
Effects of Alloying Elements on the Solution and Diffusion of Oxygen at Iron Grain Boundary Investigated by First-Principles Study
 , , and
, , and
Abstract
:1. Introduction
2. Methods
2.1. Computational Methods
2.2. Analysis Methods
3. Results
3.1. Stable Positions of Alloying and O Atoms in the Fe-GB
3.2. The Effects of Alloying Elements on the Solution of O Atom in the GB
3.3. The Effects of Alloying Elements on the Diffusion of O Atoms in the GB
4. Discussion
4.1. Insight into the Effects of Alloying Elements on the Solution and Diffusion of Oxygen
4.2. The Effects of Alloying Elements on Oxygen Concentration
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Klueh, R.L.; Gelles, D.S.; Jitsukawa, S.; Kimura, A.; Odette, G.R.; van der Schaaf, B.; Victoria, M. Ferritic/martensitic steels-overview of recent results. J. Nucl. Mater. 2002, 307–311, 455–465. [Google Scholar] [CrossRef]
- Tavassoli, A.A.F.; Diegele, E.; Lindau, R.; Luzginova, N.; Tanigawa, H. Current status and recent research achievements in ferritic/martensitic steels. J. Nucl. Mater. 2014, 455, 269–276. [Google Scholar] [CrossRef]
- Murty, K.L.; Charit, I. Structural materials for Gen-IV nuclear reactors: Challenges and opportunities. J. Nucl. Mater. 2008, 383, 189–195. [Google Scholar] [CrossRef]
- Zinkle, S.J.; Was, G.S. Materials challenges in nuclear energy. Acta Mater. 2013, 61, 735–758. [Google Scholar] [CrossRef]
- Li, Q.; Shen, Y.; Zhu, J.; Huang, X.; Shang, Z. Evaluation of Irradiation Hardening of P92 Steel under Ar Ion Irradiation. Metals 2018, 8, 94. [Google Scholar] [CrossRef]
- Alemberti, A.; Smirnov, V.; Smith, C.F.; Takahashi, M. Overview of lead-cooled fast reactor activities. Prog. Nucl. Energy 2014, 77, 300–307. [Google Scholar] [CrossRef]
- Zhang, J.; Li, N. Review of the studies on fundamental issues in LBE corrosion. J. Nucl. Mater. 2008, 373, 351–377. [Google Scholar] [CrossRef]
- Dong, Q.; Yao, Z.; Wang, Q.; Yu, H.; Kirk, M.A.; Daymond, M.R. Precipitate Stability in a Zr–2.5Nb–0.5Cu Alloy under Heavy Ion Irradiation. Metals 2017, 7, 287. [Google Scholar] [CrossRef]
- Gong, X.; Short, M.P.; Auger, T.; Charalampopoulou, E.; Lambrinou, K. Environmental degradation of structural materials in liquid lead- and lead-bismuth eutectic-cooled reactors. Prog. Mater. Sci. 2022, 126, 100920. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, L.; Shen, Z. Understanding the surface oxide evolution of T91 ferritic-martensitic steel in supercritical water through advanced characterization. Acta Mater. 2020, 194, 156–167. [Google Scholar] [CrossRef]
- Shi, Q.; Zhang, L.; Yan, W.; Wang, W.; Yin, P.; Shan, Y.; Yang, K. High-Temperature Oxidation Behavior of SIMP Steel at 800 °C. Oxid. Met. 2018, 89, 49–60. [Google Scholar] [CrossRef]
- Zhang, L.; Yan, W.; Shi, Q.; Li, Y.; Shan, Y.; Yang, K. Silicon enhances high temperature oxidation resistance of SIMP steel at 700 °C. Corros. Sci. 2020, 167, 108519. [Google Scholar] [CrossRef]
- Kurata, Y.; Futakawa, M. Excellent corrosion resistance of 18Cr–20Ni–5Si steel in liquid Pb–Bi. J. Nucl. Mater. 2004, 325, 217–222. [Google Scholar] [CrossRef]
- Zhang, J. A review of steel corrosion by liquid lead and lead–bismuth. Corros. Sci. 2009, 51, 1207–1227. [Google Scholar] [CrossRef]
- Wachowicz, E.; Ossowski, T.; Kiejna, A. Cohesive and magnetic properties of grain boundaries in bcc Fe with Cr additions. Phys. Rev. B 2010, 81, 094104. [Google Scholar] [CrossRef]
- Charalampopoulou, E.; Delville, R.; Verwerft, M.; Lambrinou, K.; Schryvers, D. Transmission electron microscopy study of complex oxide scales on DIN 1.4970 steel exposed to liquid Pb-Bi eutectic. Corros. Sci. 2019, 147, 22–31. [Google Scholar] [CrossRef]
- Hosemann, P.; Dickerson, R.; Dickerson, P.; Li, N.; Maloy, S.A. Transmission electron microscopy (TEM) on oxide layers formed on D9 stainless steel in lead bismuth eutectic (LBE). Corros. Sci. 2013, 66, 196–202. [Google Scholar] [CrossRef]
- Ma, Z.; Shen, T.; Wang, Z.; Zhou, T.; Chang, H.; Jin, P.; Wei, K.; Pang, L.; Chai, J.; Liu, C. Improving the oxidation resistance of SIMP steel to liquid Pb-Bi eutectic by shot peening treatments. Appl. Surf. Sci. 2022, 578, 151910. [Google Scholar] [CrossRef]
- Wang, H.; Yuan, Q.; Chai, L.; Zhao, K.; Guo, N.; Xiao, J.; Yin, X.; Tang, B.; Li, Y.; Qiu, S. Effects of pulsed laser surface remelting on microstructure, hardness and lead-bismuth corrosion behavior of a ferrite/martensitic steel. Nucl. Eng. Technol. 2022, 54, 1972–1981. [Google Scholar] [CrossRef]
- Zhang, H.; Xiao, Y.; Xu, Z.; Yang, M.; Zhang, L.; Yin, L.; Chai, S.; Wang, G.; Zhang, L.; Cai, X. Effects of Ni-decorated reduced graphene oxide nanosheets on the microstructural evolution and mechanical properties of Sn-3.0Ag-0.5Cu composite solders. Intermetallics 2022, 150, 107683. [Google Scholar] [CrossRef]
- Xu, H.; He, T.; Zhong, N.; Zhao, B.; Liu, Z. Transient thermomechanical analysis of micro cylindrical asperity sliding contact of SnSbCu alloy. Tribol. Int. 2022, 167, 107362. [Google Scholar] [CrossRef]
- Martinelli, L.; Balbaud-Célérier, F.; Terlain, A.; Bosonnet, S.; Picard, G.; Santarini, G. Oxidation mechanism of an Fe–9Cr–1Mo steel by liquid Pb–Bi eutectic alloy at 470 °C (Part II). Corros. Sci. 2008, 50, 2537–2548. [Google Scholar] [CrossRef]
- Wang, X.; Posselt, M.; Faßbender, J. Influence of substitutional atoms on the diffusion of oxygen in dilute iron alloys. Phys. Rev. B 2018, 98, 064103. [Google Scholar] [CrossRef]
- Wang, X.; Faßbender, J.; Posselt, M. Efficient Calculation Methods for the Diffusion Coefficient of Interstitial Solutes in Dilute Alloys. Materials 2019, 12, 1491. [Google Scholar] [CrossRef]
- Shang, S.L.; Fang, H.Z.; Wang, J.; Guo, C.P.; Wang, Y.; Jablonski, P.D.; Du, Y.; Liu, Z.K. Vacancy mechanism of oxygen diffusivity in bcc Fe: A first-principles study. Corros. Sci. 2014, 83, 94–102. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, Y.; Li, X.; Xu, Y.; Wu, X.; Liu, C.; Kong, X.; Yao, C.; Wang, Z. Investigation of the dissolution and diffusion properties of interstitial oxygen at grain boundaries in body-centered-cubic iron by the first-principles study. RSC Adv. 2021, 11, 8643–8653. [Google Scholar] [CrossRef]
- Ye, Z.; Lei, Y.; Zhang, J.; Zhang, Y.; Li, X.; Xu, Y.; Wu, X.; Liu, C.S.; Hao, T.; Wang, Z. Effects of oxygen concentration and irradiation defects on the oxidation corrosion of body-centered-cubic iron surfaces: A first-principles study. Chin. Phys. B 2022, 31, 086802. [Google Scholar] [CrossRef]
- Chentouf, S.; Raulot, J.M.; Faraoun, H.I.; Aourag, H.; Grosdidier, T. Energetics of Ti and Zr transition metals in D03-Fe3Al and its Σ5(310)[001] grain boundary. Intermetallics 2012, 22, 251–254. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Li, X.; Liu, W.; Xu, Y.; Liu, C.S.; Fang, Q.F.; Pan, B.C.; Wang, Z. Energetic and kinetic behaviors of small vacancy clusters near a symmetric Σ5(310)/[001] tilt grain boundary in bcc Fe. J. Nucl. Mater. 2013, 440, 250–256. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef]
- Hristova, E.; Janisch, R.; Drautz, R.; Hartmaier, A. Solubility of carbon in α-iron under volumetric strain and close to the Σ5(310)[001] grain boundary: Comparison of DFT and empirical potential methods. Comput. Mater. Sci. 2011, 50, 1088–1096. [Google Scholar] [CrossRef]
- Olsson, P.; Klaver, T.P.C.; Domain, C. Ab initio study of solute transition-metal interactions with point defects in bcc Fe. Phys. Rev. B 2010, 81, 054102. [Google Scholar] [CrossRef]
- Lee, K.C.; Erb, U. Grain boundaries and coincidence site lattices in the corneal nanonipple structure of the Mourning Cloak butterfly. Beilstein J. Nanotechnol. 2013, 4, 292–299. [Google Scholar] [CrossRef]
- Li, X.; Liu, W.; Xu, Y.; Liu, C.S.; Fang, Q.F.; Pan, B.C.; Chen, J.-L.; Luo, G.N.; Wang, Z. An energetic and kinetic perspective of the grain-boundary role in healing radiation damage in tungsten. Nucl. Fusion 2013, 53, 123014. Available online: http://dx.doi.org/10.1088/0029-5515/53/12/123014 (accessed on 20 January 2023). [CrossRef]
- Li, X.; Wang, Y.; Zhang, Y.; Xu, Y.; Li, X.; Wang, X.; Fang, Q.F.; Wu, X.; Liu, C.S. Towards the dependence of radiation damage on the grain boundary character and grain size in tungsten: A combined study of molecular statics and rate theory. J. Nucl. Mater. 2022, 563, 153637. [Google Scholar] [CrossRef]
- Inoue, K.; Kawahara, K.; Saito, M.; Kotani, M.; Ikuhara, Y. 3D arrangement of atomic polyhedra in tilt grain boundaries. Acta Materialia 2021, 202, 266–276. [Google Scholar] [CrossRef]
- Wang, J.; Madsen, G.K.H.; Drautz, R. Grain boundaries in bcc-Fe: A density-functional theory and tight-binding study. Modell. Simul. Mater. Sci. Eng. 2018, 26, 025008. Available online: https://doi.org/10.1088/1361-651X/aa9f81 (accessed on 15 September 2022). [CrossRef]
- Čák, M.; Šob, M.; Hafner, J. First-principles study of magnetism at grain boundaries in iron and nickel. Phys. Rev. B 2008, 78, 054418. [Google Scholar] [CrossRef]
- Lejček, P.; Šandera, P.; Horníková, J.; Řehák, P.; Pokluda, J. Grain boundary segregation of elements of groups 14 and 15 and its consequences for intergranular cohesion of ferritic iron. J. Mater. Sci. 2017, 52, 5822–5834. [Google Scholar] [CrossRef]
- Kong, X.; Wu, X.; Liu, C.S.; Fang, Q.F.; Hu, Q.M.; Chen, J.; Luo, G.N. First-principles calculations of transition metal solute interactions with hydrogen in tungsten. Nucl. Fusion 2016, 56, 026004. Available online: http://dx.doi.org/10.1088/0029-5515/56/2/026004 (accessed on 14 December 2022). [CrossRef]
- Zhang, Y.; You, Y.; Li, D.; Xu, Y.; Liu, C.S.; Pan, B.C.; Wang, Z. Effect of irradiation defects on the corrosion behaviors of steels exposed to lead bismuth eutectic in ADS: A first-principles study. Phys. Chem. Chem. Phys. 2015, 17, 12292–12298. [Google Scholar] [CrossRef]
- Huo, J.; Wei, H.; Fu, L.; Zhao, C.; He, C. Highly active Fe36Co44 bimetallic nanoclusters catalysts for hydrolysis of ammonia borane: The first-principles study. Chin. Chem. Lett. 2023, 34, 107261. [Google Scholar] [CrossRef]
- Ohnuma, T.; Soneda, N.; Iwasawa, M. First-principles calculations of vacancy–solute element interactions in body-centered cubic iron. Acta Mater. 2009, 57, 5947–5955. [Google Scholar] [CrossRef]
- Mehrer, H. Diffusion in Solids: Fundamentals, Methods, Materials, Diffusion-Controlled Processes; Springer: Berlin/Heidelberg, Germany, 2007; pp. 95–104. [Google Scholar] [CrossRef]
- Fang, H.Z.; Shang, S.L.; Wang, Y.; Liu, Z.K.; Alfonso, D.; Alman, D.E.; Shin, Y.K.; Zou, C.Y.; Duin, A.C.T.v.; Lei, Y.K.; et al. First-principles studies on vacancy-modified interstitial diffusion mechanism of oxygen in nickel, associated with large-scale atomic simulation techniques. J. Appl. Phys. 2014, 115, 043501. [Google Scholar] [CrossRef]
- Murali, D.S.K.; Panigrahi, B.K.; Valsakumar, M.C.; Chandra, S.; Sundar, C.S.; Raj, B.J.J.o.N.M. The role of minor alloying elements on the stability and dispersion of yttria nanoclusters in nanostructured ferritic alloys: An ab initio study. J. Nucl. Mater. 2010, 403, 113–116. [Google Scholar] [CrossRef]
- Jansen, A.P.J. An Introduction to Kinetic Monte Carlo Simulations of Surface Reactions; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar] [CrossRef]
- Ma, H.; Chen, X.-Q.; Li, R.; Wang, S.; Dong, J.; Ke, W. First-principles modeling of anisotropic anodic dissolution of metals and alloys in corrosive environments. Acta Mater. 2017, 130, 137–146. [Google Scholar] [CrossRef]
- Yeliseyeva, O.; Tsisar, V.; Zhou, Z. Corrosion behavior of Fe–14Cr–2W and Fe–9Cr–2W ODS steels in stagnant liquid Pb with different oxygen concentration at 550 and 650 °C. J. Nucl. Mater. 2013, 442, 434–443. [Google Scholar] [CrossRef]
- Stefano, D.D.; Nazarov, R.; Hickel, T.; Neugebauer, J.; Mrovec, M.; Elsässer, C.J.P.R.B. First-principles investigation of hydrogen interaction with TiC precipitates in α-Fe. Phys. Rev. B 2016, 93, 184108. [Google Scholar] [CrossRef]
- Lejček, P. (Ed.) Models of Equilibrium Grain Boundary Segregation. In Grain Boundary Segregation in Metals; Springer: Berlin/Heidelberg, Germany, 2010; pp. 51–102. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
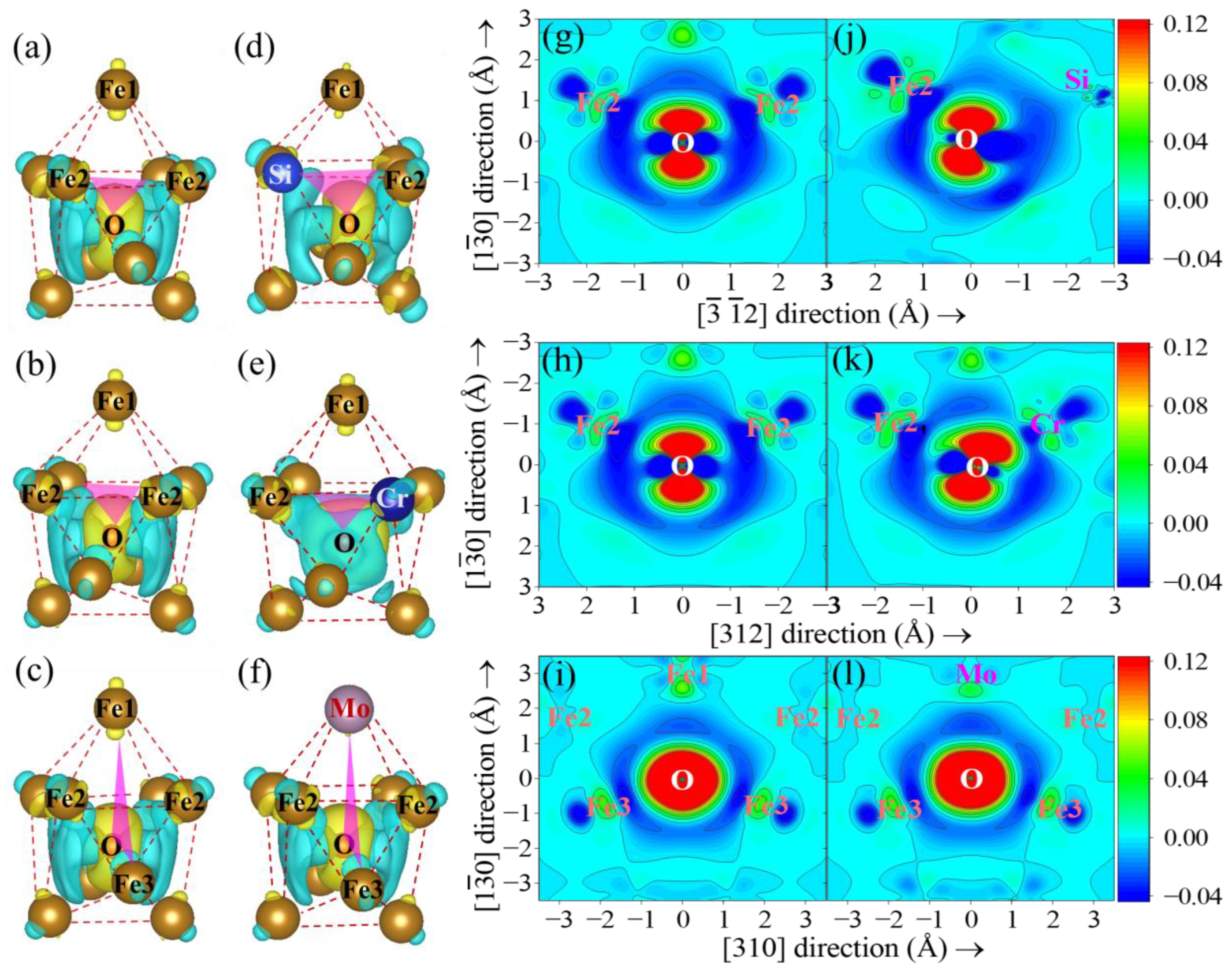{kind=link}
{kind=link}
{kind=link}
 | Initial Position | Final Position | |||
| Pure Fe | Si | Cr | Mo | ||
| c1 | c6 | c6 | c6 | c6 | |
| c2 | c2 | c2 | c2 | c2 | |
| c3 | c2 | c2 | c2 | c2 | |
| c4 | c4 | c2 | c2 | c4 | |
| c5 | c5 | c5 | c5 | c5 | |
| c6 | c6 | c6 | c6 | c6 | |
| c7 | c2 | c2 | c2 | c2 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Li, X.; Lei, Y.; Zhang, Y.; Li, X.; Xu, Y.; Wu, X.; Yang, J.; Li, B.; Liu, C. Effects of Alloying Elements on the Solution and Diffusion of Oxygen at Iron Grain Boundary Investigated by First-Principles Study. Metals 2023, 13, 789. https://doi.org/10.3390/met13040789
Zhang J, Li X, Lei Y, Zhang Y, Li X, Xu Y, Wu X, Yang J, Li B, Liu C. Effects of Alloying Elements on the Solution and Diffusion of Oxygen at Iron Grain Boundary Investigated by First-Principles Study. Metals. 2023; 13(4):789. https://doi.org/10.3390/met13040789
Chicago/Turabian StyleZhang, Jingdan, Xiaolin Li, Yawei Lei, Yange Zhang, Xiangyan Li, Yichun Xu, Xuebang Wu, Junfeng Yang, Bingsheng Li, and Changsong Liu. 2023. "Effects of Alloying Elements on the Solution and Diffusion of Oxygen at Iron Grain Boundary Investigated by First-Principles Study" Metals 13, no. 4: 789. https://doi.org/10.3390/met13040789