
The Roles of Inflammasomes in Host Defense against Mycobacterium tuberculosis

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Events in MTB-Infected Phagocytes
3. A Brief Introduction to Inflammasome
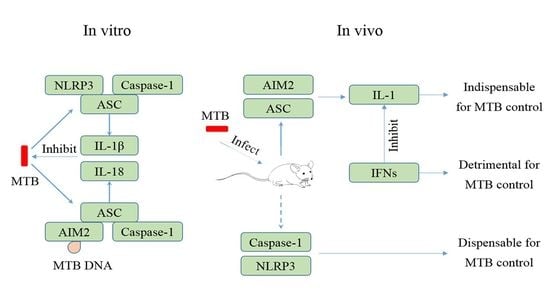
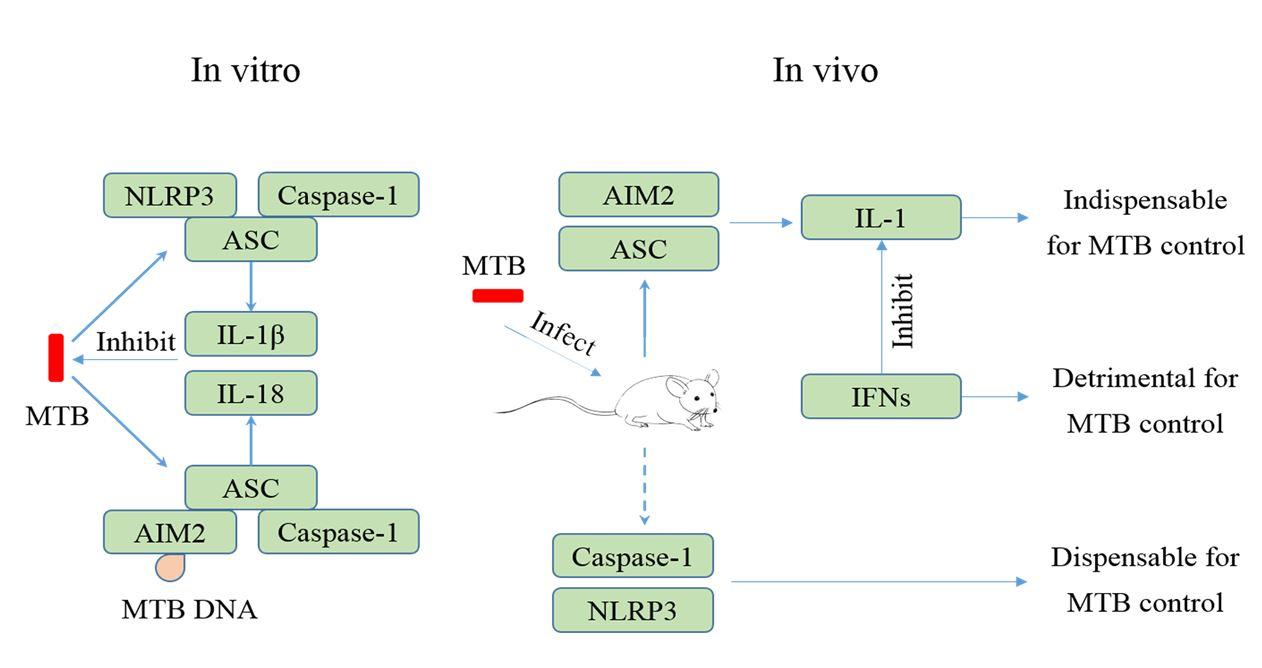
4. MTB and the NLRP3 Inflammasome
5. MTB and the AIM2 Inflammasome
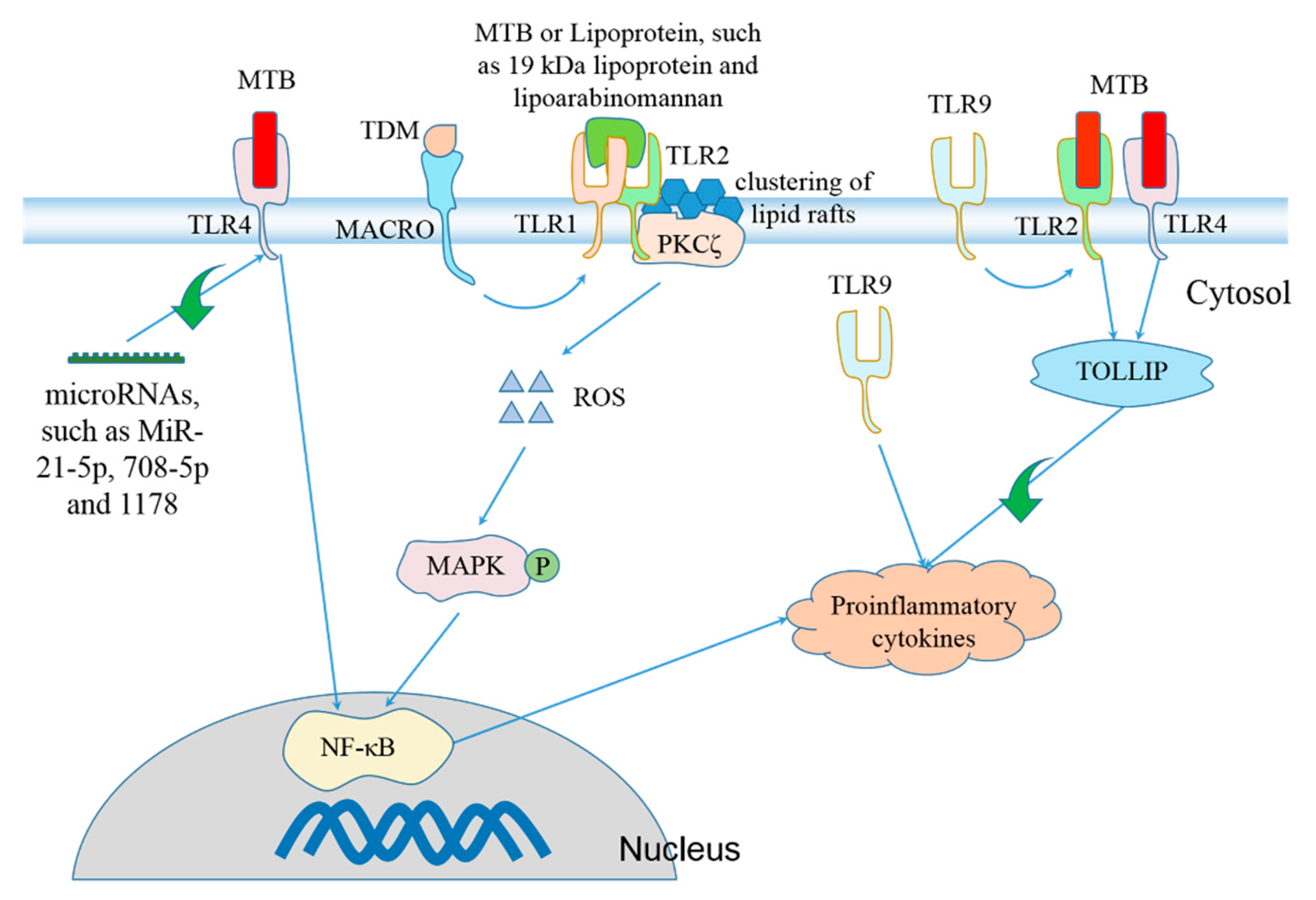
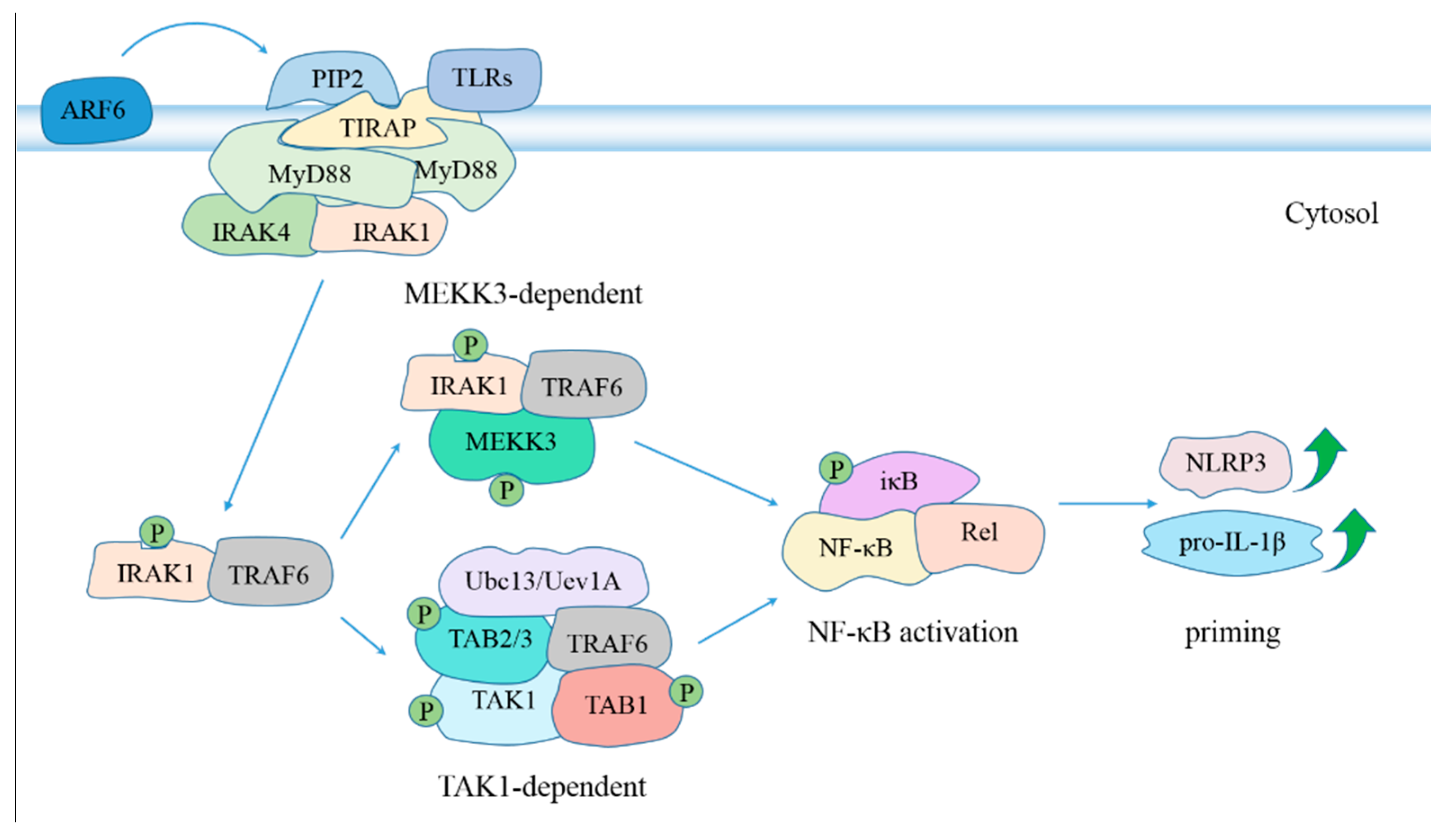
6. Regulation of Inflammasome Activation during MTB Infection
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Philips, J.A.; Ernst, J.D. Tuberculosis pathogenesis and immunity. Annu. Rev. Pathol. 2012, 7, 353–384. [Google Scholar] [CrossRef] [PubMed]
- Van Crevel, R.; Ottenhoff, T.H.; van der Meer, J.W. Innate immunity to mycobacterium tuberculosis. Clin. Microbiol. Rev. 2002, 15, 294–309. [Google Scholar] [PubMed] [Green Version]
- Huynh, K.K.; Joshi, S.A.; Brown, E.J. A delicate dance: Host response to mycobacteria. Curr. Opin. Immunol. 2011, 23, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Overview of the interleukin-1 family of ligands and receptors. Semin. Immunol. 2013, 25, 389–393. [Google Scholar] [CrossRef]
- O’Connor, K.A.; Johnson, J.D.; Hansen, M.K.; Wieseler Frank, J.L.; Maksimova, E.; Watkins, L.R.; Maier, S.F. Peripheral and central proinflammatory cytokine response to a severe acute stressor. Brain Res. 2003, 991, 123–132. [Google Scholar] [CrossRef]
- Vilarrasa, N.; Vendrell, J.; Sanchez-Santos, R.; Broch, M.; Megia, A.; Masdevall, C.; Gomez, N.; Soler, J.; Pujol, J.; Bettonica, C.; et al. Effect of weight loss induced by gastric bypass on proinflammatory interleukin-18, soluble tumour necrosis factor-alpha receptors, c-reactive protein and adiponectin in morbidly obese patients. Clin. Endocrinol. 2007, 67, 679–686. [Google Scholar] [CrossRef]
- Mayer-Barber, K.D.; Barber, D.L.; Shenderov, K.; White, S.D.; Wilson, M.S.; Cheever, A.; Kugler, D.; Hieny, S.; Caspar, P.; Nunez, G.; et al. Caspase-1 independent il-1beta production is critical for host resistance to mycobacterium tuberculosis and does not require tlr signaling in vivo. J. Immunol. 2010, 184, 3326–3330. [Google Scholar] [CrossRef] [Green Version]
- Mayer-Barber, K.D.; Andrade, B.B.; Barber, D.L.; Hieny, S.; Feng, C.G.; Caspar, P.; Oland, S.; Gordon, S.; Sher, A. Innate and adaptive interferons suppress il-1α and il-1β production by distinct pulmonary myeloid subsets during mycobacterium tuberculosis infection. Immunity 2011, 35, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- Sugawara, I.; Tuomanen, E.I.; Yamada, H.; Kaneko, H.; Mizuno, S.; Takeda, K.; Akira, S. Role of interleukin-18 (il-18) in mycobacterial infection in il-18-gene-disrupted mice. Infect. Immun. 1999, 67, 2585–2589. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Wang, M.; Huang, K.; Zhang, Z.; Shao, N.; Zhang, Y.; Wang, W.; Wang, S. Oxidized low-density lipoprotein induces secretion of interleukin-1beta by macrophages via reactive oxygen species-dependent nlrp3 inflammasome activation. Biochem. Biophys. Res. Commun. 2012, 425, 121–126. [Google Scholar] [CrossRef]
- Mezzasoma, L.; Antognelli, C.; Talesa, V.N. Atrial natriuretic peptide down-regulates lps/atp-mediated il-1beta release by inhibiting nf-kb, nlrp3 inflammasome and caspase-1 activation in thp-1 cells. Immunol. Res. 2016, 64, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Joosten, L.A.; Netea, M.G.; Fantuzzi, G.; Koenders, M.I.; Helsen, M.M.; Sparrer, H.; Pham, C.T.; van der Meer, J.W.; Dinarello, C.A.; van den Berg, W.B. Inflammatory arthritis in caspase 1 gene-deficient mice: Contribution of proteinase 3 to caspase 1-independent production of bioactive interleukin-1beta. Arthritis Rheum. 2009, 60, 3651–3662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coeshott, C.; Ohnemus, C.; Pilyavskaya, A.; Ross, S.; Wieczorek, M.; Kroona, H.; Leimer, A.H.; Cheronis, J. Converting enzyme-independent release of tumor necrosis factor alpha and il-1beta from a stimulated human monocytic cell line in the presence of activated neutrophils or purified proteinase 3. Proc. Natl. Acad. Sci. USA 1999, 96, 6261–6266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfaidi, M.; Wilson, H.; Daigneault, M.; Burnett, A.; Ridger, V.; Chamberlain, J.; Francis, S. Neutrophil elastase promotes interleukin-1β secretion from human coronary endothelium. J. Biol. Chem. 2015, 290, 24067–24078. [Google Scholar] [CrossRef] [Green Version]
- Guma, M.; Ronacher, L.; Liu-Bryan, R.; Takai, S.; Karin, M.; Corr, M. Caspase 1-independent activation of interleukin-1beta in neutrophil-predominant inflammation. Arthritis Rheum. 2009, 60, 3642–3650. [Google Scholar] [CrossRef] [Green Version]
- McLoed, A.G.; Sherrill, T.P.; Cheng, D.S.; Han, W.; Saxon, J.A.; Gleaves, L.A.; Wu, P.; Polosukhin, V.V.; Karin, M.; Yull, F.E.; et al. Neutrophil-derived il-1beta impairs the efficacy of nf-kappab inhibitors against lung cancer. Cell Rep. 2016, 16, 120–132. [Google Scholar] [CrossRef] [Green Version]
- Schonbeck, U.; Mach, F.; Libby, P. Generation of biologically active il-1 beta by matrix metalloproteinases: A novel caspase-1-independent pathway of il-1 beta processing. J. Immunol. 1998, 161, 3340–3346. [Google Scholar]
- Ito, A.; Mukaiyama, A.; Itoh, Y.; Nagase, H.; Thogersen, I.B.; Enghild, J.J.; Sasaguri, Y.; Mori, Y. Degradation of interleukin 1beta by matrix metalloproteinases. J. Biol. Chem. 1996, 271, 14657–14660. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Schlesinger, L.S.; Kaufman, T.M.; Iyer, S.; Hull, S.R.; Marchiando, L.K. Differences in mannose receptor-mediated uptake of lipoarabinomannan from virulent and attenuated strains of mycobacterium tuberculosis by human macrophages. J. Immunol. 1996, 157, 4568–4575. [Google Scholar]
- Ishikawa, E.; Ishikawa, T.; Morita, Y.S.; Toyonaga, K.; Yamada, H.; Takeuchi, O.; Kinoshita, T.; Akira, S.; Yoshikai, Y.; Yamasaki, S. Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by c-type lectin mincle. J. Exp. Med. 2009, 206, 2879–2888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tailleux, L.; Schwartz, O.; Herrmann, J.L.; Pivert, E.; Jackson, M.; Amara, A.; Legres, L.; Dreher, D.; Nicod, L.P.; Gluckman, J.C.; et al. Dc-sign is the major mycobacterium tuberculosis receptor on human dendritic cells. J. Exp. Med. 2003, 197, 121–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonilla, D.L.; Bhattacharya, A.; Sha, Y.; Xu, Y.; Xiang, Q.; Kan, A.; Jagannath, C.; Komatsu, M.; Eissa, N.T. Autophagy regulates phagocytosis by modulating the expression of scavenger receptors. Immunity 2013, 39, 537–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlesinger, L.S.; Bellingerkawahara, C.G.; Payne, N.R.; Horwitz, M.A. Phagocytosis of mycobacterium-tuberculosis is mediated by human monocyte complement receptors and complement component-c3. J. Immunol. 1990, 144, 2771–2780. [Google Scholar] [PubMed]
- Via, L.E.; Deretic, D.; Ulmer, R.J.; Hibler, N.S.; Huber, L.A.; Deretic, V. Arrest of mycobacterial phagosome maturation is caused by a block in vesicle fusion between stages controlled by rab5 and rab7. J. Biol. Chem. 1997, 272, 13326–13331. [Google Scholar] [CrossRef] [Green Version]
- Clemens, D.L.; Lee, B.Y.; Horwitz, M.A. Deviant expression of rab5 on phagosomes containing the intracellular pathogens mycobacterium tuberculosis and legionella pneumophila is associated with altered phagosomal fate. Infect. Immun. 2000, 68, 2671–2684. [Google Scholar] [CrossRef] [Green Version]
- Deretic, V. Autophagy, an immunologic magic bullet: Mycobacterium tuberculosis phagosome maturation block and how to bypass it. Future Microbiol. 2008, 3, 517–524. [Google Scholar] [CrossRef] [Green Version]
- Augenstreich, J.; Arbues, A.; Simeone, R.; Haanappel, E.; Wegener, A.; Sayes, F.; Le Chevalier, F.; Chalut, C.; Malaga, W.; Guilhot, C.; et al. Esx-1 and phthiocerol dimycocerosates of mycobacterium tuberculosis act in concert to cause phagosomal rupture and host cell apoptosis. Cell. Microbiol. 2017, 19, e12726. [Google Scholar] [CrossRef] [Green Version]
- Lienard, J.; Nobs, E.; Lovins, V.; Movert, E.; Valfridsson, C.; Carlsson, F. The mycobacterium marinum esx-1 system mediates phagosomal permeabilization and type i interferon production via separable mechanisms. Proc. Natl. Acad. Sci. USA 2020, 117, 1160–1166. [Google Scholar] [CrossRef]
- Van der Wel, N.; Hava, D.; Houben, D.; Fluitsma, D.; van Zon, M.; Pierson, J.; Brenner, M.; Peters, P.J. Tuberculosis and m. Leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 2007, 129, 1287–1298. [Google Scholar] [CrossRef] [Green Version]
- Simeone, R.; Sayes, F.; Song, O.; Groschel, M.I.; Brodin, P.; Brosch, R.; Majlessi, L. Cytosolic access of mycobacterium tuberculosis: Critical impact of phagosomal acidification control and demonstration of occurrence in vivo. PLoS Pathog. 2015, 11, e1004650. [Google Scholar] [CrossRef] [Green Version]
- Lukacs, G.L.; Rotstein, O.D.; Grinstein, S. Phagosomal acidification is mediated by a vacuolar-type h+-atpase in murine macrophages. J. Biol. Chem. 1990, 265, 21099–21107. [Google Scholar] [CrossRef]
- Ehrt, S.; Simeone, R.; Bobard, A.; Lippmann, J.; Bitter, W.; Majlessi, L.; Brosch, R.; Enninga, J. Phagosomal rupture by mycobacterium tuberculosis results in toxicity and host cell death. PLoS Pathog. 2012, 8, e1002507. [Google Scholar] [CrossRef]
- Beckwith, K.S.; Beckwith, M.S.; Ullmann, S.; Sætra, R.S.; Kim, H.; Marstad, A.; Åsberg, S.E.; Strand, T.A.; Haug, M.; Niederweis, M.; et al. Plasma membrane damage causes nlrp3 activation and pyroptosis during mycobacterium tuberculosis infection. Nat. Commun. 2020, 11, 1–18. [Google Scholar]
- McElvania Tekippe, E.; Allen, I.C.; Hulseberg, P.D.; Sullivan, J.T.; McCann, J.R.; Sandor, M.; Braunstein, M.; Ting, J.P. Granuloma formation and host defense in chronic mycobacterium tuberculosis infection requires pycard/asc but not nlrp3 or caspase-1. PLoS ONE 2010, 5, e12320. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.M.; Clay, H.; Lewis, J.L.; Ghori, N.; Herbomel, P.; Ramakrishnan, L. Real-time visualization of mycobacterium-macrophage interactions leading to initiation of granuloma formation in zebrafish embryos. Immunity 2002, 17, 693–702. [Google Scholar] [CrossRef] [Green Version]
- Puissegur, M.-P.; Botanch, C.; Duteyrat, J.-L.; Delsol, G.; Caratero, C.; Altare, F. An in vitro dual model of mycobacterial granulomas to investigate the molecular interactions between mycobacteria and human host cells. Cell. Microbiol. 2004, 6, 423–433. [Google Scholar] [CrossRef]
- Puissegur, M.P.; Lay, G.; Gilleron, M.; Botella, L.; Nigou, J.; Marrakchi, H.; Mari, B.; Duteyrat, J.L.; Guerardel, Y.; Kremer, L.; et al. Mycobacterial lipomannan induces granuloma macrophage fusion via a tlr2-dependent, adam9- and beta1 integrin-mediated pathway. J. Immunol. 2007, 178, 3161–3169. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, H.A.; Hulseberg, P.D.; Lee, J.; Prechl, J.; Barta, P.; Szlavik, N.; Harding, J.S.; Fabry, Z.; Sandor, M. Dendritic cells in chronic mycobacterial granulomas restrict local anti-bacterial t cell response in a murine model. PLoS ONE 2010, 5, e11453. [Google Scholar] [CrossRef]
- Guirado, E.; Schlesinger, L.S. Modeling the mycobacterium tuberculosis granuloma—The critical battlefield in host immunity and disease. Front. Immunol. 2013, 4, 98. [Google Scholar] [CrossRef] [Green Version]
- Bulut, Y.; Michelsen, K.S.; Hayrapetian, L.; Naiki, Y.; Spallek, R.; Singh, M.; Arditi, M. Mycobacterium tuberculosis heat shock proteins use diverse toll-like receptor pathways to activate pro-inflammatory signals. J. Biol. Chem. 2005, 280, 20961–20967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.; Zhang, M.; Barnes, P.F. Chemokine production by a human alveolar epithelial cell line in response to mycobacterium tuberculosis. Infect. Immun. 1998, 66, 1121–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez Ramirez, G.M.; Rom, W.N.; Ciotoli, C.; Talbot, A.; Martiniuk, F.; Cronstein, B.; Reibman, J. Mycobacterium tuberculosis alters expression of adhesion molecules on monocytic cells. Infect. Immun. 1994, 62, 2515–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, D.; Zlotnik, A. The biology of chemokines and their receptors. Annu. Rev. Immunol. 2000, 18, 217–242. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.K.; Kim, J.K.; Shin, D.M.; Sasakawa, C. Molecular mechanisms regulating nlrp3 inflammasome activation. Cell. Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-mediated programmed necrotic cell death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin d causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [Green Version]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial cardiolipin is required for nlrp3 inflammasome activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the nlrp3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Hara, H.; Nunez, G. Mechanism and regulation of nlrp3 inflammasome activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Bauernfeind, F.; Bartok, E.; Rieger, A.; Franchi, L.; Nunez, G.; Hornung, V. Cutting edge: Reactive oxygen species inhibitors block priming, but not activation, of the nlrp3 inflammasome. J. Immunol. 2011, 187, 613–617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in nlrp3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tong, Z.Z.; Jiang, S.H.; Zheng, W.Y.; Zhao, J.J.; Zhou, X.M. The roles of endoplasmic reticulum in nlrp3 inflammasome activation. Cells 2020, 9, 1219. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Hwang, I.; Gim, E.; Yang, J.; Park, S.; Yoon, S.H.; Lee, W.W.; Yu, J.W. Brefeldin a-sensitive er-golgi vesicle trafficking contributes to nlrp3-dependent caspase-1 activation. FASEB J. 2019, 33, 4547–4558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Meszaros, G.; He, W.T.; Xu, Y.; de Fatima Magliarelli, H.; Mailly, L.; Mihlan, M.; Liu, Y.; Puig Gamez, M.; Goginashvili, A.; et al. Protein kinase d at the golgi controls nlrp3 inflammasome activation. J. Exp. Med. 2017, 214, 2671–2693. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Perry, A.; Jiang, J.; Smith, P.; Curry, J.A.; Unterholzner, L.; Jiang, Z.; Horvath, G.; Rathinam, V.A.; Johnstone, R.W.; et al. Structures of the hin domain:DNA complexes reveal ligand binding and activation mechanisms of the aim2 inflammasome and ifi16 receptor. Immunity 2012, 36, 561–571. [Google Scholar] [CrossRef] [Green Version]
- Fernandes-Alnemri, T.; Yu, J.W.; Datta, P.; Wu, J.; Alnemri, E.S. Aim2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef] [Green Version]
- Morrone, S.R.; Matyszewski, M.; Yu, X.; Delannoy, M.; Egelman, E.H.; Sohn, J. Assembly-driven activation of the aim2 foreign-dsdna sensor provides a polymerization template for downstream asc. Nat. Commun. 2015, 6, 7827. [Google Scholar] [CrossRef]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. Aim2 recognizes cytosolic dsdna and forms a caspase-1-activating inflammasome with asc. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [Green Version]
- Mishra, B.B.; Moura-Alves, P.; Sonawane, A.; Hacohen, N.; Griffiths, G.; Moita, L.F.; Anes, E. Mycobacterium tuberculosis protein esat-6 is a potent activator of the nlrp3/asc inflammasome. Cell. Microbiol. 2010, 12, 1046–1063. [Google Scholar] [CrossRef]
- Wong, K.W.; Jacobs, W.R., Jr. Critical role for nlrp3 in necrotic death triggered by mycobacterium tuberculosis. Cell. Microbiol. 2011, 13, 1371–1384. [Google Scholar] [CrossRef] [PubMed]
- Amaral, E.P.; Riteau, N.; Moayeri, M.; Maier, N.; Mayer-Barber, K.D.; Pereira, R.M.; Lage, S.L.; Kubler, A.; Bishai, W.R.; D’Imperio-Lima, M.R.; et al. Lysosomal cathepsin release is required for nlrp3-inflammasome activation by mycobacterium tuberculosis in infected macrophages. Front. Immunol. 2018, 9, 1427. [Google Scholar] [CrossRef] [PubMed]
- Dorhoi, A.; Nouailles, G.; Jorg, S.; Hagens, K.; Heinemann, E.; Pradl, L.; Oberbeck-Muller, D.; Duque-Correa, M.A.; Reece, S.T.; Ruland, J.; et al. Activation of the nlrp3 inflammasome by mycobacterium tuberculosis is uncoupled from susceptibility to active tuberculosis. Eur. J. Immunol. 2012, 42, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Fowler, B.J.; Kerur, N.; Arnvig, K.B.; Rao, N.A. Nlrp3 inflammasome activation by mycobacterial esat-6 and dsrna in intraocular tuberculosis. Microb. Pathog. 2018, 114, 219–224. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.M.; Kang, J.; Lee, S.J.; Jo, E.K. Microglial activation of the nlrp3 inflammasome by the priming signals derived from macrophages infected with mycobacteria. Glia 2013, 61, 441–452. [Google Scholar] [CrossRef]
- Bermudez, L.E.; Goodman, J. Mycobacterium tuberculosis invades and replicates within type ii alveolar cells. Infect. Immun. 1996, 64, 1400–1406. [Google Scholar] [CrossRef] [Green Version]
- Mvubu, N.E.; Pillay, B.; McKinnon, L.R.; Pillay, M. Mycobacterium tuberculosis strains induce strain-specific cytokine and chemokine response in pulmonary epithelial cells. Cytokine 2018, 104, 53–64. [Google Scholar] [CrossRef]
- Zhang, Q.; Jiang, X.; He, W.; Wei, K.; Sun, J.; Qin, X.; Zheng, Y.; Jiang, X. Mcl plays an anti-inflammatory role inmycobacterium tuberculosis-induced immune response by inhibiting nf-κb and nlrp3 inflammasome activation. Mediat. Inflamm. 2017, 2017, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Ma, W.; Gao, W.; Xing, Y.; Chen, L.; Xia, Z.; Zhang, Z.; Dai, Z. Propofol directly induces caspase-1-dependent macrophage pyroptosis through the nlrp3-asc inflammasome. Cell Death Dis. 2019, 10, 542. [Google Scholar] [CrossRef] [Green Version]
- Hara, H.; Tsuchiya, K.; Kawamura, I.; Fang, R.; Hernandez-Cuellar, E.; Shen, Y.; Mizuguchi, J.; Schweighoffer, E.; Tybulewicz, V.; Mitsuyama, M. Phosphorylation of the adaptor asc acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat. Immunol. 2013, 14, 1247–1255. [Google Scholar] [CrossRef] [Green Version]
- Verma, D.; Lerm, M.; Julinder, R.B.; Eriksson, P.; Soderkvist, P.; Sarndahl, E. Gene polymorphisms in the nalp3 inflammasome are associated with interleukin-1 production and severe inflammation: Relation to common inflammatory diseases? Arthritis Rheum. 2008, 58, 888–894. [Google Scholar] [PubMed]
- Eklund, D.; Welin, A.; Andersson, H.; Verma, D.; Soderkvist, P.; Stendahl, O.; Sarndahl, E.; Lerm, M. Human gene variants linked to enhanced nlrp3 activity limit intramacrophage growth of mycobacterium tuberculosis. J. Infect. Dis. 2014, 209, 749–753. [Google Scholar] [PubMed]
- Chen, C.C.; Tsai, S.H.; Lu, C.C.; Hu, S.T.; Wu, T.S.; Huang, T.T.; Said-Sadier, N.; Ojcius, D.M.; Lai, H.C. Activation of an nlrp3 inflammasome restricts mycobacterium kansasii infection. PLoS ONE 2012, 7, e36292. [Google Scholar]
- Lee, H.M.; Yuk, J.M.; Kim, K.H.; Jang, J.; Kang, G.; Park, J.B.; Son, J.W.; Jo, E.K. Mycobacterium abscessus activates the nlrp3 inflammasome via dectin-1-syk and p62/sqstm1. Immunol. Cell Biol. 2012, 90, 601–610. [Google Scholar]
- Carlsson, F.; Kim, J.; Dumitru, C.; Barck, K.H.; Carano, R.A.; Sun, M.; Diehl, L.; Brown, E.J. Host-detrimental role of esx-1-mediated inflammasome activation in mycobacterial infection. PLoS Pathog. 2010, 6, e1000895. [Google Scholar]
- Lin, K.M.; Hu, W.; Troutman, T.D.; Jennings, M.; Brewer, T.; Li, X.; Nanda, S.; Cohen, P.; Thomas, J.A.; Pasare, C. Irak-1 bypasses priming and directly links tlrs to rapid nlrp3 inflammasome activation. Proc. Natl. Acad. Sci. USA 2014, 111, 775–780. [Google Scholar]
- Fernandes-Alnemri, T.; Kang, S.; Anderson, C.; Sagara, J.; Fitzgerald, K.A.; Alnemri, E.S. Cutting edge: Tlr signaling licenses irak1 for rapid activation of the nlrp3 inflammasome. J. Immunol. 2013, 191, 3995–3999. [Google Scholar] [CrossRef]
- Qiao, Y.; Wang, P.; Qi, J.; Zhang, L.; Gao, C. Tlr-induced nf-kappab activation regulates nlrp3 expression in murine macrophages. FEBS Lett. 2012, 586, 1022–1026. [Google Scholar]
- Stenger, S.; Modlin, R.L. Control of mycobacterium tuberculosis through mammalian toll-like receptors. Curr. Opin. Immunol. 2002, 14, 452–457. [Google Scholar]
- Schenk, M.; Belisle, J.T.; Modlin, R.L. Tlr2 looks at lipoproteins. Immunity 2009, 31, 847–849. [Google Scholar]
- Reiling, N.; Holscher, C.; Fehrenbach, A.; Kroger, S.; Kirschning, C.J.; Goyert, S.; Ehlers, S. Cutting edge: Toll-like receptor (tlr)2- and tlr4-mediated pathogen recognition in resistance to airborne infection with mycobacterium tuberculosis. J. Immunol. 2002, 169, 3480–3484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bafica, A.; Scanga, C.A.; Feng, C.G.; Leifer, C.; Cheever, A.; Sher, A. Tlr9 regulates th1 responses and cooperates with tlr2 in mediating optimal resistance to mycobacterium tuberculosis. J. Exp. Med. 2005, 202, 1715–1724. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszynski, A.; et al. Noncanonical inflammasome activation by intracellular lps independent of tlr4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef] [PubMed]
- Branger, J.; Leemans, J.C.; Florquin, S.; Weijer, S.; Speelman, P.; Van Der Poll, T. Toll-like receptor 4 plays a protective role in pulmonary tuberculosis in mice. Int. Immunol. 2004, 16, 509–516. [Google Scholar] [CrossRef]
- Abel, B.; Thieblemont, N.; Quesniaux, V.J.; Brown, N.; Mpagi, J.; Miyake, K.; Bihl, F.; Ryffel, B. Toll-like receptor 4 expression is required to control chronic mycobacterium tuberculosis infection in mice. J. Immunol. 2002, 169, 3155–3162. [Google Scholar] [CrossRef] [Green Version]
- Latz, E.; Schoenemeyer, A.; Visintin, A.; Fitzgerald, K.A.; Monks, B.G.; Knetter, C.F.; Lien, E.; Nilsen, N.J.; Espevik, T.; Golenbock, D.T. Tlr9 signals after translocating from the er to cpg DNA in the lysosome. Nat. Immunol. 2004, 5, 190–198. [Google Scholar] [CrossRef]
- Krieg, A.M. Cpg motifs in bacterial DNA and their immune effects. Annu. Rev. Immunol. 2002, 20, 709–760. [Google Scholar] [CrossRef]
- Uciechowski, P.; Imhoff, H.; Lange, C.; Meyer, C.G.; Browne, E.N.; Kirsten, D.K.; Schroder, A.K.; Schaaf, B.; Al-Lahham, A.; Reinert, R.R.; et al. Susceptibility to tuberculosis is associated with tlr1 polymorphisms resulting in a lack of tlr1 cell surface expression. J. Leukoc. Biol. 2011, 90, 377–388. [Google Scholar] [CrossRef]
- Sugawara, I.; Yamada, H.; Li, C.; Mizuno, S.; Takeuchi, O.; Akira, S. Mycobacterial infection in tlr2 and tlr6 knockout mice. Microbiol. Immunol. 2003, 47, 327–336. [Google Scholar] [CrossRef]
- Carlos, D.; Frantz, F.G.; Souza-Junior, D.A.; Jamur, M.C.; Oliver, C.; Ramos, S.G.; Quesniaux, V.F.; Ryffel, B.; Silva, C.L.; Bozza, M.T.; et al. Tlr2-dependent mast cell activation contributes to the control of mycobacterium tuberculosis infection. Microbes Infect. 2009, 11, 770–778. [Google Scholar] [CrossRef]
- Means, T.K.; Lien, E.; Yoshimura, A.; Wang, S.Y.; Golenbock, D.T.; Fenton, M.J. The cd14 ligands lipoarabinomannan and lipopolysaccharide differ in their requirement for toll-like receptors. J. Immunol. 1999, 163, 6748–6755. [Google Scholar] [PubMed]
- Shin, D.M.; Yang, C.S.; Lee, J.Y.; Lee, S.J.; Choi, H.H.; Lee, H.M.; Yuk, J.M.; Harding, C.V.; Jo, E.K. Mycobacterium tuberculosis lipoprotein-induced association of tlr2 with protein kinase c zeta in lipid rafts contributes to reactive oxygen species-dependent inflammatory signalling in macrophages. Cell. Microbiol. 2008, 10, 1893–1905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, M.S.; Kim, S.E.; Heo, J.Y.; Lee, M.E.; Kim, H.M.; Paik, S.G.; Lee, H.; Lee, J.O. Crystal structure of the tlr1-tlr2 heterodimer induced by binding of a tri-acylated lipopeptide. Cell 2007, 130, 1071–1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancioni, C.L.; Li, Q.; Thomas, J.J.; Ding, X.; Thiel, B.; Drage, M.G.; Pecora, N.D.; Ziady, A.G.; Shank, S.; Harding, C.V.; et al. Mycobacterium tuberculosis lipoproteins directly regulate human memory cd4(+) t cell activation via toll-like receptors 1 and 2. Infect. Immun. 2011, 79, 663–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakon, S.; Xue, X.; Takekawa, M.; Sasazuki, T.; Okazaki, T.; Kojima, Y.; Piao, J.H.; Yagita, H.; Okumura, K.; Doi, T.; et al. Nf-kappab inhibits tnf-induced accumulation of ros that mediate prolonged mapk activation and necrotic cell death. EMBO J. 2003, 22, 3898–3909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bragado, P.; Armesilla, A.; Silva, A.; Porras, A. Apoptosis by cisplatin requires p53 mediated p38alpha mapk activation through ros generation. Apoptosis 2007, 12, 1733–1742. [Google Scholar] [CrossRef]
- Kaminska, B. Mapk signalling pathways as molecular targets for anti-inflammatory therapy--from molecular mechanisms to therapeutic benefits. Biochim. Biophys. Acta Proteins Proteom. 2005, 1754, 253–262. [Google Scholar] [CrossRef]
- Hawn, T.R.; Misch, E.A.; Dunstan, S.J.; Thwaites, G.E.; Lan, N.T.; Quy, H.T.; Chau, T.T.; Rodrigues, S.; Nachman, A.; Janer, M.; et al. A common human tlr1 polymorphism regulates the innate immune response to lipopeptides. Eur. J. Immunol. 2007, 37, 2280–2289. [Google Scholar] [CrossRef]
- Chambers, M.A.; Whelan, A.O.; Spallek, R.; Singh, M.; Coddeville, B.; Guerardel, Y.; Elass, E. Non-acylated mycobacterium bovis glycoprotein mpb83 binds to tlr1/2 and stimulates production of matrix metalloproteinase 9. Biochem. Biophys. Res. Commun. 2010, 400, 403–408. [Google Scholar] [CrossRef]
- Bowdish, D.M.; Sakamoto, K.; Kim, M.J.; Kroos, M.; Mukhopadhyay, S.; Leifer, C.A.; Tryggvason, K.; Gordon, S.; Russell, D.G. Marco, tlr2, and cd14 are required for macrophage cytokine responses to mycobacterial trehalose dimycolate and mycobacterium tuberculosis. PLoS Pathog. 2009, 5, e1000474. [Google Scholar] [CrossRef] [Green Version]
- Shah, J.A.; Vary, J.C.; Chau, T.T.; Bang, N.D.; Yen, N.T.; Farrar, J.J.; Dunstan, S.J.; Hawn, T.R. Human tollip regulates tlr2 and tlr4 signaling and its polymorphisms are associated with susceptibility to tuberculosis. J. Immunol. 2012, 189, 1737–1746. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, D.; Rojas, M.; Hernandez, I.; Radzioch, D.; Garcia, L.F.; Barrera, L.F. Role of tlr2- and tlr4-mediated signaling in mycobacterium tuberculosis-induced macrophage death. Cell Immunol. 2010, 260, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Guijarro-Munoz, I.; Compte, M.; Alvarez-Cienfuegos, A.; Alvarez-Vallina, L.; Sanz, L. Lipopolysaccharide activates toll-like receptor 4 (tlr4)-mediated nf-kappab signaling pathway and proinflammatory response in human pericytes. J. Biol. Chem. 2014, 289, 2457–2468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Hao, J.; Li, X.; Chen, Y.; Qi, X. Mir-21-5p regulates mycobacterial survival and inflammatory responses by targeting bcl-2 and tlr4 in mycobacterium tuberculosis-infected macrophages. FEBS Lett. 2019, 593, 1326–1335. [Google Scholar] [CrossRef]
- Li, W.T.; Zhang, Q. Microrna-708-5p regulates mycobacterial vitality and the secretion of inflammatory factors in mycobacterium tuberculosis-infected macrophages by targeting tlr4. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 8028–8038. [Google Scholar]
- Shi, G.; Mao, G.; Xie, K.; Wu, D.; Wang, W. Mir-1178 regulates mycobacterial survival and inflammatory responses in mycobacterium tuberculosis-infected macrophages partly via tlr4. J. Cell. Biochem. 2018, 119, 7449–7457. [Google Scholar] [CrossRef]
- Medzhitov, R.; Preston-Hurlburt, P.; Kopp, E.; Stadlen, A.; Chen, C.; Ghosh, S.; Janeway, C.A. Myd88 is an adaptor protein in the htoll/il-1 receptor family signaling pathways. Mol. Cell 1998, 2, 253–258. [Google Scholar] [CrossRef]
- Warner, N.; Nunez, G. Myd88: A critical adaptor protein in innate immunity signal transduction. J. Immunol. 2013, 190, 3–4. [Google Scholar] [CrossRef] [Green Version]
- Holscher, C.; Reiling, N.; Schaible, U.E.; Holscher, A.; Bathmann, C.; Korbel, D.; Lenz, I.; Sonntag, T.; Kroger, S.; Akira, S.; et al. Containment of aerogenic mycobacterium tuberculosis infection in mice does not require myd88 adaptor function for tlr2, -4 and -9. Eur. J. Immunol. 2008, 38, 680–694. [Google Scholar] [CrossRef]
- Fremond, C.M.; Yeremeev, V.; Nicolle, D.M.; Jacobs, M.; Quesniaux, V.F.; Ryffel, B. Fatal mycobacterium tuberculosis infection despite adaptive immune response in the absence of myd88. J. Clin. Investig. 2004, 114, 1790–1799. [Google Scholar] [CrossRef] [Green Version]
- Nagpal, K.; Plantinga, T.S.; Wong, J.; Monks, B.G.; Gay, N.J.; Netea, M.G.; Fitzgerald, K.A.; Golenbock, D.T. A tir domain variant of myd88 adapter-like (mal)/tirap results in loss of myd88 binding and reduced tlr2/tlr4 signaling. J. Biol. Chem. 2009, 284, 25742–25748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaloye, J.; Roger, T.; Steiner-Tardivel, Q.G.; Le Roy, D.; Knaup Reymond, M.; Akira, S.; Petrilli, V.; Gomez, C.E.; Perdiguero, B.; Tschopp, J.; et al. Innate immune sensing of modified vaccinia virus ankara (mva) is mediated by tlr2-tlr6, mda-5 and the nalp3 inflammasome. PLoS Pathog. 2009, 5, e1000480. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Choi, R.J.; Shehzad, O.; Kim, H.P.; Islam, M.N.; Choi, J.S.; Kim, Y.S. Molecular mechanism of capillarisin-mediated inhibition of myd88/tirap inflammatory signaling in in vitro and in vivo experimental models. J. Ethnopharmacol. 2013, 145, 626–637. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.; Li, L.; Sun, Y.; Yang, H.; Ye, Z.; Zhao, J. Effects of the tlr4/myd88/nf-kappab signaling pathway on nlrp3 inflammasome in coronary microembolization-induced myocardial injury. Cell. Physiol. Biochem. 2018, 47, 1497–1508. [Google Scholar] [CrossRef] [PubMed]
- Lim, R.; Barker, G.; Lappas, M. The tlr2 ligand fsl-1 and the tlr5 ligand flagellin mediate pro-inflammatory and pro-labour response via myd88/traf6/nf-kappab-dependent signalling. Am. J. Reprod. Immunol. 2014, 71, 401–417. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Akira, S. Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 2009, 388, 621–625. [Google Scholar] [CrossRef]
- Daniele, S.G.; Beraud, D.; Davenport, C.; Cheng, K.; Yin, H.; Maguire-Zeiss, K.A. Activation of myd88-dependent tlr1/2 signaling by misfolded alpha-synuclein, a protein linked to neurodegenerative disorders. Sci. Signal. 2015, 8, ra45. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Karmakar, M.; Roy, S.; Ramadan, R.T.; Williams, S.R.; Howell, S.; Shive, C.L.; Han, Y.; Stopford, C.M.; Rietsch, A.; et al. Tlr4 and tlr5 on corneal macrophages regulate pseudomonas aeruginosa keratitis by signaling through myd88-dependent and -independent pathways. J. Immunol. 2010, 185, 4272–4283. [Google Scholar] [CrossRef] [Green Version]
- Hussain, S.; Johnson, C.G.; Sciurba, J.; Meng, X.; Stober, V.P.; Liu, C.; Cyphert-Daly, J.M.; Bulek, K.; Qian, W.; Solis, A.; et al. Tlr5 participates in the tlr4 receptor complex and promotes myd88-dependent signaling in environmental lung injury. eLife 2020, 9, e50458. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting edge: Nf-kappab activating pattern recognition and cytokine receptors license nlrp3 inflammasome activation by regulating nlrp3 expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef]
- Honda, A.; Nogami, M.; Yokozeki, T.; Yamazaki, M.; Nakamura, H.; Watanabe, H.; Kawamoto, K.; Nakayama, K.; Morris, A.J.; Frohman, M.A.; et al. Phosphatidylinositol 4-phosphate 5-kinase α is a downstream effector of the small g protein arf6 in membrane ruffle formation. Cell 1999, 99, 521–532. [Google Scholar] [CrossRef] [Green Version]
- Kagan, J.C.; Medzhitov, R. Phosphoinositide-mediated adaptor recruitment controls toll-like receptor signaling. Cell 2006, 125, 943–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Sanjo, H.; Uematsu, S.; Kaisho, T.; Hoshino, K.; Takeuchi, O.; Kobayashi, M.; Fujita, T.; et al. Essential role for tirap in activation of the signalling cascade shared by tlr2 and tlr4. Nature 2002, 420, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Jung, J.; Chung, H.K.; Im, E.; Rhee, S.H. Pten regulates tlr5-induced intestinal inflammation by controlling mal/tirap recruitment. FASEB J. 2013, 27, 243–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenny, E.F.; Talbot, S.; Gong, M.; Golenbock, D.T.; Bryant, C.E.; O’Neill, L.A. Myd88 adaptor-like is not essential for tlr2 signaling and inhibits signaling by tlr3. J. Immunol. 2009, 183, 3642–3651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burns, K.; Janssens, S.; Brissoni, B.; Olivos, N.; Beyaert, R.; Tschopp, J. Inhibition of interleukin 1 receptor/toll-like receptor signaling through the alternatively spliced, short form of myd88 is due to its failure to recruit irak-4. J. Exp. Med. 2003, 197, 263–268. [Google Scholar] [CrossRef] [Green Version]
- Motshwene, P.G.; Moncrieffe, M.C.; Grossmann, J.G.; Kao, C.; Ayaluru, M.; Sandercock, A.M.; Robinson, C.V.; Latz, E.; Gay, N.J. An oligomeric signaling platform formed by the toll-like receptor signal transducers myd88 and irak-4. J. Biol. Chem. 2009, 284, 25404–25411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kollewe, C.; Mackensen, A.C.; Neumann, D.; Knop, J.; Cao, P.; Li, S.; Wesche, H.; Martin, M.U. Sequential autophosphorylation steps in the interleukin-1 receptor-associated kinase-1 regulate its availability as an adapter in interleukin-1 signaling. J. Biol. Chem. 2004, 279, 5227–5236. [Google Scholar] [CrossRef] [Green Version]
- Cao, Z.; Henzel, W.J.; Gao, X. Irak: A kinase associated with the interleukin-1 receptor. Science 1996, 271, 1128–1131. [Google Scholar] [CrossRef]
- Burns, K.; Clatworthy, J.; Martin, L.; Martinon, F.; Plumpton, C.; Maschera, B.; Lewis, A.; Ray, K.; Tschopp, J.; Volpe, F. Tollip, a new component of the il-1ri pathway, links irak to the il-1 receptor. Nat. Cell Biol. 2000, 2, 346–351. [Google Scholar] [CrossRef]
- Yao, J.; Kim, T.W.; Qin, J.; Jiang, Z.; Qian, Y.; Xiao, H.; Lu, Y.; Qian, W.; Gulen, M.F.; Sizemore, N.; et al. Interleukin-1 (il-1)-induced tak1-dependent versus mekk3-dependent nfkappab activation pathways bifurcate at il-1 receptor-associated kinase modification. J. Biol. Chem. 2007, 282, 6075–6089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvaraj, P.; Harishankar, M.; Singh, B.; Jawahar, M.S.; Banurekha, V.V. Toll-like receptor and tirap gene polymorphisms in pulmonary tuberculosis patients of south india. Tuberculosis 2010, 90, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.X.; Xue, Y.; Liu, J.Y.; Zhao, M.Y.; Li, F.J.; Zhou, J.M.; Wang, H.J.; Li, J.C. Association of tirap (mal) gene polymorhisms with susceptibility to tuberculosis in a chinese population. Genet. Mol. Res. 2011, 10, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Hawn, T.R.; Dunstan, S.J.; Thwaites, G.E.; Simmons, C.P.; Thuong, N.T.; Lan, N.T.N.; Quy, H.T.; Chau, T.T.H.; Hieu, N.T.; Rodrigues, S.; et al. A polymorphism in toll-interleukin 1 receptor domain containing adaptor protein is associated with susceptibility to meningeal tuberculosis. J. Infect. Dis. 2006, 194, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Khor, C.C.; Chapman, S.J.; Vannberg, F.O.; Dunne, A.; Murphy, C.; Ling, E.Y.; Frodsham, A.J.; Walley, A.J.; Kyrieleis, O.; Khan, A.; et al. A mal functional variant is associated with protection against invasive pneumococcal disease, bacteremia, malaria and tuberculosis. Nat. Genet. 2007, 39, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Castiblanco, J.; Varela, D.C.; Castano-Rodriguez, N.; Rojas-Villarraga, A.; Hincapie, M.E.; Anaya, J.M. Tirap (mal) s180l polymorphism is a common protective factor against developing tuberculosis and systemic lupus erythematosus. Infect. Genet. Evol. 2008, 8, 541–544. [Google Scholar] [CrossRef]
- Capparelli, R.; De Chiara, F.; Di Matteo, A.; Medaglia, C.; Iannelli, D. The myd88 rs6853 and tirap rs8177374 polymorphic sites are associated with resistance to human pulmonary tuberculosis. Genes Immun. 2013, 14, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Dissanayeke, S.R.; Levin, S.; Pienaar, S.; Wood, K.; Eley, B.; Beatty, D.; Henderson, H.; Anderson, S.; Levin, M. Polymorphic variation in tirap is not associated with susceptibility to childhood tb but may determine susceptibility to tbm in some ethnic groups. PLoS ONE 2009, 4, e6698. [Google Scholar] [CrossRef] [Green Version]
- Nejentsev, S.; Thye, T.; Szeszko, J.S.; Stevens, H.; Balabanova, Y.; Chinbuah, A.M.; Hibberd, M.; van de Vosse, E.; Alisjahbana, B.; van Crevel, R.; et al. Analysis of association of the tirap (mal) s180l variant and tuberculosis in three populations. Nat. Genet. 2008, 40, 261–262. [Google Scholar] [CrossRef]
- Sanchez, D.; Lefebvre, C.; Rioux, J.; Garcia, L.F.; Barrera, L.F. Evaluation of toll-like receptor and adaptor molecule polymorphisms for susceptibility to tuberculosis in a colombian population. Int. J. Immunogenet. 2012, 39, 216–223. [Google Scholar] [CrossRef]
- Yang, Y.; Li, X.; Cui, W.; Guan, L.; Shen, F.; Xu, J.; Zhou, F.; Li, M.; Gao, C.; Jin, Q.; et al. Potential association of pulmonary tuberculosis with genetic polymorphisms of toll-like receptor 9 and interferon-gamma in a chinese population. BMC Infect. Dis. 2013, 13, 511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fremond, C.M.; Togbe, D.; Doz, E.; Rose, S.; Vasseur, V.; Maillet, I.; Jacobs, M.; Ryffel, B.; Quesniaux, V.F. Il-1 receptor-mediated signal is an essential component of myd88-dependent innate response to mycobacterium tuberculosis infection. J. Immunol. 2007, 179, 1178–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Q.; Li, W.; Li, D.; Feng, Y.; Tao, C. Tirap c539t polymorphism contributes to tuberculosis susceptibility: Evidence from a meta-analysis. Infect. Genet. Evol. 2014, 27, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Qu, Z.; Zhou, J.; Zhou, Y.; Xie, Y.; Jiang, Y.; Wu, J.; Luo, Z.; Liu, G.; Yin, L.; Zhang, X.L. Mycobacterial est12 activates a rack1-nlrp3-gasdermin d pyroptosis-il-1beta immune pathway. Sci. Adv. 2020, 6, eaba4733. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xu, P.; He, P.; Shi, F.; Tang, Y.; Guan, C.; Zeng, H.; Zhou, Y.; Song, Q.; Zhou, B.; et al. Mycobacterial ppe13 activates inflammasome by interacting with the natch and lrr domains of nlrp3. FASEB J. 2020, 34, 12820–12833. [Google Scholar] [CrossRef]
- Smith, J.; Manoranjan, J.; Pan, M.; Bohsali, A.; Xu, J.; Liu, J.; McDonald, K.L.; Szyk, A.; LaRonde-LeBlanc, N.; Gao, L.Y. Evidence for pore formation in host cell membranes by esx-1-secreted esat-6 and its role in mycobacterium marinum escape from the vacuole. Infect. Immun. 2008, 76, 5478–5487. [Google Scholar] [CrossRef] [Green Version]
- Boggaram, V.; Gottipati, K.R.; Wang, X.; Samten, B. Early secreted antigenic target of 6 kda (esat-6) protein of mycobacterium tuberculosis induces interleukin-8 (il-8) expression in lung epithelial cells via protein kinase signaling and reactive oxygen species. J. Biol. Chem. 2013, 288, 25500–25511. [Google Scholar] [CrossRef] [Green Version]
- Manzanillo, P.S.; Shiloh, M.U.; Portnoy, D.A.; Cox, J.S. Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe 2012, 11, 469–480. [Google Scholar] [CrossRef] [Green Version]
- Saiga, H.; Kitada, S.; Shimada, Y.; Kamiyama, N.; Okuyama, M.; Makino, M.; Yamamoto, M.; Takeda, K. Critical role of aim2 in mycobacterium tuberculosis infection. Int. Immunol. 2012, 24, 637–644. [Google Scholar] [CrossRef] [Green Version]
- Garnier, T.; Eiglmeier, K.; Camus, J.C.; Medina, N.; Mansoor, H.; Pryor, M.; Duthoy, S.; Grondin, S.; Lacroix, C.; Monsempe, C.; et al. The complete genome sequence of mycobacterium bovis. Proc. Natl. Acad. Sci. USA 2003, 100, 7877–7882. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhou, X.; Kouadir, M.; Shi, F.; Ding, T.; Liu, C.; Liu, J.; Wang, M.; Yang, L.; Yin, X.; et al. The aim2 inflammasome is involved in macrophage activation during infection with virulent mycobacterium bovis strain. J. Infect. Dis. 2013, 208, 1849–1858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.; Bohsali, A.; Ahlbrand, S.E.; Srinivasan, L.; Rathinam, V.A.; Vogel, S.N.; Fitzgerald, K.A.; Sutterwala, F.S.; Briken, V. Cutting edge: Mycobacterium tuberculosis but not nonvirulent mycobacteria inhibits ifn-beta and aim2 inflammasome-dependent il-1beta production via its esx-1 secretion system. J. Immunol. 2013, 191, 3514–3518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.R.; Kim, B.J.; Kook, Y.H.; Kim, B.J. Mycobacterium abscessus infection leads to enhanced production of type 1 interferon and nlrp3 inflammasome activation in murine macrophages via mitochondrial oxidative stress. PLoS Pathog. 2020, 16, e1008294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, R.O.; Bell, S.L.; MacDuff, D.A.; Kimmey, J.M.; Diner, E.J.; Olivas, J.; Vance, R.E.; Stallings, C.L.; Virgin, H.W.; Cox, J.S. The cytosolic sensor cgas detects mycobacterium tuberculosis DNA to induce type i interferons and activate autophagy. Cell Host Microbe 2015, 17, 811–819. [Google Scholar] [CrossRef] [Green Version]
- Collins, A.C.; Cai, H.; Li, T.; Franco, L.H.; Li, X.D.; Nair, V.R.; Scharn, C.R.; Stamm, C.E.; Levine, B.; Chen, Z.J.; et al. Cyclic gmp-amp synthase is an innate immune DNA sensor for mycobacterium tuberculosis. Cell Host Microbe 2015, 17, 820–828. [Google Scholar] [CrossRef] [Green Version]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. Ifi16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Liu, C.; Yue, R.; El-Ashram, S.; Wang, J.; He, X.; Zhao, D.; Zhou, X.; Xu, L. Cgas/sting/tbk1/irf3 signaling pathway activates bmdcs maturation following mycobacterium bovis infection. Int. J. Mol. Sci. 2019, 20, 895. [Google Scholar] [CrossRef] [Green Version]
- Stanley, S.A.; Johndrow, J.E.; Manzanillo, P.; Cox, J.S. The type i ifn response to infection with mycobacterium tuberculosis requires esx-1-mediated secretion and contributes to pathogenesis. J. Immunol. 2007, 178, 3143–3152. [Google Scholar] [CrossRef] [Green Version]
- Manca, C.; Tsenova, L.; Bergtold, A.; Freeman, S.; Tovey, M.; Musser, J.M.; Barry, C.E., 3rd; Freedman, V.H.; Kaplan, G. Virulence of a mycobacterium tuberculosis clinical isolate in mice is determined by failure to induce th1 type immunity and is associated with induction of ifn-alpha /beta. Proc. Natl. Acad. Sci. USA 2001, 98, 5752–5757. [Google Scholar] [CrossRef] [Green Version]
- Novikov, A.; Cardone, M.; Thompson, R.; Shenderov, K.; Kirschman, K.D.; Mayer-Barber, K.D.; Myers, T.G.; Rabin, R.L.; Trinchieri, G.; Sher, A.; et al. Mycobacterium tuberculosis triggers host type i ifn signaling to regulate il-1beta production in human macrophages. J. Immunol. 2011, 187, 2540–2547. [Google Scholar] [CrossRef] [Green Version]
- Guarda, G.; Braun, M.; Staehli, F.; Tardivel, A.; Mattmann, C.; Forster, I.; Farlik, M.; Decker, T.; Du Pasquier, R.A.; Romero, P.; et al. Type i interferon inhibits interleukin-1 production and inflammasome activation. Immunity 2011, 34, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer-Barber, K.D.; Andrade, B.B.; Oland, S.D.; Amaral, E.P.; Barber, D.L.; Gonzales, J.; Derrick, S.C.; Shi, R.; Kumar, N.P.; Wei, W.; et al. Host-directed therapy of tuberculosis based on interleukin-1 and type i interferon crosstalk. Nature 2014, 511, 99–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, J.L.; Chan, J. Immunology of tuberculosis. Annu. Rev. Immunol. 2001, 19, 93–129. [Google Scholar] [CrossRef] [PubMed]
- Abreu, R.; Essler, L.; Giri, P.; Quinn, F. Interferon-gamma promotes iron export in human macrophages to limit intracellular bacterial replication. PLoS ONE 2020, 15, e0240949. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, J.; Zhao, S.; Gao, X.; Wang, R.; Liu, J.; Zhou, X.; Zhou, Y. The Roles of Inflammasomes in Host Defense against Mycobacterium tuberculosis. Pathogens 2021, 10, 120. https://doi.org/10.3390/pathogens10020120
Ma J, Zhao S, Gao X, Wang R, Liu J, Zhou X, Zhou Y. The Roles of Inflammasomes in Host Defense against Mycobacterium tuberculosis. Pathogens. 2021; 10(2):120. https://doi.org/10.3390/pathogens10020120
Chicago/Turabian StyleMa, Jialu, Shasha Zhao, Xiao Gao, Rui Wang, Juan Liu, Xiangmei Zhou, and Yang Zhou. 2021. "The Roles of Inflammasomes in Host Defense against Mycobacterium tuberculosis" Pathogens 10, no. 2: 120. https://doi.org/10.3390/pathogens10020120