespoli, A.; Isopi, E.; Mattoscio, D.; Arita, M.; Bragonzi, A.; Iezzi, M.; et al. Resolvin D1 Enhances the Resolution of Lung Inflammation Caused by Long-Term Pseudomonas Aeruginosa Infection. Mucosal Immunol. 2018, 11, 35–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duvall, M.G.; Bruggemann, T.R.; Levy, B.D. Bronchoprotective Mechanisms for Specialized Pro-Resolving Mediators in the Resolution of Lung Inflammation. Mol. Aspects Med. 2017, 58, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Werz, O.; Gerstmeier, J.; Libreros, S.; De la Rosa, X.; Werner, M.; Norris, P.C.; Chiang, N.; Serhan, C.N. Human Macrophages Differentially Produce Specific Resolvin or Leukotriene Signals That Depend on Bacterial Pathogenicity. Nat. Commun. 2018, 9, 59. [Google Scholar] [CrossRef]
- Pirault, J.; Bäck, M. Lipoxin and Resolvin Receptors Transducing the Resolution of Inflammation in Cardiovascular Disease. Front. Pharmacol. 2018, 9, 1273. [Google Scholar] [CrossRef]
- Bondue, B.; Vosters, O.; de Nadai, P.; Glineur, S.; De Henau, O.; Luangsay, S.; Van Gool, F.; Communi, D.; De Vuyst, P.; Desmecht, D.; et al. ChemR23 Dampens Lung Inflammation and Enhances Anti-Viral Immunity in a Mouse Model of Acute Viral Pneumonia. PLoS Pathog. 2011, 7, e1002358. [Google Scholar] [CrossRef]
- Bozinovski, S.; Uddin, M.; Vlahos, R.; Thompson, M.; McQualter, J.L.; Merritt, A.-S.; Wark, P.A.B.; Hutchinson, A.; Irving, L.B.; Levy, B.D.; et al. Serum Amyloid A Opposes Lipoxin A4 to Mediate Glucocorticoid Refractory Lung Inflammation in Chronic Obstructive Pulmonary Disease. Proc. Natl. Acad. Sci. USA 2012, 109, 935–940. [Google Scholar] [CrossRef] [Green Version]
- Fujimori, T.; Grabiec, A.M.; Kaur, M.; Bell, T.J.; Fujino, N.; Cook, P.C.; Svedberg, F.R.; MacDonald, A.S.; Maciewicz, R.A.; Singh, D.; et al. The Axl Receptor Tyrosine Kinase Is a Discriminator of Macrophage Function in the Inflamed Lung. Mucosal Immunol. 2015, 8, 1021–1030. [Google Scholar] [CrossRef] [Green Version]
- Hussell, T.; Bell, T.J. Alveolar Macrophages: Plasticity in a Tissue-Specific Context. Nat. Rev. Immunol. 2014, 14, 81–93. [Google Scholar] [CrossRef]
- Mukherjee, S.; Subramaniam, R.; Chen, H.; Smith, A.; Keshava, S.; Shams, H. Boosting Efferocytosis in Alveolar Space Using BCG Vaccine to Protect Host against Influenza Pneumonia. PLoS ONE 2017, 12, e0180143. [Google Scholar] [CrossRef]
- Kasahara, Y.; Tuder, R.M.; Cool, C.D.; Lynch, D.A.; Flores, S.C.; Voelkel, N.F. Endothelial Cell Death and Decreased Expression of Vascular Endothelial Growth Factor and Vascular Endothelial Growth Factor Receptor 2 in Emphysema. Am. J. Respir. Crit. Care Med. 2001, 163, 737–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knapp, S.; Leemans, J.C.; Florquin, S.; Branger, J.; Maris, N.A.; Pater, J.; van Rooijen, N.; van der Poll, T. Alveolar Macrophages Have a Protective Antiinflammatory Role during Murine Pneumococcal Pneumonia. Am. J. Respir. Crit. Care Med. 2003, 167, 171–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holt, P.G. Inhibitory Activity of Unstimulated Alveolar Macrophages on T-Lymphocyte Blastogenic Response. Am. Rev. Respir. Dis. 1978, 118, 791–793. [Google Scholar] [CrossRef] [PubMed]
- Neupane, A.S.; Willson, M.; Chojnacki, A.K.; Vargas E Silva Castanheira, F.; Morehouse, C.; Carestia, A.; Keller, A.E.; Peiseler, M.; DiGiandomenico, A.; Kelly, M.M.; et al. Patrolling Alveolar Macrophages Conceal Bacteria from the Immune System to Maintain Homeostasis. Cell 2020, 183, 110–125.e11. [Google Scholar] [CrossRef]
- Summers, K.M.; Bush, S.J.; Hume, D.A. Network Analysis of Transcriptomic Diversity amongst Resident Tissue Macrophages and Dendritic Cells in the Mouse Mononuclear Phagocyte System. PLoS Biol. 2020, 18, e3000859. [Google Scholar] [CrossRef] [PubMed]
- Emam, M.; Cánovas, A.; Islas-Trejo, A.D.; Fonseca, P.A.S.; Medrano, J.F.; Mallard, B. Transcriptomic Profiles of Monocyte-Derived Macrophages in Response to Escherichia Coli Is Associated with the Host Genetics. Sci. Rep. 2020, 10, 271. [Google Scholar] [CrossRef]
- Janssen, W.J.; McPhillips, K.A.; Dickinson, M.G.; Linderman, D.J.; Morimoto, K.; Xiao, Y.Q.; Oldham, K.M.; Vandivier, R.W.; Henson, P.M.; Gardai, S.J. Surfactant Proteins A and D Suppress Alveolar Macrophage Phagocytosis via Interaction with SIRP Alpha. Am. J. Respir. Crit. Care Med. 2008, 178, 158–167. [Google Scholar] [CrossRef]
- Shimizu, T.; Nishitani, C.; Mitsuzawa, H.; Ariki, S.; Takahashi, M.; Ohtani, K.; Wakamiya, N.; Kuroki, Y. Mannose Binding Lectin and Lung Collectins Interact with Toll-like Receptor 4 and MD-2 by Different Mechanisms. Biochim. Biophys. Acta 2009, 1790, 1705–1710. [Google Scholar] [CrossRef]
- Henning, L.N.; Azad, A.K.; Parsa, K.V.L.; Crowther, J.E.; Tridandapani, S.; Schlesinger, L.S. Pulmonary Surfactant Protein A Regulates TLR Expression and Activity in Human Macrophages. J. Immunol. 2008, 180, 7847–7858. [Google Scholar] [CrossRef] [Green Version]
- Ohya, M.; Nishitani, C.; Sano, H.; Yamada, C.; Mitsuzawa, H.; Shimizu, T.; Saito, T.; Smith, K.; Crouch, E.; Kuroki, Y. Human Pulmonary Surfactant Protein D Binds the Extracellular Domains of Toll-like Receptors 2 and 4 through the Carbohydrate Recognition Domain by a Mechanism Different from Its Binding to Phosphatidylinositol and Lipopolysaccharide. Biochemistry 2006, 45, 8657–8664. [Google Scholar] [CrossRef]
- Fernandez, S.; Jose, P.; Avdiushko, M.G.; Kaplan, A.M.; Cohen, D.A. Inhibition of IL-10 Receptor Function in Alveolar Macrophages by Toll-like Receptor Agonists. J. Immunol. 2004, 172, 2613–2620. [Google Scholar] [CrossRef] [PubMed]
- Mihrshahi, R.; Barclay, A.N.; Brown, M.H. Essential Roles for Dok2 and RasGAP in CD200 Receptor-Mediated Regulation of Human Myeloid Cells. J. Immunol. 2009, 183, 4879–4886. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Griffiths, M.J.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The Integrin Alpha v Beta 6 Binds and Activates Latent TGF Beta 1: A Mechanism for Regulating Pulmonary Inflammation and Fibrosis. Cell 1999, 96, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Van der Sluijs, K.F.; van Elden, L.J.R.; Nijhuis, M.; Schuurman, R.; Pater, J.M.; Florquin, S.; Goldman, M.; Jansen, H.M.; Lutter, R.; van der Poll, T. IL-10 Is an Important Mediator of the Enhanced Susceptibility to Pneumococcal Pneumonia after Influenza Infection. J. Immunol. 2004, 172, 7603–7609. [Google Scholar] [CrossRef] [Green Version]
- Snelgrove, R.J.; Goulding, J.; Didierlaurent, A.M.; Lyonga, D.; Vekaria, S.; Edwards, L.; Gwyer, E.; Sedgwick, J.D.; Barclay, A.N.; Hussell, T. A Critical Function for CD200 in Lung Immune Homeostasis and the Severity of Influenza Infection. Nat. Immunol. 2008, 9, 1074–1083. [Google Scholar] [CrossRef]
- Evans, H.G.; Roostalu, U.; Walter, G.J.; Gullick, N.J.; Frederiksen, K.S.; Roberts, C.A.; Sumner, J.; Baeten, D.L.; Gerwien, J.G.; Cope, A.P.; et al. TNF-α Blockade Induces IL-10 Expression in Human CD4+ T Cells. Nat. Commun. 2014, 5, 3199. [Google Scholar] [CrossRef]
- Ural, B.B.; Yeung, S.T.; Damani-Yokota, P.; Devlin, J.C.; de Vries, M.; Vera-Licona, P.; Samji, T.; Sawai, C.M.; Jang, G.; Perez, O.A.; et al. Identification of a Nerve-Associated, Lung-Resident Interstitial Macrophage Subset with Distinct Localization and Immunoregulatory Properties. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Cheng, L.; Li, J.; Wang, X.; Wang, F.; et al. Single-Cell Landscape of Bronchoalveolar Immune Cells in Patients with COVID-19. Nat. Med. 2020, 26, 842–844. [Google Scholar] [CrossRef]
- Krysko, O.; Vandenabeele, P.; Krysko, D.V.; Bachert, C. Impairment of Phagocytosis of Apoptotic Cells and Its Role in Chronic Airway Diseases. Apoptosis 2010, 15, 1137–1146. [Google Scholar] [CrossRef]
- Hodge, S.; Hodge, G.; Scicchitano, R.; Reynolds, P.N.; Holmes, M. Alveolar Macrophages from Subjects with Chronic Obstructive Pulmonary Disease Are Deficient in Their Ability to Phagocytose Apoptotic Airway Epithelial Cells. Immunol. Cell Biol. 2003, 81, 289–296. [Google Scholar] [CrossRef]
- Hodge, S.; Hodge, G.; Ahern, J.; Jersmann, H.; Holmes, M.; Reynolds, P.N. Smoking Alters Alveolar Macrophage Recognition and Phagocytic Ability: Implications in Chronic Obstructive Pulmonary Disease. Am. J. Respir. Cell Mol. Biol. 2007, 37, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.P.; Tuder, R.M. Role of Apoptosis in Amplifying Inflammatory Responses in Lung Diseases. J. Cell Death 2010, 2010, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, C.; Zhao, H.; Liu, L.; Li, W.; Zhou, X.; Lv, Y.; Shen, X.; Liang, Z.; Cai, S.; Zou, F. High Mobility Group Protein B1 (HMGB1) in Asthma: Comparison of Patients with Chronic Obstructive Pulmonary Disease and Healthy Controls. Mol. Med. 2011, 17, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Schagat, T.L.; Wofford, J.A.; Wright, J.R. Surfactant Protein A Enhances Alveolar Macrophage Phagocytosis of Apoptotic Neutrophils. J. Immunol. 2001, 166, 2727–2733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, M.-L.N.; Malcolm, K.C.; Kotaru, C.; Tilstra, J.A.; Westcott, J.Y.; Fadok, V.A.; Wenzel, S.E. Defective Apoptotic Cell Phagocytosis Attenuates Prostaglandin E2 and 15-Hydroxyeicosatetraenoic Acid in Severe Asthma Alveolar Macrophages. Am. J. Respir. Crit. Care Med. 2005, 172, 972–979. [Google Scholar] [CrossRef]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L. Identification of the Cystic Fibrosis Gene: Cloning and Characterization of Complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.V.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-Induced NETosis Is a Dynamic Process Involving Neutrophil Multitasking in Vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef] [Green Version]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. A Myeloperoxidase-Containing Complex Regulates Neutrophil Elastase Release and Actin Dynamics during NETosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef] [Green Version]
- Roquilly, A.; Jacqueline, C.; Davieau, M.; Mollé, A.; Sadek, A.; Fourgeux, C.; Rooze, P.; Broquet, A.; Misme-Aucouturier, B.; Chaumette, T.; et al. Alveolar Macrophages Are Epigenetically Altered after Inflammation, Leading to Long-Term Lung Immunoparalysis. Nat. Immunol. 2020, 21, 636–648. [Google Scholar] [CrossRef]
- Kreiselmeier, N.E.; Kraynack, N.C.; Corey, D.A.; Kelley, T.J. Statin-Mediated Correction of STAT1 Signaling and Inducible Nitric Oxide Synthase Expression in Cystic Fibrosis Epithelial Cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2003, 285, 1286–1295. [Google Scholar] [CrossRef] [Green Version]
- Corvol, H.; Rousselet, N.; Thompson, K.E.; Berdah, L.; Cottin, G.; Foussigniere, T.; Longchampt, E.; Fiette, L.; Sage, E.; Prunier, C.; et al. FAM13A Is a Modifier Gene of Cystic Fibrosis Lung Phenotype Regulating Rhoa Activity, Actin Cytoskeleton Dynamics and Epithelial-Mesenchymal Transition. J. Cyst Fibros 2018, 17, 190–203. [Google Scholar] [CrossRef] [Green Version]
- Vaught, D.B.; Stanford, J.C.; Cook, R.S. Efferocytosis Creates a Tumor Microenvironment Supportive of Tumor Survival and Metastasis. Cancer Cell Microenviron. 2015, 2. [Google Scholar] [CrossRef] [Green Version]
- Birge, R.B.; Boeltz, S.; Kumar, S.; Carlson, J.; Wanderley, J.; Calianese, D.; Barcinski, M.; Brekken, R.A.; Huang, X.; Hutchins, J.T.; et al. Phosphatidylserine Is a Global Immunosuppressive Signal in Efferocytosis, Infectious Disease, and Cancer. Cell Death Differ. 2016, 23, 962–978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Wang, H.; Wu, X.; Liu, B.; Liu, W.; Wang, R.; Liang, X.; Ma, C.; Gao, L. TIM-4 Promotes the Growth of Non-Small-Cell Lung Cancer in a RGD Motif-Dependent Manner. Br. J. Cancer 2015, 113, 1484–1492. [Google Scholar] [CrossRef] [Green Version]
- Salina, A.C.; Souza, T.P.; Serezani, C.H.; Medeiros, A.I. Efferocytosis-Induced Prostaglandin E2 Production Impairs Alveolar Macrophage Effector Functions during Streptococcus Pneumoniae Infection. Innate Immun. 2017, 23, 219–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medeiros, A.I.; Serezani, C.H.; Lee, S.P.; Peters-Golden, M. Efferocytosis Impairs Pulmonary Macrophage and Lung Antibacterial Function via PGE2/EP2 Signaling. J. Exp. Med. 2009, 206, 61–68. [Google Scholar] [CrossRef] [Green Version]
- Serezani, C.H.; Chung, J.; Ballinger, M.N.; Moore, B.B.; Aronoff, D.M.; Peters-Golden, M. Prostaglandin E2 Suppresses Bacterial Killing in Alveolar Macrophages by Inhibiting NADPH Oxidase. Am. J. Respir. Cell Mol. Biol. 2007, 37, 562–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Losick, V.P.; Isberg, R.R. NF-KappaB Translocation Prevents Host Cell Death after Low-Dose Challenge by Legionella Pneumophila. J. Exp. Med. 2006, 203, 2177–2189. [Google Scholar] [CrossRef] [Green Version]
- Finn, C.E.; Chong, A.; Cooper, K.G.; Starr, T.; Steele-Mortimer, O. A Second Wave of Salmonella T3SS1 Activity Prolongs the Lifespan of Infected Epithelial Cells. PLoS Pathog. 2017, 13, e1006354. [Google Scholar] [CrossRef]
- Miller, J.L.; Velmurugan, K.; Cowan, M.J.; Briken, V. The Type I NADH Dehydrogenase of Mycobacterium Tuberculosis Counters Phagosomal NOX2 Activity to Inhibit TNF-Alpha-Mediated Host Cell Apoptosis. PLoS Pathog. 2010, 6, e1000864. [Google Scholar] [CrossRef]
- Behar, S.M.; Martin, C.J.; Booty, M.G.; Nishimura, T.; Zhao, X.; Gan, H.-X.; Divangahi, M.; Remold, H.G. Apoptosis Is an Innate Defense Function of Macrophages against Mycobacterium Tuberculosis. Mucosal Immunol. 2011, 4, 279–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jondle, C.N.; Gupta, K.; Mishra, B.B.; Sharma, J. Klebsiella Pneumoniae Infection of Murine Neutrophils Impairs Their Efferocytic Clearance by Modulating Cell Death Machinery. PLoS Pathog. 2018, 14, e1007338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, D.; Vince, J.E. Pyroptosis versus Necroptosis: Similarities, Differences, and Crosstalk. Cell Death Differ. 2019, 26, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Karki, R.; Kanneganti, T.-D. Molecular Mechanisms and Functions of Pyroptosis, Inflammatory Caspases and Inflammasomes in Infectious Diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef] [Green Version]
- Viganò, E.; Diamond, C.E.; Spreafico, R.; Balachander, A.; Sobota, R.M.; Mortellaro, A. Human Caspase-4 and Caspase-5 Regulate the One-Step Non-Canonical Inflammasome Activation in Monocytes. Nat. Commun. 2015, 6, 8761. [Google Scholar] [CrossRef] [Green Version]
- Baroja-Mazo, A.; Martín-Sánchez, F.; Gomez, A.I.; Martínez, C.M.; Amores-Iniesta, J.; Compan, V.; Barberà-Cremades, M.; Yagüe, J.; Ruiz-Ortiz, E.; Antón, J.; et al. The NLRP3 Inflammasome Is Released as a Particulate Danger Signal That Amplifies the Inflammatory Response. Nat. Immunol. 2014, 15, 738–748. [Google Scholar] [CrossRef]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.-C.; Shao, F. Pore-Forming Activity and Structural Autoinhibition of the Gasdermin Family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef]
- Codo, A.C.; Saraiva, A.C.; Dos Santos, L.L.; Visconde, M.F.; Gales, A.C.; Zamboni, D.S.; Medeiros, A.I. Inhibition of Inflammasome Activation by a Clinical Strain of Klebsiella Pneumoniae Impairs Efferocytosis and Leads to Bacterial Dissemination. Cell Death Dis. 2018, 9, 1182. [Google Scholar] [CrossRef]
- Lu, J.; Shi, W.; Liang, B.; Chen, C.; Wu, R.; Lin, H.; Zhang, Y.; Han, J. Efficient Engulfment of Necroptotic and Pyroptotic Cells by Nonprofessional and Professional Phagocytes. Cell Discov. 2019, 5, 39. [Google Scholar] [CrossRef]
- Self, W.H.; Wunderink, R.G.; Williams, D.J.; Zhu, Y.; Anderson, E.J.; Balk, R.A.; Fakhran, S.S.; Chappell, J.D.; Casimir, G.; Courtney, D.M.; et al. Staphylococcus Aureus Community-Acquired Pneumonia: Prevalence, Clinical Characteristics, and Outcomes. Clin. Infect. Dis. 2016, 63, 300–309. [Google Scholar] [CrossRef] [Green Version]
- Greenlee-Wacker, M.C.; Rigby, K.M.; Kobayashi, S.D.; Porter, A.R.; DeLeo, F.R.; Nauseef, W.M. Phagocytosis of Staphylococcus Aureus by Human Neutrophils Prevents Macrophage Efferocytosis and Induces Programmed Necrosis. J. Immunol. 2014, 192, 4709–4717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, T.S.; Jones-Nelson, O.; Hotz, M.; Cheng, L.; Miller, L.S.; Suzich, J.; Stover, C.K.; Sellman, B.R.S. Aureus Blocks Efferocytosis of Neutrophils by Macrophages through the Activity of Its Virulence Factor Alpha Toxin. Sci. Rep. 2016, 6, 35466. [Google Scholar] [CrossRef]
- Flannagan, R.S.; Heit, B.; Heinrichs, D.E. Intracellular Replication of Staphylococcus Aureus in Mature Phagolysosomes in Macrophages Precedes Host Cell Death, and Bacterial Escape and Dissemination. Cell. Microbiol. 2015. [Google Scholar] [CrossRef] [Green Version]
- Mares, C.A.; Sharma, J.; Li, Q.; Rangel, E.L.; Morris, E.G.; Enriquez, M.I.; Teale, J.M. Defect in Efferocytosis Leads to Alternative Activation of Macrophages in Francisella Infections. Immunol. Cell Biol. 2011, 89, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Roth, K.M.; Oghumu, S.; Satoskar, A.A.; Gunn, J.S.; van Rooijen, N.; Satoskar, A.R. Respiratory Infection with Francisella Novicida Induces Rapid Dystrophic Cardiac Calcinosis (DCC). FEMS Immunol. Med. Microbiol. 2008, 53, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.; Dumas, E.K.; Lawrence, C.; Pate, L.; Longobardi, S.; Wang, X.; James, J.A.; Kovats, S.; Farris, A.D. Bacillus Anthracis Edema Toxin Inhibits Efferocytosis in Human Macrophages and Alters Efferocytic Receptor Signaling. Int. J. Mol. Sci. 2019, 20, 1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyei, G.B.; Vergne, I.; Chua, J.; Roberts, E.; Harris, J.; Junutula, J.R.; Deretic, V. Rab14 Is Critical for Maintenance of Mycobacterium Tuberculosis Phagosome Maturation Arrest. EMBO J. 2006, 25, 5250–5259. [Google Scholar] [CrossRef] [Green Version]
- Malik, Z.A.; Denning, G.M.; Kusner, D.J. Inhibition of Ca(2+) Signaling by Mycobacterium Tuberculosis Is Associated with Reduced Phagosome-Lysosome Fusion and Increased Survival within Human Macrophages. J. Exp. Med. 2000, 191, 287–302. [Google Scholar] [CrossRef]
- Mehra, A.; Zahra, A.; Thompson, V.; Sirisaengtaksin, N.; Wells, A.; Porto, M.; Köster, S.; Penberthy, K.; Kubota, Y.; Dricot, A.; et al. Mycobacterium Tuberculosis Type VII Secreted Effector EsxH Targets Host ESCRT to Impair Trafficking. PLoS Pathog. 2013, 9, e1003734. [Google Scholar] [CrossRef] [Green Version]
- Wong, D.; Bach, H.; Sun, J.; Hmama, Z.; Av-Gay, Y. Mycobacterium Tuberculosis Protein Tyrosine Phosphatase (PtpA) Excludes Host Vacuolar-H+-ATPase to Inhibit Phagosome Acidification. Proc. Natl Acad Sci USA 2011, 108, 19371–19376. [Google Scholar] [CrossRef] [Green Version]
- Bach, H.; Papavinasasundaram, K.G.; Wong, D.; Hmama, Z.; Av-Gay, Y. Mycobacterium Tuberculosis Virulence Is Mediated by PtpA Dephosphorylation of Human Vacuolar Protein Sorting 33B. Cell Host Microbe 2008, 3, 316–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poirier, V.; Av-Gay, Y. Intracellular Growth of Bacterial Pathogens: The Role of Secreted Effector Proteins in the Control of Phagocytosed Microorganisms. Microbiol. Spectr. 2015, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amara, A.; Mercer, J. Viral Apoptotic Mimicry. Nat. Rev. Microbiol. 2015, 13, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.L.; Blackburn, J.W.D.; Taruc, K.; Kipp, A.; Dirk, B.S.; Hunt, N.R.; Barr, S.D.; Dikeakos, J.D.; Heit, B. Antagonistic Coevolution of MER Tyrosine Kinase Expression and Function. Mol. Biol. Evol. 2017, 34, 1613–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabiec, A.M.; Hussell, T. The Role of Airway Macrophages in Apoptotic Cell Clearance Following Acute and Chronic Lung Inflammation. Semin. Immunopathol. 2016. [Google Scholar] [CrossRef] [Green Version]
- Czuczman, M.A.; Fattouh, R.; van Rijn, J.M.; Canadien, V.; Osborne, S.; Muise, A.M.; Kuchroo, V.K.; Higgins, D.E.; Brumell, J.H. Listeria Monocytogenes Exploits Efferocytosis to Promote Cell-to-Cell Spread. Nature 2014. [Google Scholar] [CrossRef] [Green Version]
- Arifuzzaman, M.; Ang, W.X.G.; Choi, H.W.; Nilles, M.L.; St John, A.L.; Abraham, S.N. Necroptosis of Infiltrated Macrophages Drives Yersinia Pestis Dispersal within Buboes. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- McCubbrey, A.L.; Sonstein, J.; Ames, T.M.; Freeman, C.M.; Curtis, J.L. Glucocorticoids Relieve Collectin-Driven Suppression of Apoptotic Cell Uptake in Murine Alveolar Macrophages through Downregulation of SIRPα. J. Immunol. 2012, 189, 112–119. [Google Scholar] [CrossRef] [Green Version]
- Park, H.-J.; Baen, J.-Y.; Lee, Y.-J.; Choi, Y.-H.; Kang, J.L. The TAM-Family Receptor Mer Mediates Production of HGF through the RhoA-Dependent Pathway in Response to Apoptotic Cells. Mol. Biol. Cell 2012, 23, 3254–3265. [Google Scholar] [CrossRef]
- Molnár, G.; Dagher, M.C.; Geiszt, M.; Settleman, J.; Ligeti, E. Role of Prenylation in the Interaction of Rho-Family Small GTPases with GTPase Activating Proteins. Biochemistry 2001, 40, 10542–10549. [Google Scholar] [CrossRef]
- Morimoto, K.; Janssen, W.J.; Fessler, M.B.; McPhillips, K.A.; Borges, V.M.; Bowler, R.P.; Xiao, Y.-Q.; Kench, J.A.; Henson, P.M.; Vandivier, R.W. Lovastatin Enhances Clearance of Apoptotic Cells (Efferocytosis) with Implications for Chronic Obstructive Pulmonary Disease. J. Immunol. 2006, 176, 7657–7665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friesen, J.A.; Rodwell, V.W. The 3-Hydroxy-3-Methylglutaryl Coenzyme-A (HMG-CoA) Reductases. Genome Biol. 2004, 5, 248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akula, M.K.; Ibrahim, M.X.; Ivarsson, E.G.; Khan, O.M.; Kumar, I.T.; Erlandsson, M.; Karlsson, C.; Xu, X.; Brisslert, M.; Brakebusch, C.; et al. Protein Prenylation Restrains Innate Immunity by Inhibiting Rac1 Effector Interactions. Nat. Commun. 2019, 10, 3975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Q.; Xu, J.; Ma, L. Simvastatin Enhances Chemotherapy in Cervical Cancer via Inhibition of Multiple Prenylation-Dependent GTPases-Regulated Pathways. Fundam. Clin. Pharmacol. 2020, 34, 32–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.-J.; Kim, M.-J.; Yoon, Y.-S.; Choi, Y.-H.; Kim, H.-S.; Kang, J.L. Simvastatin Treatment Boosts Benefits of Apoptotic Cell Infusion in Murine Lung Fibrosis. Cell Death Dis. 2017, 8, e2860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, K.-S.; Cushman, H.J.; Akaike, M.; Woo, C.-H.; Wang, X.; Qiu, X.; Fujiwara, K.; Abe, J.-I. ERK5 Activation in Macrophages Promotes Efferocytosis and Inhibits Atherosclerosis. Circulation 2014, 130, 180–191. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Xiong, R.; Chen, X.; Li, P.; Ning, Y.; Peng, Y.; Zhao, Y.; Yang, N.; Zhou, Y. The Glucocorticoid Dexamethasone Inhibits U937 Cell Adhesion and Neutrophil Release via RhoA/ROCK1-Dependent and Independent Pathways. Cell Physiol. Biochem. 2014, 33, 1654–1662. [Google Scholar] [CrossRef]
- Higham, A.; Scott, T.; Li, J.; Gaskell, R.; Dikwa, A.B.; Shah, R.; Montero-Fernandez, M.A.; Lea, S.; Singh, D. Effects of Corticosteroids on COPD Lung Macrophage Phenotype and Function. Clin. Sci. 2020, 134, 751–763. [Google Scholar] [CrossRef]
- Garabuczi, É.; Sarang, Z.; Szondy, Z. Glucocorticoids Enhance Prolonged Clearance of Apoptotic Cells by Upregulating Liver X Receptor, Peroxisome Proliferator-Activated Receptor-δ and UCP2. Biochim. Biophys. Acta 2015, 1853, 573–582. [Google Scholar] [CrossRef] [Green Version]
- Zizzo, G.; Hilliard, B.A.; Monestier, M.; Cohen, P.L. Efficient Clearance of Early Apoptotic Cells by Human Macrophages Requires M2c Polarization and MerTK Induction. J. Immunol. 2012, 189, 3508–3520. [Google Scholar] [CrossRef] [Green Version]
- Cai, B.; Thorp, E.B.; Doran, A.C.; Sansbury, B.E.; Daemen, M.J.A.P.; Dorweiler, B.; Spite, M.; Fredman, G.; Tabas, I. MerTK Receptor Cleavage Promotes Plaque Necrosis and Defective Resolution in Atherosclerosis. J. Clin. Investing. 2017, 127, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Thorp, E.; Cui, D.; Schrijvers, D.M.; Kuriakose, G.; Tabas, I. Mertk Receptor Mutation Reduces Efferocytosis Efficiency and Promotes Apoptotic Cell Accumulation and Plaque Necrosis in Atherosclerotic Lesions of Apoe-/- Mice. Arter. Thromb. Vasc. Biol. 2008, 28, 1421–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Wittchen, E.S.; Monaghan-Benson, E.; Hahn, C.; Earp, H.S.; Doerschuk, C.M.; Burridge, K. The Role of Endothelial MERTK during the Inflammatory Response in Lungs. PLoS ONE 2019, 14, e0225051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohning, M.P.; Thomas, S.M.; Barthel, L.; Mould, K.J.; McCubbrey, A.L.; Frasch, S.C.; Bratton, D.; Henson, P.M.; Janssen, W.J. Phagocytosis of Microparticles by Alveolar Macrophages during Acute Lung Injury Requires MerTK. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 63. [Google Scholar] [CrossRef]
- Healy, L.M.; Perron, G.; Won, S.-Y.; Michell-Robinson, M.A.; Rezk, A.; Ludwin, S.K.; Moore, C.S.; Hall, J.A.; Bar-Or, A.; Antel, J.P. MerTK Is a Functional Regulator of Myelin Phagocytosis by Human Myeloid Cells. J. Immunol. 2016. [Google Scholar] [CrossRef] [Green Version]
- Hodrea, J.; Majai, G.; Doró, Z.; Zahuczky, G.; Pap, A.; Rajnavölgyi, É.; Fésüs, L. The Glucocorticoid Dexamethasone Programs Human Dendritic Cells for Enhanced Phagocytosis of Apoptotic Neutrophils and Inflammatory Response. J. Leukoc. Biol. 2012, 91, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Zeiser, R. Immune Modulatory Effects of Statins. Immunology 2018, 154, 69–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beltowski, J.; Jamroz-Wisniewska, G.W. Adverse Effects of Statins—Mechanisms and Consequences. Available online: https://www.eurekaselect.com/69865/article (accessed on 8 December 2020).
- Deichmann, R.; Lavie, C.; Andrews, S. Coenzyme Q10 and Statin-Induced Mitochondrial Dysfunction. Ochsner J. 2010, 10, 16–21. [Google Scholar] [PubMed]
- Sharma, U. Statin-Induced Delayed Rhabdomyolysis. BMJ Case Rep. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Tulbah, A.S.; Ong, H.X.; Lee, W.-H.; Colombo, P.; Young, P.M.; Traini, D. Biological Effects of Simvastatin Formulated as PMDI on Pulmonary Epithelial Cells. Pharm. Res. 2016, 33, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Zeki, A.A.; Bratt, J.M.; Chang, K.Y.; Franzi, L.M.; Ott, S.; Silveria, M.; Fiehn, O.; Last, J.A.; Kenyon, N.J. Intratracheal Instillation of Pravastatin for the Treatment of Murine Allergic Asthma: A Lung-Targeted Approach to Deliver Statins. Physiol. Rep. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Zeki, A.A.; Elbadawi-Sidhu, M. Innovations in Asthma Therapy: Is There a Role for Inhaled Statins? Expert Rev. Respir. Med. 2018, 12, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Giron, D.J.; Schmidt, J.P.; Pindak, F.F. Effect of Progesterone and Testosterone on Interferon Production and on the Viral Infection-Enhancing Activity of Estrone and Hydrocortisone. Infect. Immun. 1971, 4, 537–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patterson, C.M.; Morrison, R.L.; D’Souza, A.; Teng, X.S.; Happel, K.I. Inhaled Fluticasone Propionate Impairs Pulmonary Clearance of Klebsiella Pneumoniae in Mice. Respir. Res. 2012, 13, 40. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Wang, S.; Dong, X.; Leanse, L.G.; Dai, T.; Wang, Z. Co-Delivery of Resolvin D1 and Antibiotics with Nanovesicles to Lungs Resolves Inflammation and Clears Bacteria in Mice. Commun. Biol. 2020, 3, 680. [Google Scholar] [CrossRef] [PubMed]
- Rymut, N.; Heinz, J.; Sadhu, S.; Hosseini, Z.; Riley, C.O.; Marinello, M.; Maloney, J.; MacNamara, K.C.; Spite, M.; Fredman, G. Resolvin D1 Promotes Efferocytosis in Aging by Limiting Senescent Cell-Induced MerTK Cleavage. FASEB J. 2020, 34, 597–609. [Google Scholar] [CrossRef] [Green Version]
- Isopi, E.; Mattoscio, D.; Codagnone, M.; Mari, V.C.; Lamolinara, A.; Patruno, S.; D’Aurora, M.; Cianci, E.; Nespoli, A.; Franchi, S.; et al. Resolvin D1 Reduces Lung Infection and Inflammation Activating Resolution in Cystic Fibrosis. Front. Immunol. 2020, 11, 581. [Google Scholar] [CrossRef]
- Uddin, M.; Levy, B.D. Resolvins: Natural Agonists for Resolution of Pulmonary Inflammation. Prog. Lipid Res. 2011, 50, 75–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.J.; Luther, J.; Bohr, S.; Iracheta-Vellve, A.; Li, M.; King, K.R.; Chung, R.T.; Yarmush, M.L. A Novel Resolvin-Based Strategy for Limiting Acetaminophen Hepatotoxicity. Clin. Transl. Gastroenterol. 2016, 7, e153. [Google Scholar] [CrossRef] [PubMed]
- Hecker, M.; Linder, T.; Ott, J.; Walmrath, H.-D.; Lohmeyer, J.; Vadász, I.; Marsh, L.M.; Herold, S.; Reichert, M.; Buchbinder, A.; et al. Immunomodulation by Lipid Emulsions in Pulmonary Inflammation: A Randomized Controlled Trial. Crit. Care 2015, 19, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooke, G.P.; Parsons, K.R.; Howard, C.J. Cloning of Two Members of the SIRP Alpha Family of Protein Tyrosine Phosphatase Binding Proteins in Cattle That Are Expressed on Monocytes and a Subpopulation of Dendritic Cells and Which Mediate Binding to CD4 T Cells. Eur. J. Immunol. 1998, 28, 1–11. [Google Scholar] [CrossRef]
- Vernon-Wilson, E.F.; Kee, W.J.; Willis, A.C.; Barclay, A.N.; Simmons, D.L.; Brown, M.H. CD47 Is a Ligand for Rat Macrophage Membrane Signal Regulatory Protein SIRP (OX41) and Human SIRPalpha 1. Eur. J. Immunol. 2000, 30, 2130–2137. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Volkmer, J.-P.; McKenna, K.; Civelek, M.; Lusis, A.J.; Miller, C.L.; Direnzo, D.; Nanda, V.; Ye, J.; Connolly, A.J.; et al. CD47-Blocking Antibodies Restore Phagocytosis and Prevent Atherosclerosis. Nature 2016, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Wang, J.; Willingham, S.B.; Martin, R.; Wernig, G.; Weissman, I.L. Anti-CD47 Antibodies Promote Phagocytosis and Inhibit the Growth of Human Myeloma Cells. Leukemia 2012, 26, 2538–2545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cham, L.B.; Torrez Dulgeroff, L.B.; Tal, M.C.; Adomati, T.; Li, F.; Bhat, H.; Huang, A.; Lang, P.A.; Moreno, M.E.; Rivera, J.M.; et al. Immunotherapeutic Blockade of CD47 Inhibitory Signaling Enhances Innate and Adaptive Immune Responses to Viral Infection. Cell Rep. 2020, 31, 107494. [Google Scholar] [CrossRef] [PubMed]
- Cham, L.B.; Adomati, T.; Li, F.; Ali, M.; Lang, K.S. CD47 as a Potential Target to Therapy for Infectious Diseases. Antibodies 2020, 9, 44. [Google Scholar] [CrossRef]
- Johnson, L.D.S.; Banerjee, S.; Kruglov, O.; Viller, N.N.; Horwitz, S.M.; Lesokhin, A.; Zain, J.; Querfeld, C.; Chen, R.; Okada, C.; et al. Targeting CD47 in Sézary Syndrome with SIRPαFc. Blood Adv. 2019, 3, 1145–1153. [Google Scholar] [CrossRef] [Green Version]
- Brierley, C.K.; Staves, J.; Roberts, C.; Johnson, H.; Vyas, P.; Goodnough, L.T.; Murphy, M.F. The Effects of Monoclonal Anti-CD47 on RBCs, Compatibility Testing, and Transfusion Requirements in Refractory Acute Myeloid Leukemia. Transfusion 2019, 59, 2248–2254. [Google Scholar] [CrossRef]
- Liu, J.; Wang, L.; Zhao, F.; Tseng, S.; Narayanan, C.; Shura, L.; Willingham, S.; Howard, M.; Prohaska, S.; Volkmer, J.; et al. Pre-Clinical Development of a Humanized Anti-CD47 Antibody with Anti-Cancer Therapeutic Potential. PLoS ONE 2015, 10, e0137345. [Google Scholar] [CrossRef] [Green Version]
- Khandelwal, S.; van Rooijen, N.; Saxena, R.K. Reduced Expression of CD47 during Murine Red Blood Cell (RBC) Senescence and Its Role in RBC Clearance from the Circulation. Transfusion 2007, 47, 1725–1732. [Google Scholar] [CrossRef]
- Guo, Y.-L.; Liu, D.-Q.; Bian, Z.; Zhang, C.-Y.; Zen, K. Down-Regulation of Platelet Surface CD47 Expression in Escherichia Coli O157:H7 Infection-Induced Thrombocytopenia. PLoS ONE 2009, 4, e7131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansell, S.; Chen, R.W.; Flinn, I.W.; Maris, M.B.; O’Connor, O.A.; Johnson, L.D.; Irwin, M.; Petrova, P.S.; Uger, R.A.; Sievers, E.L. A Phase 1 Study of TTI-621, a Novel Immune Checkpoint Inhibitor Targeting CD47, in Patients with Relapsed or Refractory Hematologic Malignancies. Blood 2016, 128, 1812. [Google Scholar] [CrossRef]
- Su, X.; Johansen, M.; Looney, M.R.; Brown, E.J.; Matthay, M.A. CD47 Deficiency Protects Mice from Lipopolysaccharide-Induced Acute Lung Injury and Escherichia Coli Pneumonia. J. Immunol. 2008, 180, 6947–6953. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, D.J.; Abou Taka, M.; Heit, B. Role of Apoptotic Cell Clearance in Pneumonia and Inflammatory Lung Disease. Pathogens 2021, 10, 134. https://doi.org/10.3390/pathogens10020134
Zheng DJ, Abou Taka M, Heit B. Role of Apoptotic Cell Clearance in Pneumonia and Inflammatory Lung Disease. Pathogens. 2021; 10(2):134. https://doi.org/10.3390/pathogens10020134
Chicago/Turabian StyleZheng, David Jiao, Maria Abou Taka, and Bryan Heit. 2021. "Role of Apoptotic Cell Clearance in Pneumonia and Inflammatory Lung Disease" Pathogens 10, no. 2: 134. https://doi.org/10.3390/pathogens10020134