

Out of the Silence: Insights into How Genes Escape X-Chromosome Inactivation

Abstract
:
1. Introduction
2. Identification of Genes That Escape from XCI
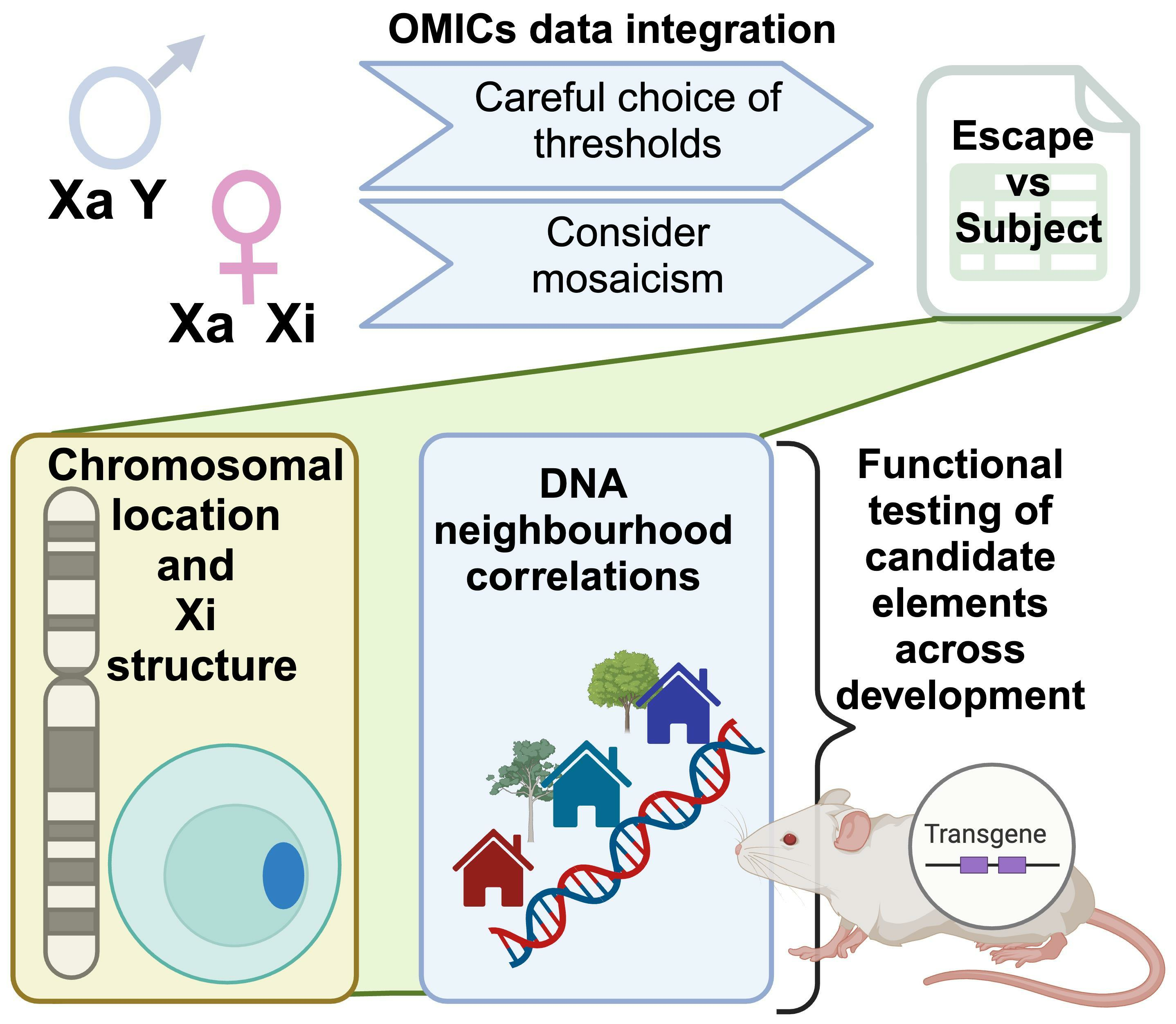
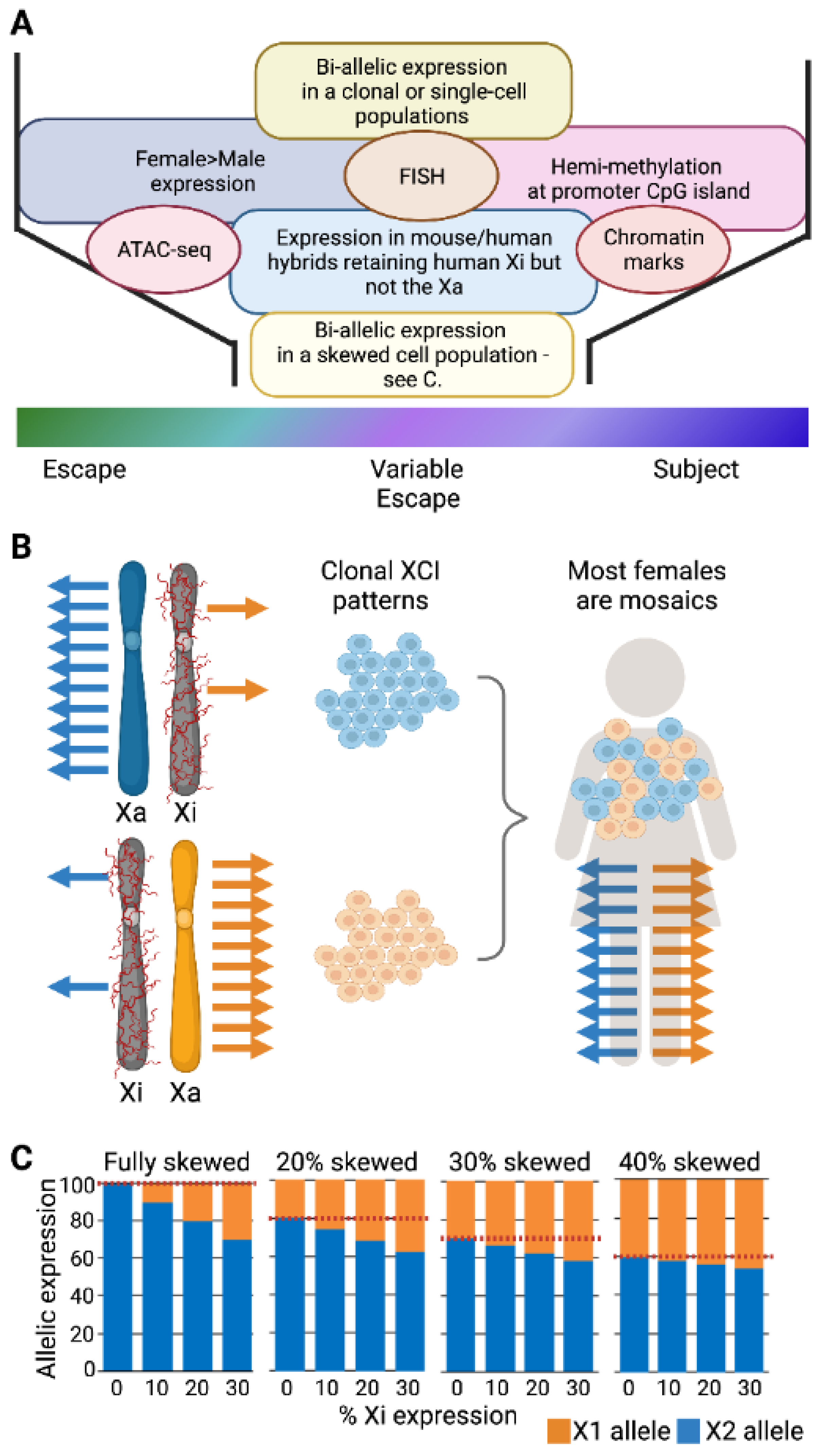
2.1. The Challenges to Determining XCI Status in Humans
2.2. Which Genes Escape XCI?
3. Clinical Implications of Escapees

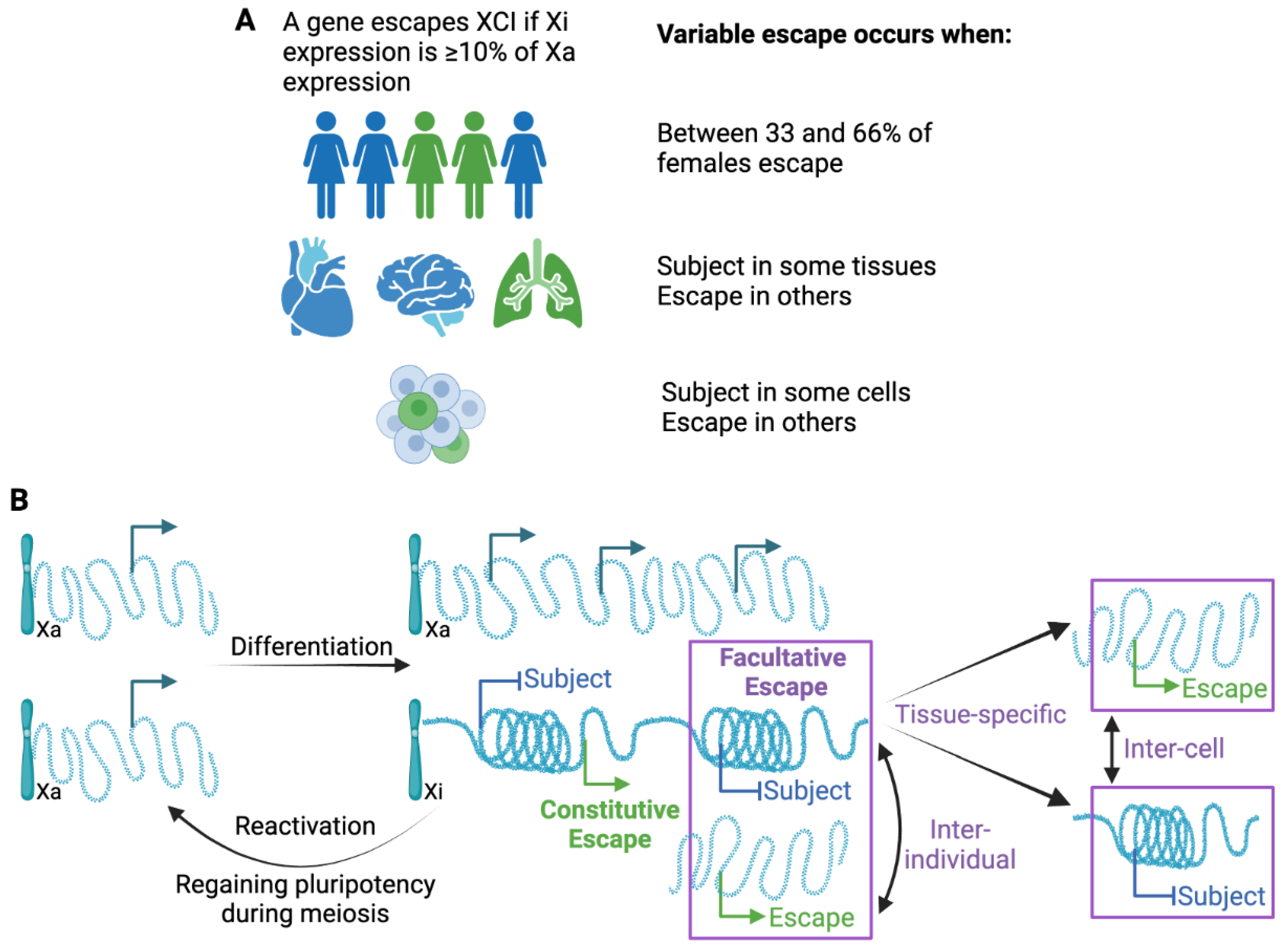
4. Variability in Escape from XCI
4.1. Inter-Individual Differences in Escape from XCI
4.2. Tissue-Specific Variable Escape from XCI
4.3. Reactivation of Inactivated Genes
4.4. Impact of Age
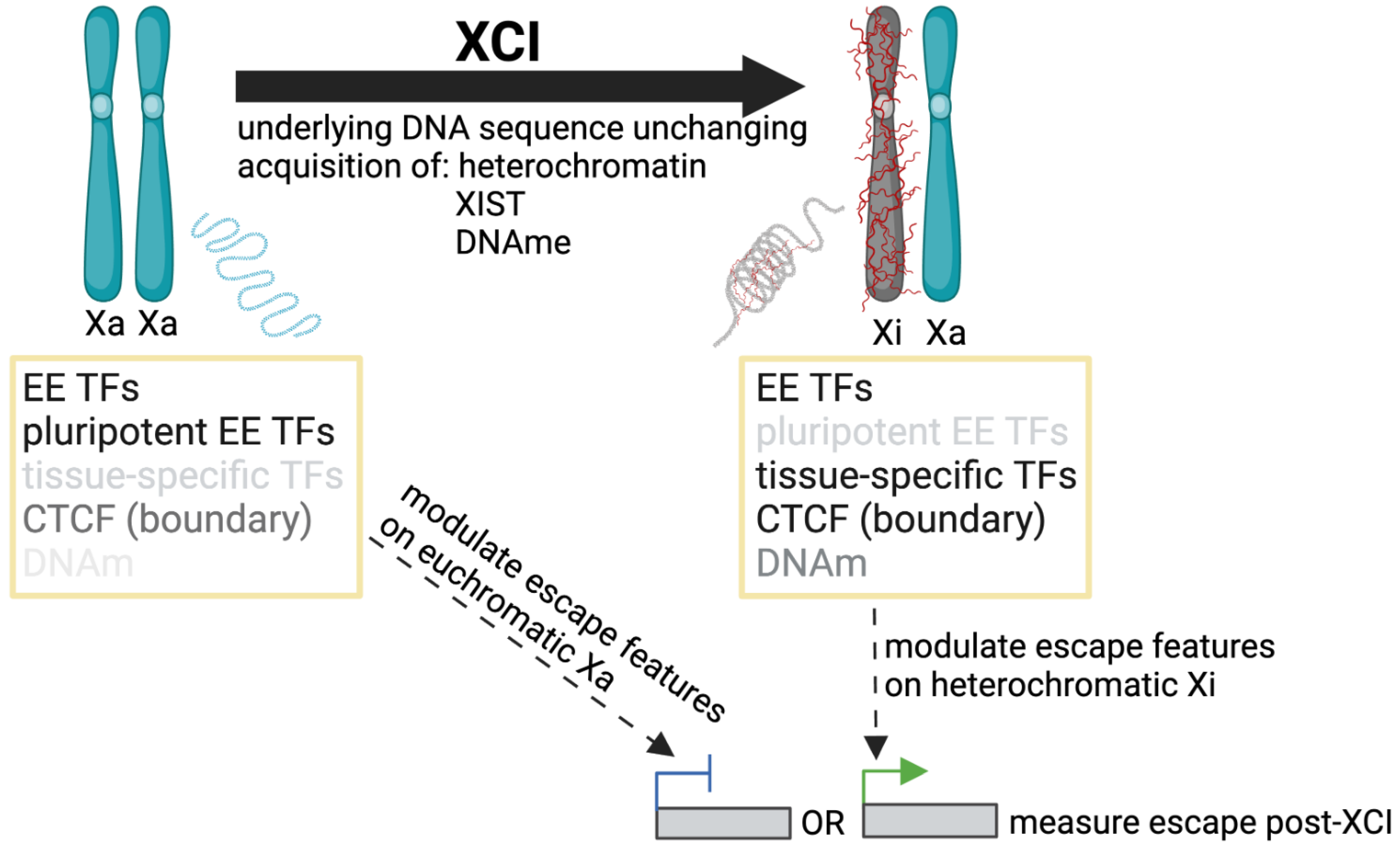
5. Elements Associated with Escape from XCI
5.1. The Physical Structure of the Inactive X Chromosome
5.2. Identifying Genomic Features Associated with Escape from XCI
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genomic Element | Source of XCI Status | Source of Element | Window Size | Key Results |
|---|---|---|---|---|
| Repetitive elements: LINEs (L1s) enriched on X and in regions of genes subject to inactivation; SINEs (Alus) less prevalent on X but enriched at genes escaping from XCI | XCI status from [149] | RepeatMasker for X to autosomes | Genomic segments ~200 kb | X was enriched 2-fold for L1 repetitive elements; escapee-containing regions were significantly reduced in L1 content compared with subject-containing regions [150] |
| XCI status from [3,149] | RepeatMasker for X across evolutionary strata | TSS ± 50, 100 kb 1 | LINEs were significantly enriched around subject genes, while Alu elements and short motifs containing ACG/CGT were significantly enriched around escapees 2 [151] | |
| XCI status from [3] | Oligomer enrichment analysis focused on Xp22 | TSS ± 50, 100, 250 kb | 38% of oligomers overrepresented in escapee-containing regions were located in Alu repeats, while 64% of the oligomers overrepresented in subject-containing regions were within L1 repeats 2 [152] | |
| DNAm predicted XCI status of autosomal genes in X; auto some translocations | DNA sequence features and epigenetic markers from ENCODE | TSS ±5, 15 kb, transcribed genic region | Alu elements were enriched at autosomal genes that escaped from inactivation while L1s were enriched at subject genes 2 [132] | |
| DNAm predicted XCI status of autosomal genes in X; autosome translocations | Motif enrichment plus RepeatMasker for inactivated vs active autosome genes | Gene ±50 kb | Motifs mapping to primate-specific L1s were enriched around both autosomal and X-linked genes silenced by XCI; Alu elements were enriched around autosomal genes that escape XCI 2 [133] | |
| RNA-seq detected “not-silenced” mouse genes with inducible Xist | RepeatMasker for mouse chromosomes X, 8, and 12 | TSS ± 500 kb | Escape genes clustered in LINE-poor regions of chromosomes X, 12, and 8; SINE enrichment correlated with escape genes on both chromosomes X and 12 [134] | |
| Boundary elements: play a role at transition regions involved in maintaining both inactive and escape domains (CTCF, TAD-like structures) | RNA-seq in mouse Patski cells and F1 hybrid brain with skewed XCI | ChIP-seq in mouse Patski cells and F1 hybrid brain with skewed XCI | Sliding 500 kb, step size 1 kb | CTCF binding was reduced on the Xi vs Xa except at escape regions, and was denser in brain compared to the Patski cell line, possibly contributing to a more compartmentalized structure of the Xi and fewer escape genes in brain compared to the cell line [144] |
| RNA-seq in mouse Patski cells and F1 hybrid brain with skewed XCI | TF motifs and Chip-seq peaks in mouse Patski cells and F1 hybrid brain with skewed XCI | Escape domains | Escape gene domains are flanked by convergent arrays of CTCF binding sites present on both the Xa and Xi, often with cohesin subunit RAD21; facultative escapees showed differences in Xi CTCF binding dependent on their XCI status in various cells/tissues [139] | |
| RNA-seq in mouse trophoblast stem cells (TSCs, imprinted XCI) | ChIP-seq in TSCs | Gene ± 5 kb | Moderate positive correlation between CTCF binding and Xi expression; the majority of Xi CTCF peaks were located at sites shared with the Xa [153] | |
| Escape elements: Local control of escape from XCI (CTCF and other transcription-related factors) | RNA-seq detected “not-silenced” mouse genes with inducible Xist | ChIP-seq of male mouse ESC X to autosomes | TSS ± 4 kb | Enrichment of CTCF at the TSSs of “not silenced” X-linked genes relative to partially affected and fully silenced genes; enrichment was notably absent for autosomal genes escaping XCI [134] |
| RNA-seq of mouse ESC to NPC differentiation | ChIP-seq and ATAC-seq in mouse ESCs | ≤5 kb from TSS, and >5 kb from TSS | 51% of Xi accessible sites were <5 kb from a promoter compared to ~35% on the Xa, suggesting that escape is often regulated through promoter-proximal sites; most ATAC–seq peaks on the Xi were found at CTCF sites [112] | |
| PRO-seq in inducible Xist female mouse ESC line | Random forest model to predict XCI status based on biological peaks and genome features in male ESCs | Promoters or gene body regions | Top 10 features important for classifying escape included distance to Xist locus, gene density, RING1B, TAF1, MLL2, HDAC2, ESRRB, HDAC1, TET1, and E2F1; CTCF was not a discriminating feature at promoter, but signal was significantly enriched at enhancers of escape genes [154] | |
| Escape TSSs called using FANTOM5 CAGE data in multiple cell types; PAR excluded | TF motif and ChIP-seq peaks from primary cells, tissues and transformed cell lines | TSS ± 500 bp | Over-representation of YY1 around escape TSSs (X-linked and autosomal from X;autosome translocations); additional over-represented TFs at escape include PAX5, MYC, TBLR1, FOXM1, MAZ, ATF2, CTCF, CHD1 and SIN3A when compared to subject genes [155] | |
| 55 constitutive escape genes [40]; variable escape and PAR were excluded | ReMAP ChIP-seq peaks in female GM12878 | TSS ± 500 bp 1 | Five TFs (ZFP36, NIPBL, MYB, STAT1, and HSF1) were enriched around escape compared to subject genes, and in general the number of TFs binding per gene was depleted for subject genes compared to escape and autosomal genes [142] |
References
- Balaton, B.P.; Fornes, O.; Wasserman, W.W.; Brown, C.J. Cross-Species Examination of X-Chromosome Inactivation Highlights Domains of Escape from Silencing. Epigenet. Chromatin 2021, 14, 12. [Google Scholar] [CrossRef]
- GTEx Consortium; Tukiainen, T.; Villani, A.-C.; Yen, A.; Rivas, M.A.; Marshall, J.L.; Satija, R.; Aguirre, M.; Gauthier, L.; Fleharty, M.; et al. Landscape of X Chromosome Inactivation across Human Tissues. Nature 2017, 550, 244–248. [Google Scholar] [CrossRef]
- Carrel, L.; Willard, H.F. X-Inactivation Profile Reveals Extensive Variability in X-Linked Gene Expression in Females. Nature 2005, 434, 400–404. [Google Scholar] [CrossRef]
- Willard, H.F.; Brown, C.J.; Carrel, L.; Hendrich, B.; Miller, A.P. Epigenetic and Chromosomal Control of Gene Expression: Molecular and Genetic Analysis of X Chromosome Inactivation. Cold Spring Harb. Symp. Quant. Biol. 1993, 58, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Cobos, M.J.; Balaton, B.P.; Brown, C.J. Genes That Escape from X-Chromosome Inactivation: Potential Contributors to Klinefelter Syndrome. Am. J. Med. Genet. Part C Semin. Med. Genet. 2020, 184, 226–238. [Google Scholar] [CrossRef]
- Sudbrak, R.; Wieczorek, G.; Nuber, U.A.; Mann, W.; Kirchner, R.; Erdogan, F.; Brown, C.J.; Wöhrle, D.; Sterk, P.; Kalscheuer, V.M.; et al. X Chromosome-Specific cDNA Arrays: Identification of Genes That Escape from X-Inactivation and Other Applications. Hum. Mol. Genet. 2001, 10, 77–83. [Google Scholar] [CrossRef] [PubMed]
- San Roman, A.K.; Godfrey, A.K.; Skaletsky, H.; Bellott, D.W.; Groff, A.F.; Harris, H.L.; Blanton, L.V.; Hughes, J.F.; Brown, L.; Phou, S.; et al. The Human Inactive X Chromosome Modulates Expression of the Active X Chromosome. Cell Genom. 2023, 3, 100259. [Google Scholar] [CrossRef]
- San Roman, A.K.; Skaletsky, H.; Godfrey, A.K.; Bokil, N.V.; Teitz, L.; Singh, I.; Blanton, L.V.; Bellott, D.W.; Pyntikova, T.; Lange, J.; et al. The Human Y and Inactive X Chromosomes Similarly Modulate Autosomal Gene Expression. bioRxiv 2023. [Google Scholar] [CrossRef]
- Tallaksen, H.B.L.; Johannsen, E.B.; Just, J.; Viuff, M.H.; Gravholt, C.H.; Skakkebæk, A. The Multi-Omic Landscape of Sex Chromosome Abnormalities: Current Status and Future Directions. Endocr. Connect. 2023, 12, e230011. [Google Scholar] [CrossRef]
- Balaton, B.P.; Brown, C.J. Contribution of Genetic and Epigenetic Changes to Escape from X-Chromosome Inactivation. Epigenet. Chromatin 2021, 14, 30. [Google Scholar] [CrossRef]
- Cotton, A.M.; Price, E.M.; Jones, M.J.; Balaton, B.P.; Kobor, M.S.; Brown, C.J. Landscape of DNA Methylation on the X Chromosome Reflects CpG Density, Functional Chromatin State and X-Chromosome Inactivation. Hum. Mol. Genet. 2015, 24, 1528–1539. [Google Scholar] [CrossRef]
- Gershman, A.; Sauria, M.E.G.; Guitart, X.; Vollger, M.R.; Hook, P.W.; Hoyt, S.J.; Jain, M.; Shumate, A.; Razaghi, R.; Koren, S.; et al. Epigenetic Patterns in a Complete Human Genome. Science 2022, 376, eabj5089. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global Epigenomic Reconfiguration During Mammalian Brain Development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef]
- Kucera, K.S.; Reddy, T.E.; Pauli, F.; Gertz, J.; Logan, J.E.; Myers, R.M.; Willard, H.F. Allele-Specific Distribution of RNA Polymerase II on Female X Chromosomes. Hum. Mol. Genet. 2011, 20, 3964–3973. [Google Scholar] [CrossRef] [PubMed]
- Qu, K.; Zaba, L.C.; Giresi, P.G.; Li, R.; Longmire, M.; Kim, Y.H.; Greenleaf, W.J.; Chang, H.Y. Individuality and Variation of Personal Regulomes in Primary Human T Cells. Cell Syst. 2015, 1, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Peñaherrera, M.S.; Jiang, R.; Avila, L.; Yuen, R.K.C.; Brown, C.J.; Robinson, W.P. Patterns of Placental Development Evaluated by X Chromosome Inactivation Profiling Provide a Basis to Evaluate the Origin of Epigenetic Variation. Hum. Reprod. 2012, 27, 1745–1753. [Google Scholar] [CrossRef]
- Swierczek, S.I.; Piterkova, L.; Jelinek, J.; Agarwal, N.; Hammoud, S.; Wilson, A.; Hickman, K.; Parker, C.J.; Cairns, B.R.; Prchal, J.T. Methylation of AR Locus Does Not Always Reflect X Chromosome Inactivation State. Blood 2012, 119, e100–e109. [Google Scholar] [CrossRef]
- Clemson, C.M.; Hall, L.L.; Byron, M.; McNeil, J.; Lawrence, J.B. The X Chromosome Is Organized into a Gene-Rich Outer Rim and an Internal Core Containing Silenced Nongenic Sequences. Proc. Natl. Acad. Sci. USA 2006, 103, 7688–7693. [Google Scholar] [CrossRef]
- Żylicz, J.J.; Bousard, A.; Žumer, K.; Dossin, F.; Mohammad, E.; da Rocha, S.T.; Schwalb, B.; Syx, L.; Dingli, F.; Loew, D.; et al. The Implication of Early Chromatin Changes in X Chromosome Inactivation. Cell 2019, 176, 182–197.e23. [Google Scholar] [CrossRef]
- Nesterova, T.B.; Wei, G.; Coker, H.; Pintacuda, G.; Bowness, J.S.; Zhang, T.; Almeida, M.; Bloechl, B.; Moindrot, B.; Carter, E.J.; et al. Systematic Allelic Analysis Defines the Interplay of Key Pathways in X Chromosome Inactivation. Nat. Commun. 2019, 10, 3129. [Google Scholar] [CrossRef]
- Sauteraud, R.; Stahl, J.M.; James, J.; Englebright, M.; Chen, F.; Zhan, X.; Carrel, L.; Liu, D.J. Inferring Genes That Escape X-Chromosome Inactivation Reveals Important Contribution of Variable Escape Genes to Sex-Biased Diseases. Genome Res. 2021, 31, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, Z.; Lu, T.; Manolio, T.A.; Paterson, A.D. eXclusionarY: 10 Years Later, Where Are the Sex Chromosomes in GWASs? Am. J. Hum. Genet. 2023, 110, 903–912. [Google Scholar] [CrossRef]
- Plenge, R.M.; Stevenson, R.A.; Lubs, H.A.; Schwartz, C.E.; Willard, H.F. Skewed X-Chromosome Inactivation Is a Common Feature of X-Linked Mental Retardation Disorders. Am. J. Hum. Genet. 2002, 71, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Amos-Landgraf, J.M.; Cottle, A.; Plenge, R.M.; Friez, M.; Schwartz, C.E.; Longshore, J.; Willard, H.F. X Chromosome–Inactivation Patterns of 1,005 Phenotypically Unaffected Females. Am. J. Hum. Genet. 2006, 79, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, C.; Anderson, C.; Beever, C.; Peñaherrera, M.; Brown, C.; Robinson, W. The Dynamics of X-Inactivation Skewing as Women Age. Clin. Genet. 2004, 66, 327–332. [Google Scholar] [CrossRef]
- Werner, J.M.; Ballouz, S.; Hover, J.; Gillis, J. Variability of Cross-Tissue X-Chromosome Inactivation Characterizes Timing of Human Embryonic Lineage Specification Events. Dev. Cell 2022, 57, 1995–2008.e5. [Google Scholar] [CrossRef] [PubMed]
- Phung, T.N.; Olney, K.C.; Pinto, B.J.; Silasi, M.; Perley, L.; O’Bryan, J.; Kliman, H.J.; Wilson, M.A. X Chromosome Inactivation in the Human Placenta Is Patchy and Distinct from Adult Tissues. Hum. Genet. Genom. Adv. 2022, 3, 100121. [Google Scholar] [CrossRef] [PubMed]
- Borel, C.; Ferreira, P.G.; Santoni, F.; Delaneau, O.; Fort, A.; Popadin, K.Y.; Garieri, M.; Falconnet, E.; Ribaux, P.; Guipponi, M.; et al. Biased Allelic Expression in Human Primary Fibroblast Single Cells. Am. J. Hum. Genet. 2015, 96, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Garieri, M.; Stamoulis, G.; Blanc, X.; Falconnet, E.; Ribaux, P.; Borel, C.; Santoni, F.; Antonarakis, S.E. Extensive Cellular Heterogeneity of X Inactivation Revealed by Single-Cell Allele-Specific Expression in Human Fibroblasts. Proc. Natl. Acad. Sci. USA 2018, 115, 13015–13020. [Google Scholar] [CrossRef]
- Lentini, A.; Cheng, H.; Noble, J.C.; Papanicolaou, N.; Coucoravas, C.; Andrews, N.; Deng, Q.; Enge, M.; Reinius, B. Elastic Dosage Compensation by X-Chromosome Upregulation. Nat. Commun. 2022, 13, 1854. [Google Scholar] [CrossRef]
- Bowness, J.S.; Nesterova, T.B.; Wei, G.; Rodermund, L.; Almeida, M.; Coker, H.; Carter, E.J.; Kadaster, A.; Brockdorff, N. Xist-Mediated Silencing Requires Additive Functions of SPEN and Polycomb Together with Differentiation-Dependent Recruitment of SmcHD1. Cell Rep. 2022, 39, 110830. [Google Scholar] [CrossRef] [PubMed]
- Markaki, Y. Xist Nucleates Local Protein Gradients to Propagate Silencing across the X Chromosome. Cell 2021, 52, 6174–6192.e32. [Google Scholar] [CrossRef] [PubMed]
- Pandya-Jones, A.; Markaki, Y.; Serizay, J.; Chitiashvili, T.; Mancia Leon, W.R.; Damianov, A.; Chronis, C.; Papp, B.; Chen, C.-K.; McKee, R.; et al. A Protein Assembly Mediates Xist Localization and Gene Silencing. Nature 2020, 587, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Lahn, B.T.; Page, D.C. Four Evolutionary Strata on the Human X Chromosome. Science 1999, 286, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Ning, Z.; McLellan, A.S.; Ball, M.; Wynne, F.; O’Neill, C.; Mills, W.; Quinn, J.P.; Kleinjan, D.A.; Anney, R.J.; Carmody, R.J.; et al. Regulation of SPRY3 by X Chromosome and PAR2-Linked Promoters in an Autism Susceptibility Region. Hum. Mol. Genet. 2015, 24, 5126–5141. [Google Scholar] [CrossRef]
- De Bonis, M.L.; Cerase, A.; Matarazzo, M.R.; Ferraro, M.; Strazzullo, M.; Hansen, R.S.; Chiurazzi, P.; Neri, G.; D’Esposito, M. Maintenance of X- and Y-Inactivation of the Pseudoautosomal (PAR2) Gene SPRY3 Is Independent from DNA Methylation and Associated to Multiple Layers of Epigenetic Modifications. Hum. Mol. Genet. 2006, 15, 1123–1132. [Google Scholar] [CrossRef]
- Matarazzo, M.R.; De Bonis, M.L.; Gregory, R.I.; Vacca, M.; Hansen, R.S.; Mercadante, G.; D’Urso, M.; Feil, R.; D’Esposito, M. Allelic Inactivation of the Pseudoautosomal Gene SYBL1 Is Controlled by Epigenetic Mechanisms Common to the X and Y Chromosomes. Hum. Mol. Genet. 2002, 11, 3191–3198. [Google Scholar] [CrossRef]
- D’Esposito, M.; Matarazzo, M.R.; Ciccodicola, A.; Strazzullo, M.; Mazzarella, R.; Quaderi, N.A.; Fujiwara, H.; Ko, M.S.H.; Rowe, L.B.; Ricco, A.; et al. Differential Expression Pattern of XqPAR-Linked Genes SYBL1 and IL9R Correlates with the Structure and Evolution of the Region. Hum. Mol. Genet. 1997, 6, 1917–1923. [Google Scholar] [CrossRef]
- Bellott, D.W.; Hughes, J.F.; Skaletsky, H.; Brown, L.G.; Pyntikova, T.; Cho, T.-J.; Koutseva, N.; Zaghlul, S.; Graves, T.; Rock, S.; et al. Mammalian Y Chromosomes Retain Widely Expressed Dosage-Sensitive Regulators. Nature 2014, 508, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Balaton, B.P.; Cotton, A.M.; Brown, C.J. Derivation of Consensus Inactivation Status for X-Linked Genes from Genome-Wide Studies. Biol. Sex Differ. 2015, 6, 35. [Google Scholar] [CrossRef]
- Naqvi, S.; Bellott, D.W.; Lin, K.S.; Page, D.C. Conserved microRNA Targeting Reveals Preexisting Gene Dosage Sensitivities That Shaped Amniote Sex Chromosome Evolution. Genome Res. 2018, 28, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Veerappa, A.M.; Padakannaya, P.; Ramachandra, N.B. Copy Number Variation-Based Polymorphism in a New Pseudoautosomal Region 3 (PAR3) of a Human X-Chromosome-Transposed Region (XTR) in the Y Chromosome. Funct. Integr. Genom. 2013, 13, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Balaton, B.P.; Brown, C.J. Escape Artists of the X Chromosome. Trends Genet. 2016, 32, 348–359. [Google Scholar] [CrossRef]
- Posynick, B.J.; Brown, C.J. Escape From X-Chromosome Inactivation: An Evolutionary Perspective. Front. Cell Dev. Biol. 2019, 7, 241. [Google Scholar] [CrossRef]
- Gravholt, C.H.; Viuff, M.; Just, J.; Sandahl, K.; Brun, S.; van der Velden, J.; Andersen, N.H.; Skakkebaek, A. The Changing Face of Turner Syndrome. Endocr. Rev. 2023, 44, 33–69. [Google Scholar] [CrossRef]
- Fukami, M.; Seki, A.; Ogata, T. SHOX Haploinsufficiency as a Cause of Syndromic and Nonsyndromic Short Stature. Mol. Syndr. 2016, 7, 3–11. [Google Scholar] [CrossRef]
- Ottesen, A.M.; Aksglaede, L.; Garn, I.; Tartaglia, N.; Tassone, F.; Gravholt, C.H.; Bojesen, A.; Sørensen, K.; Jørgensen, N.; Meyts, E.R.-D.; et al. Increased Number of Sex Chromosomes Affects Height in a Nonlinear Fashion: A Study of 305 Patients with Sex Chromosome Aneuploidy. Am. J. Med. Genet. A 2010, 152A, 1206–1212. [Google Scholar] [CrossRef]
- Migeon, B.R. X-Linked Diseases: Susceptible Females. Genet. Med. 2020, 22, 1156–1174. [Google Scholar] [CrossRef]
- Schwartz, C.E.; Louie, R.J.; Toutain, A.; Skinner, C.; Friez, M.J.; Stevenson, R.E. X-Linked Intellectual Disability Update 2022. Am. J. Med. Genet. Part A 2023, 191, 144–159. [Google Scholar] [CrossRef]
- Mo, J.; Liang, H.; Su, C.; Li, P.; Chen, J.; Zhang, B. DDX3X: Structure, Physiologic Functions and Cancer. Mol. Cancer 2021, 20, 38. [Google Scholar] [CrossRef]
- Khan, O.M.; Carvalho, J.; Spencer-Dene, B.; Mitter, R.; Frith, D.; Snijders, A.P.; Wood, S.A.; Behrens, A. The Deubiquitinase USP9X Regulates FBW7 Stability and Suppresses Colorectal Cancer. J. Clin. Investig. 2018, 128, 1326–1337. [Google Scholar] [CrossRef]
- Dunford, A.; Weinstock, D.M.; Savova, V.; Schumacher, S.E.; Cleary, J.P.; Yoda, A.; Sullivan, T.J.; Hess, J.M.; Gimelbrant, A.A.; Beroukhim, R.; et al. Tumor-Suppressor Genes That Escape from X-Inactivation Contribute to Cancer Sex Bias. Nat. Genet. 2017, 49, 10–16. [Google Scholar] [CrossRef]
- Ricketts, C.J.; Linehan, W.M. Gender Specific Mutation Incidence and Survival Associations in Clear Cell Renal Cell Carcinoma (CCRCC). PLoS ONE 2015, 10, e0140257. [Google Scholar] [CrossRef]
- Van der Meulen, J.; Sanghvi, V.; Mavrakis, K.; Durinck, K.; Fang, F.; Matthijssens, F.; Rondou, P.; Rosen, M.; Pieters, T.; Vandenberghe, P.; et al. The H3K27me3 Demethylase UTX Is a Gender-Specific Tumor Suppressor in T-Cell Acute Lymphoblastic Leukemia. Blood 2015, 125, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, T.F.; Gonçalves, A.P.; Fintelman Rodrigues, N.; dos Santos, J.M.; Pimentel, M.M.G.; Santos-Rebouças, C.B. KDM5C Mutational Screening among Males with Intellectual Disability Suggestive of X-Linked Inheritance and Review of the Literature. Eur. J. Med. Genet. 2014, 57, 138–144. [Google Scholar] [CrossRef]
- Lederer, D.; Grisart, B.; Digilio, M.C.; Benoit, V.; Crespin, M.; Ghariani, S.C.; Maystadt, I.; Dallapiccola, B.; Verellen-Dumoulin, C. Deletion of KDM6A, a Histone Demethylase Interacting with MLL2, in Three Patients with Kabuki Syndrome. Am. J. Hum. Genet. 2012, 90, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, P.S.; Arnold, A.P. A Primer on the Use of Mouse Models for Identifying Direct Sex Chromosome Effects That Cause Sex Differences in Non-Gonadal Tissues. Biol. Sex Differ. 2016, 7, 68. [Google Scholar] [CrossRef]
- Link, J.C.; Wiese, C.B.; Chen, X.; Avetisyan, R.; Ronquillo, E.; Ma, F.; Guo, X.; Yao, J.; Allison, M.; Chen, Y.-D.I.; et al. X Chromosome Dosage of Histone Demethylase KDM5C Determines Sex Differences in Adiposity. J. Clin. Investig. 2020, 130, 5688–5702. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Zhong, M.B.; Zhang, L.; Zhang, B.; Cai, D. Sex Differences in Alzheimer’s Disease: Insights from the Multiomics Landscape. Biol. Psychiatry 2022, 91, 61–71. [Google Scholar] [CrossRef]
- Davis, E.J.; Solsberg, C.W.; White, C.C.; Miñones-Moyano, E.; Sirota, M.; Chibnik, L.; Bennett, D.A.; De Jager, P.L.; Yokoyama, J.S.; Dubal, D.B. Sex-Specific Association of the X Chromosome with Cognitive Change and Tau Pathology in Aging and Alzheimer Disease. JAMA Neurol. 2021, 78, 1249–1254. [Google Scholar] [CrossRef]
- Fazazi, M.R.; Ruda, G.F.; Brennan, P.E.; Rangachari, M. The X-Linked Histone Demethylases KDM5C and KDM6A as Regulators of T Cell-Driven Autoimmunity in the Central Nervous System. Brain Res. Bull. 2023, 202, 110748. [Google Scholar] [CrossRef]
- Itoh, Y.; Golden, L.C.; Itoh, N.; Matsukawa, M.A.; Ren, E.; Tse, V.; Arnold, A.P.; Voskuhl, R.R. The X-Linked Histone Demethylase Kdm6a in CD4+ T Lymphocytes Modulates Autoimmunity. J. Clin. Investig. 2019, 129, 3852–3863. [Google Scholar] [CrossRef]
- Wilkinson, N.M.; Chen, H.-C.; Lechner, M.G.; Su, M.A. Sex Differences in Immunity. Annu. Rev. Immunol. 2022, 40, 75–94. [Google Scholar] [CrossRef] [PubMed]
- Scully, E.P.; Haverfield, J.; Ursin, R.L.; Tannenbaum, C.; Klein, S.L. Considering How Biological Sex Impacts Immune Responses and COVID-19 Outcomes. Nat. Rev. Immunol. 2020, 20, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Iwasaki, A. Sex Differences in Immune Responses. Science 2021, 371, 347–348. [Google Scholar] [CrossRef] [PubMed]
- Souyris, M.; Cenac, C.; Azar, P.; Daviaud, D.; Canivet, A.; Grunenwald, S.; Pienkowski, C.; Chaumeil, J.; Mejía, J.E.; Guéry, J.-C. TLR7 Escapes X Chromosome Inactivation in Immune Cells. Sci. Immunol. 2018, 3, eaap8855. [Google Scholar] [CrossRef]
- Yu, B.; Qi, Y.; Li, R.; Shi, Q.; Satpathy, A.T.; Chang, H.Y. B Cell-Specific XIST Complex Enforces X-Inactivation and Restrains Atypical B Cells. Cell 2021, 184, 1790–1803.e17. [Google Scholar] [CrossRef] [PubMed]
- Zito, A.; Roberts, A.L.; Visconti, A.; Rossi, N.; Andres-Ejarque, R.; Nardone, S.; Moustafa, J.S.E.-S.; Falchi, M.; Small, K.S. Escape from X-Inactivation in Twins Exhibits Intra- and Inter-Individual Variability across Tissues and Is Heritable. PLoS Genet. 2023, 19, e1010556. [Google Scholar] [CrossRef]
- Mizuno, Y.; Nonaka, I.; Hirai, S.; Ozawa, E. Reciprocal Expression of Dystrophin and Utrophin in Muscles of Duchenne Muscular Dystrophy Patients, Female DMD-Carriers and Control Subjects. J. Neurol. Sci. 1993, 119, 43–52. [Google Scholar] [CrossRef]
- Carrel, L.; Willard, H.F. Heterogeneous Gene Expression from the Inactive X Chromosome: An X-Linked Gene That Escapes X Inactivation in Some Human Cell Lines but Is Inactivated in Others. Proc. Natl. Acad. Sci. USA 1999, 96, 7364–7369. [Google Scholar] [CrossRef]
- Anderson, C.L.; Brown, C.J. Polymorphic X-Chromosome Inactivation of the Human TIMP1 Gene. Am. J. Hum. Genet. 1999, 65, 699–708. [Google Scholar] [CrossRef]
- Luijk, R.; Wu, H.; Ward-Caviness, C.K.; Hannon, E.; Carnero-Montoro, E.; Min, J.L.; Mandaviya, P.; Müller-Nurasyid, M.; Mei, H.; van der Maarel, S.M.; et al. Autosomal Genetic Variation Is Associated with DNA Methylation in Regions Variably Escaping X-Chromosome Inactivation. Nat. Commun. 2018, 9, 3738. [Google Scholar] [CrossRef] [PubMed]
- Gendrel, A.-V.; Apedaile, A.; Coker, H.; Termanis, A.; Zvetkova, I.; Godwin, J.; Tang, Y.A.; Huntley, D.; Montana, G.; Taylor, S.; et al. Smchd1-Dependent and -Independent Pathways Determine Developmental Dynamics of CpG Island Methylation on the Inactive X Chromosome. Dev. Cell 2012, 23, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Cotton, A.M.; Ge, B.; Light, N.; Adoue, V.; Pastinen, T.; Brown, C.J. Analysis of Expressed SNPs Identifies Variable Extents of Expression from the Human Inactive X Chromosome. Genome Biol. 2013, 14, R122. [Google Scholar] [CrossRef] [PubMed]
- Gylemo, B.; Nestor, C.E. A Whole Organism Map of X-Inactivation Reveals Tissue-Specific Escape as an Exceptionally Rare Event in Humans. bioRxiv 2023. [Google Scholar] [CrossRef]
- Anderson, C.L.; Brown, C.J. Variability of X Chromosome Inactivation: Effect on Levels of TIMP1 RNA and Role of DNA Methylation. Hum. Genet. 2002, 110, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.L.; Brown, C.J. Epigenetic Predisposition to Expression of TIMP1 from the Human Inactive X Chromosome. BMC Genet. 2005, 6, 48. [Google Scholar] [CrossRef]
- Patrat, C.; Ouimette, J.-F.; Rougeulle, C. X Chromosome Inactivation in Human Development. Development 2020, 147, dev183095. [Google Scholar] [CrossRef]
- Brenes, A.J.; Yoshikawa, H.; Bensaddek, D.; Mirauta, B.; Seaton, D.; Hukelmann, J.L.; Jiang, H.; Stegle, O.; Lamond, A.I. Erosion of Human X Chromosome Inactivation Causes Major Remodeling of the iPSC Proteome. Cell Rep. 2021, 35, 109032. [Google Scholar] [CrossRef]
- Bansal, P.; Ahern, D.T.; Kondaveeti, Y.; Qiu, C.W.; Pinter, S.F. Contiguous Erosion of the Inactive X in Human Pluripotency Concludes with Global DNA Hypomethylation. Cell Rep. 2021, 35, 109215. [Google Scholar] [CrossRef]
- Linder, D.; Gartler, S.M. Glucose-6-Phosphate Dehydrogenase Mosaicism: Utilization as a Cell Marker in the Study of Leiomyomas. Science 1965, 150, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Migeon, B.R.; Axelman, J.; Beggs, A.H. Effect of Ageing on Reactivation of the Human X-Linked HPRT Locus. Nature 1988, 335, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Käseberg, S.; Bertin, M.; Menon, R.; Gabassi, E.; Todorov, H.; Frank, S.; Brennenstuhl, H.; Lohrer, B.; Winter, J.; Krummeich, J.; et al. Dynamic X-Chromosomal Reactivation Enhances Female Brain Resilience. bioRxiv 2023. [Google Scholar] [CrossRef]
- Pyfrom, S.; Paneru, B.; Knox, J.J.; Cancro, M.P.; Posso, S.; Buckner, J.H.; Anguera, M.C. The Dynamic Epigenetic Regulation of the Inactive X Chromosome in Healthy Human B Cells Is Dysregulated in Lupus Patients. Proc. Natl. Acad. Sci. USA 2021, 118, e2024624118. [Google Scholar] [CrossRef] [PubMed]
- Syrett, C.M.; Paneru, B.; Sandoval-Heglund, D.; Wang, J.; Banerjee, S.; Sindhava, V.; Behrens, E.M.; Atchison, M.; Anguera, M.C. Altered X-Chromosome Inactivation in T Cells May Promote Sex-Biased Autoimmune Diseases. JCI Insight 2019, 4, e126751. [Google Scholar] [CrossRef]
- Syrett, C.M.; Sierra, I.; Beethem, Z.T.; Dubin, A.H.; Anguera, M.C. Loss of Epigenetic Modifications on the Inactive X Chromosome and Sex-Biased Gene Expression Profiles in B Cells from NZB/W F1 Mice with Lupus-like Disease. J. Autoimmun. 2020, 107, 102357. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Syrett, C.M.; Kramer, M.C.; Basu, A.; Atchison, M.L.; Anguera, M.C. Unusual Maintenance of X Chromosome Inactivation Predisposes Female Lymphocytes for Increased Expression from the Inactive X. Proc. Natl. Acad. Sci. USA 2016, 113, E2029–E2038. [Google Scholar] [CrossRef]
- Lee, H.J.; Gopalappa, R.; Sunwoo, H.; Choi, S.-W.; Ramakrishna, S.; Lee, J.T.; Kim, H.H.; Nam, J.-W. En Bloc and Segmental Deletions of Human XIST Reveal X Chromosome Inactivation-Involving RNA Elements. Nucleic Acids Res. 2019, 47, 3875–3887. [Google Scholar] [CrossRef]
- Richart, L.; Picod-Chedotel, M.-L.; Wassef, M.; Macario, M.; Aflaki, S.; Salvador, M.A.; Héry, T.; Dauphin, A.; Wicinski, J.; Chevrier, V.; et al. XIST Loss Impairs Mammary Stem Cell Differentiation and Increases Tumorigenicity through Mediator Hyperactivation. Cell 2022, 185, 2164–2183.e25. [Google Scholar] [CrossRef]
- Westervelt, N.; Yoest, A.; Sayed, S.; Von Zimmerman, M.; Kaps, K.; Chadwick, B.P. Deletion of the XIST Promoter from the Human Inactive X Chromosome Compromises Polycomb Heterochromatin Maintenance. Chromosoma 2021, 130, 177–197. [Google Scholar] [CrossRef]
- Grigoryan, A.; Pospiech, J.; Krämer, S.; Lipka, D.; Liehr, T.; Geiger, H.; Kimura, H.; Mulaw, M.A.; Florian, M.C. Attrition of X Chromosome Inactivation in Aged Hematopoietic Stem Cells. Stem Cell Rep. 2021, 16, 708–716. [Google Scholar] [CrossRef]
- Riggs, A.D. X Inactivation, Differentiation, and DNA Methylation. Cytogenet. Cell Genet. 1975, 14, 9–25. [Google Scholar] [CrossRef]
- Horvath, S.; Raj, K. DNA Methylation-Based Biomarkers and the Epigenetic Clock Theory of Ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Bell, C.G.; Lowe, R.; Adams, P.D.; Baccarelli, A.A.; Beck, S.; Bell, J.T.; Christensen, B.C.; Gladyshev, V.N.; Heijmans, B.T.; Horvath, S.; et al. DNA Methylation Aging Clocks: Challenges and Recommendations. Genome Biol. 2019, 20, 249. [Google Scholar] [CrossRef] [PubMed]
- McCartney, D.L.; Zhang, F.; Hillary, R.F.; Zhang, Q.; Stevenson, A.J.; Walker, R.M.; Bermingham, M.L.; Boutin, T.; Morris, S.W.; Campbell, A.; et al. An Epigenome-Wide Association Study of Sex-Specific Chronological Ageing. Genome Med. 2019, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Lund, J.B.; Christensen, K.; Baumbach, J.; Mengel-From, J.; Kruse, T.; Li, W.; Mohammadnejad, A.; Pattie, A.; Marioni, R.E.; et al. Exploratory Analysis of Age and Sex Dependent DNA Methylation Patterns on the X-Chromosome in Whole Blood Samples. Genome Med. 2020, 12, 39. [Google Scholar] [CrossRef] [PubMed]
- Kananen, L.; Marttila, S. Ageing-Associated Changes in DNA Methylation in X and Y Chromosomes. Epigenet. Chromatin 2021, 14, 33. [Google Scholar] [CrossRef]
- Liu, Y.; Sinke, L.; Jonkman, T.H.; Slieker, R.C.; BIOS Consortium; van Zwet, E.W.; Daxinger, L.; Heijmans, B.T. The Inactive X Chromosome Accumulates Widespread Epigenetic Variability with Age. Clin. Epigenet. 2023, 15, 135. [Google Scholar] [CrossRef]
- Barr, M.L.; Bertram, E.G. A Morphological Distinction between Neurones of the Male and Female, and the Behaviour of the Nucleolar Satellite during Accelerated Nucleoprotein Synthesis. Nature 1949, 163, 676–677. [Google Scholar] [CrossRef]
- Ohno, S.; Hauschka, T.S. Allocycly of the X-Chromosome in Tumors and Normal Tissues. Cancer Res. 1960, 20, 541–545. [Google Scholar]
- Lyon, M. Gene Action in the X-chromosome of the Mouse (Mus musculus L.). Nature 1961, 190, 372–373. [Google Scholar] [CrossRef]
- Bischoff, A.; Albers, J.; Kharboush, I.; Stelzer, E.; Cremer, T.; Cremer, C. Differences of Size and Shape of Active and Inactive X-Chromosome Domains in Human Amniotic Fluid Cell Nuclei. Microsc. Res. Tech. 1993, 25, 68–77. [Google Scholar] [CrossRef]
- Eils, R.; Dietzel, S.; Bertin, E.; Schröck, E.; Speicher, M.R.; Ried, T.; Robert-Nicoud, M.; Cremer, C.; Cremer, T. Three-Dimensional Reconstruction of Painted Human Interphase Chromosomes: Active and Inactive X Chromosome Territories Have Similar Volumes but Differ in Shape and Surface Structure. J. Cell Biol. 1996, 135, 1427–1440. [Google Scholar] [CrossRef]
- Teller, K.; Illner, D.; Thamm, S.; Casas-Delucchi, C.S.; Versteeg, R.; Indemans, M.; Cremer, T.; Cremer, M. A Top-down Analysis of Xa- and Xi-Territories Reveals Differences of Higher Order Structure at ≥ 20 Mb Genomic Length Scales. Nucleus 2011, 2, 465–477. [Google Scholar] [CrossRef]
- Jégu, T.; Aeby, E.; Lee, J.T. The X Chromosome in Space. Nat. Rev. Genet. 2017, 18, 377–389. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological Domains in Mammalian Genomes Identified by Analysis of Chromatin Interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef]
- Nora, E.P.; Lajoie, B.R.; Schulz, E.G.; Giorgetti, L.; Okamoto, I.; Servant, N.; Piolot, T.; van Berkum, N.L.; Meisig, J.; Sedat, J.; et al. Spatial Partitioning of the Regulatory Landscape of the X-Inactivation Center. Nature 2012, 485, 381–385. [Google Scholar] [CrossRef]
- Horakova, A.H.; Moseley, S.C.; McLaughlin, C.R.; Tremblay, D.C.; Chadwick, B.P. The Macrosatellite DXZ4 Mediates CTCF-Dependent Long-Range Intrachromosomal Interactions on the Human Inactive X Chromosome. Hum. Mol. Genet. 2012, 21, 4367–4377. [Google Scholar] [CrossRef]
- Darrow, E.M.; Huntley, M.H.; Dudchenko, O.; Stamenova, E.K.; Durand, N.C.; Sun, Z.; Huang, S.-C.; Sanborn, A.L.; Machol, I.; Shamim, M.; et al. Deletion of DXZ4 on the Human Inactive X Chromosome Alters Higher-Order Genome Architecture. Proc. Natl. Acad. Sci. USA 2016, 113, E4504–E4512. [Google Scholar] [CrossRef]
- Loda, A.; Collombet, S.; Heard, E. Gene Regulation in Time and Space during X-Chromosome Inactivation. Nat. Rev. Mol. Cell Biol. 2022, 23, 231–249. [Google Scholar] [CrossRef]
- Giorgetti, L.; Lajoie, B.R.; Carter, A.C.; Attia, M.; Zhan, Y.; Xu, J.; Chen, C.J.; Kaplan, N.; Chang, H.Y.; Heard, E.; et al. Structural Organization of the Inactive X Chromosome in the Mouse. Nature 2016, 535, 575–579. [Google Scholar] [CrossRef]
- Froberg, J.E.; Pinter, S.F.; Kriz, A.J.; Jégu, T.; Lee, J.T. Megadomains and Superloops Form Dynamically but Are Dispensable for X-Chromosome Inactivation and Gene Escape. Nat. Commun. 2018, 9, 5004. [Google Scholar] [CrossRef]
- Marks, H.; Kerstens, H.H.D.; Barakat, T.S.; Splinter, E.; Dirks, R.A.M.; van Mierlo, G.; Joshi, O.; Wang, S.-Y.; Babak, T.; Albers, C.A.; et al. Dynamics of Gene Silencing during X Inactivation Using Allele-Specific RNA-Seq. Genome Biol. 2015, 16, 149. [Google Scholar] [CrossRef]
- Collombet, S.; Ranisavljevic, N.; Nagano, T.; Varnai, C.; Shisode, T.; Leung, W.; Piolot, T.; Galupa, R.; Borensztein, M.; Servant, N.; et al. Parental-to-Embryo Switch of Chromosome Organization in Early Embryogenesis. Nature 2020, 580, 142–146. [Google Scholar] [CrossRef]
- Chaumeil, J. A Novel Role for Xist RNA in the Formation of a Repressive Nuclear Compartment into Which Genes Are Recruited When Silenced. Genes. Dev. 2006, 20, 2223–2237. [Google Scholar] [CrossRef]
- Rego, A.; Sinclair, P.B.; Tao, W.; Kireev, I.; Belmont, A.S. The Facultative Heterochromatin of the Inactive X Chromosome Has a Distinctive Condensed Ultrastructure. J. Cell Sci. 2008, 121, 1119–1127. [Google Scholar] [CrossRef]
- Smeets, D.; Markaki, Y.; Schmid, V.J.; Kraus, F.; Tattermusch, A.; Cerase, A.; Sterr, M.; Fiedler, S.; Demmerle, J.; Popken, J.; et al. Three-Dimensional Super-Resolution Microscopy of the Inactive X Chromosome Territory Reveals a Collapse of Its Active Nuclear Compartment Harboring Distinct Xist RNA Foci. Epigenet. Chromatin 2014, 7, 8. [Google Scholar] [CrossRef]
- Collombet, S.; Rall, I.; Dugast-Darzacq, C.; Heckert, A.; Halavatyi, A.; Le Saux, A.; Dailey, G.; Darzacq, X.; Heard, E. RNA Polymerase II Depletion from the Inactive X Chromosome Territory Is Not Mediated by Physical Compartmentalization. Nat. Struct. Mol. Biol. 2023, 30, 1216–1223. [Google Scholar] [CrossRef]
- Cerase, A.; Armaos, A.; Neumayer, C.; Avner, P.; Guttman, M.; Tartaglia, G.G. Phase Separation Drives X-Chromosome Inactivation: A Hypothesis. Nat. Struct. Mol. Biol. 2019, 26, 331–334. [Google Scholar] [CrossRef]
- Cerase, A.; Calabrese, J.M.; Tartaglia, G.G. Phase Separation Drives X-Chromosome Inactivation. Nat. Struct. Mol. Biol. 2022, 29, 183–185. [Google Scholar] [CrossRef]
- Cerase, A.; Smeets, D.; Tang, Y.A.; Gdula, M.; Kraus, F.; Spivakov, M.; Moindrot, B.; Leleu, M.; Tattermusch, A.; Demmerle, J.; et al. Spatial Separation of Xist RNA and Polycomb Proteins Revealed by Superresolution Microscopy. Proc. Natl. Acad. Sci. USA 2014, 111, 2235–2240. [Google Scholar] [CrossRef]
- Sunwoo, H.; Wu, J.Y.; Lee, J.T. The Xist RNA-PRC2 Complex at 20-Nm Resolution Reveals a Low Xist Stoichiometry and Suggests a Hit-and-Run Mechanism in Mouse Cells. Proc. Natl. Acad. Sci. USA 2015, 112, E4216–E4225. [Google Scholar] [CrossRef]
- Jachowicz, J.W.; Strehle, M.; Banerjee, A.K.; Blanco, M.R.; Thai, J.; Guttman, M. Xist Spatially Amplifies SHARP/SPEN Recruitment to Balance Chromosome-Wide Silencing and Specificity to the X Chromosome. Nat. Struct. Mol. Biol. 2022, 29, 239–249. [Google Scholar] [CrossRef]
- Dixon-McDougall, T.; Brown, C.J. Independent Domains for Recruitment of PRC1 and PRC2 by Human XIST. PLoS Genet. 2021, 17, e1009123. [Google Scholar] [CrossRef]
- Li, N.; Carrel, L. Escape from X Chromosome Inactivation Is an Intrinsic Property of the Jarid1c Locus. Proc. Natl. Acad. Sci. USA 2008, 105, 17055–17060. [Google Scholar] [CrossRef]
- Peeters, S.B.; Korecki, A.J.; Simpson, E.M.; Brown, C.J. Human Cis-Acting Elements Regulating Escape from X-Chromosome Inactivation Function in Mouse. Hum. Mol. Genet. 2018, 27, 1252–1262. [Google Scholar] [CrossRef]
- Yang, C.; McLeod, A.J.; Cotton, A.M.; de Leeuw, C.N.; Laprise, S.; Banks, K.G.; Simpson, E.M.; Brown, C.J. Targeting of >1.5 Mb of Human DNA into the Mouse X Chromosome Reveals Presence of Cis-Acting Regulators of Epigenetic Silencing. Genetics 2012, 192, 1281–1293. [Google Scholar] [CrossRef]
- Ciavatta, D.; Kalantry, S.; Magnuson, T.; Smithies, O. A DNA Insulator Prevents Repression of a Targeted X-Linked Transgene but Not Its Random or Imprinted X Inactivation. Proc. Natl. Acad. Sci. USA 2006, 103, 9958–9963. [Google Scholar] [CrossRef]
- Peeters, S.B.; Korecki, A.J.; Baldry, S.E.L.; Yang, C.; Tosefsky, K.; Balaton, B.P.; Simpson, E.M.; Brown, C.J. How Do Genes That Escape from X-Chromosome Inactivation Contribute to Turner Syndrome? Am. J. Med. Genet. Part C Semin. Med. Genet. 2019, 181, 84–91. [Google Scholar] [CrossRef]
- Lyon, M.F. X-Chromosome Inactivation: A Repeat Hypothesis. Cytogenet. Genome Res. 1998, 80, 133–137. [Google Scholar] [CrossRef]
- Cotton, A.M.; Chen, C.-Y.; Lam, L.L.; Wasserman, W.W.; Kobor, M.S.; Brown, C.J. Spread of X-Chromosome Inactivation into Autosomal Sequences: Role for DNA Elements, Chromatin Features and Chromosomal Domains. Hum. Mol. Genet. 2014, 23, 1211–1223. [Google Scholar] [CrossRef] [PubMed]
- Bala Tannan, N.; Brahmachary, M.; Garg, P.; Borel, C.; Alnefaie, R.; Watson, C.T.; Thomas, N.S.; Sharp, A.J. DNA Methylation Profiling in X;Autosome Translocations Supports a Role for L1 Repeats in the Spread of X Chromosome Inactivation. Hum. Mol. Genet. 2014, 23, 1224–1236. [Google Scholar] [CrossRef] [PubMed]
- Loda, A.; Brandsma, J.H.; Vassilev, I.; Servant, N.; Loos, F.; Amirnasr, A.; Splinter, E.; Barillot, E.; Poot, R.A.; Heard, E.; et al. Genetic and Epigenetic Features Direct Differential Efficiency of Xist-Mediated Silencing at X-Chromosomal and Autosomal Locations. Nat. Commun. 2017, 8, 690. [Google Scholar] [CrossRef]
- Polak, P.; Domany, E. Alu Elements Contain Many Binding Sites for Transcription Factors and May Play a Role in Regulation of Developmental Processes. BMC Genom. 2006, 7, 133. [Google Scholar] [CrossRef] [PubMed]
- Bourque, G.; Leong, B.; Vega, V.B.; Chen, X.; Lee, Y.L.; Srinivasan, K.G.; Chew, J.-L.; Ruan, Y.; Wei, C.-L.; Ng, H.H.; et al. Evolution of the Mammalian Transcription Factor Binding Repertoire via Transposable Elements. Genome Res. 2008, 18, 1752–1762. [Google Scholar] [CrossRef]
- Schmidt, D.; Schwalie, P.C.; Wilson, M.D.; Ballester, B.; Gonçalves, Â.; Kutter, C.; Brown, G.D.; Marshall, A.; Flicek, P.; Odom, D.T. Waves of Retrotransposon Expansion Remodel Genome Organization and CTCF Binding in Multiple Mammalian Lineages. Cell 2012, 148, 335–348. [Google Scholar] [CrossRef]
- Filippova, G.N.; Cheng, M.K.; Moore, J.M.; Truong, J.-P.; Hu, Y.J.; Nguyen, D.K.; Tsuchiya, K.D.; Disteche, C.M. Boundaries between Chromosomal Domains of X Inactivation and Escape Bind CTCF and Lack CpG Methylation during Early Development. Dev. Cell 2005, 8, 31–42. [Google Scholar] [CrossRef]
- Fang, H.; Tronco, A.R.; Bonora, G.; Nguyen, T.; Thakur, J.; Berletch, J.B.; Filippova, G.N.; Henikoff, S.; Shendure, J.; Noble, W.S.; et al. CTCF-Mediated Insulation and Chromatin Environment Modulate Car5b Escape from X Inactivation. bioRxiv 2023. [Google Scholar] [CrossRef]
- Horvath, L.M.; Li, N.; Carrel, L. Deletion of an X-Inactivation Boundary Disrupts Adjacent Gene Silencing. PLoS Genet. 2013, 9, e1003952. [Google Scholar] [CrossRef]
- Dickson, J.; Gowher, H.; Strogantsev, R.; Gaszner, M.; Hair, A.; Felsenfeld, G.; West, A.G. VEZF1 Elements Mediate Protection from DNA Methylation. PLoS Genet. 2010, 6, e1000804. [Google Scholar] [CrossRef] [PubMed]
- Peeters, S.; Leung, T.; Fornes, O.; Farkas, R.A.; Wasserman, W.W.; Brown, C.J. Refining the Genomic Determinants Underlying Escape from X-Chromosome Inactivation. NAR Genom. Bioinform. 2023, 5, lqad052. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Guan, X.; Xie, B.; Xu, C.; Niu, J.; Tang, X.; Li, C.H.; Colecraft, H.M.; Jaenisch, R.; Liu, X.S. Multiplex Epigenome Editing of MECP2 to Rescue Rett Syndrome Neurons. Sci. Transl. Med. 2023, 15, eadd4666. [Google Scholar] [CrossRef] [PubMed]
- Berletch, J.B.; Ma, W.; Yang, F.; Shendure, J.; Noble, W.S.; Disteche, C.M.; Deng, X. Escape from X Inactivation Varies in Mouse Tissues. PLoS Genet. 2015, 11, e1005079. [Google Scholar] [CrossRef]
- Rücklé, C.; Körtel, N.; Basilicata, M.F.; Busch, A.; Zhou, Y.; Hoch-Kraft, P.; Tretow, K.; Kielisch, F.; Bertin, M.; Pradhan, M.; et al. RNA Stability Controlled by m6A Methylation Contributes to X-to-Autosome Dosage Compensation in Mammals. Nat. Struct. Mol. Biol. 2023, 30, 1207–1215. [Google Scholar] [CrossRef]
- Ohno, S. Sex Chromosomes and Sex-Linked Genes; Springer: Berlin/Heidelberg, Germany, 1967; ISBN 978-3-642-88180-0. [Google Scholar]
- Mugford, J.W.; Starmer, J.; Williams, R.L., Jr.; Calabrese, J.M.; Mieczkowski, P.; Yee, D.; Magnuson, T. Evidence for Local Regulatory Control of Escape from Imprinted X Chromosome Inactivation. Genetics 2014, 197, 715–723. [Google Scholar] [CrossRef]
- Johansson, J.; Lidéus, S.; Höijer, I.; Ameur, A.; Gudmundsson, S.; Annerén, G.; Bondeson, M.-L.; Wilbe, M. A Novel Quantitative Targeted Analysis of X-Chromosome Inactivation (XCI) Using Nanopore Sequencing. Sci. Rep. 2023, 13, 12856. [Google Scholar] [CrossRef]
- Carrel, L.; Cottle, A.A.; Goglin, K.C.; Willard, H.F. A First-Generation X-Inactivation Profile of the Human X Chromosome. Proc. Natl. Acad. Sci. USA 1999, 96, 14440–14444. [Google Scholar] [CrossRef]
- Bailey, J.A.; Carrel, L.; Chakravarti, A.; Eichler, E.E. Molecular Evidence for a Relationship between LINE-1 Elements and X Chromosome Inactivation: The Lyon Repeat Hypothesis. Proc. Natl. Acad. Sci. USA 2000, 97, 6634–6639. [Google Scholar] [CrossRef]
- Wang, Z.; Willard, H.F.; Mukherjee, S.; Furey, T.S. Evidence of Influence of Genomic DNA Sequence on Human X Chromosome Inactivation. PLoS Comput. Biol. 2006, 2, e113. [Google Scholar] [CrossRef]
- Carrel, L.; Park, C.; Tyekucheva, S.; Dunn, J.; Chiaromonte, F.; Makova, K.D. Genomic Environment Predicts Expression Patterns on the Human Inactive X Chromosome. PLoS Genet. 2006, 2, e151. [Google Scholar] [CrossRef]
- Calabrese, J.M.; Sun, W.; Song, L.; Mugford, J.W.; Williams, L.; Yee, D.; Starmer, J.; Mieczkowski, P.; Crawford, G.E.; Magnuson, T. Site-Specific Silencing of Regulatory Elements as a Mechanism of X Inactivation. Cell 2012, 151, 951–963. [Google Scholar] [CrossRef] [PubMed]
- de Andrade e Sousa, L.B.; Jonkers, I.; Syx, L.; Dunkel, I.; Chaumeil, J.; Picard, C.; Foret, B.; Chen, C.-J.; Lis, J.T.; Heard, E.; et al. Kinetics of Xist-Induced Gene Silencing Can Be Predicted from Combinations of Epigenetic and Genomic Features. Genome Res. 2019, 29, 1087–1099. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Shi, W.; Balaton, B.P.; Matthews, A.M.; Li, Y.; Arenillas, D.J.; Mathelier, A.; Itoh, M.; Kawaji, H.; Lassmann, T.; et al. YY1 Binding Association with Sex-Biased Transcription Revealed through X-Linked Transcript Levels and Allelic Binding Analyses. Sci. Rep. 2016, 6, 37324. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peeters, S.B.; Posynick, B.J.; Brown, C.J. Out of the Silence: Insights into How Genes Escape X-Chromosome Inactivation. Epigenomes 2023, 7, 29. https://doi.org/10.3390/epigenomes7040029
Peeters SB, Posynick BJ, Brown CJ. Out of the Silence: Insights into How Genes Escape X-Chromosome Inactivation. Epigenomes. 2023; 7(4):29. https://doi.org/10.3390/epigenomes7040029
Chicago/Turabian StylePeeters, Samantha B., Bronwyn J. Posynick, and Carolyn J. Brown. 2023. "Out of the Silence: Insights into How Genes Escape X-Chromosome Inactivation" Epigenomes 7, no. 4: 29. https://doi.org/10.3390/epigenomes7040029