

IndiSPENsable for X Chromosome Inactivation and Gene Silencing


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
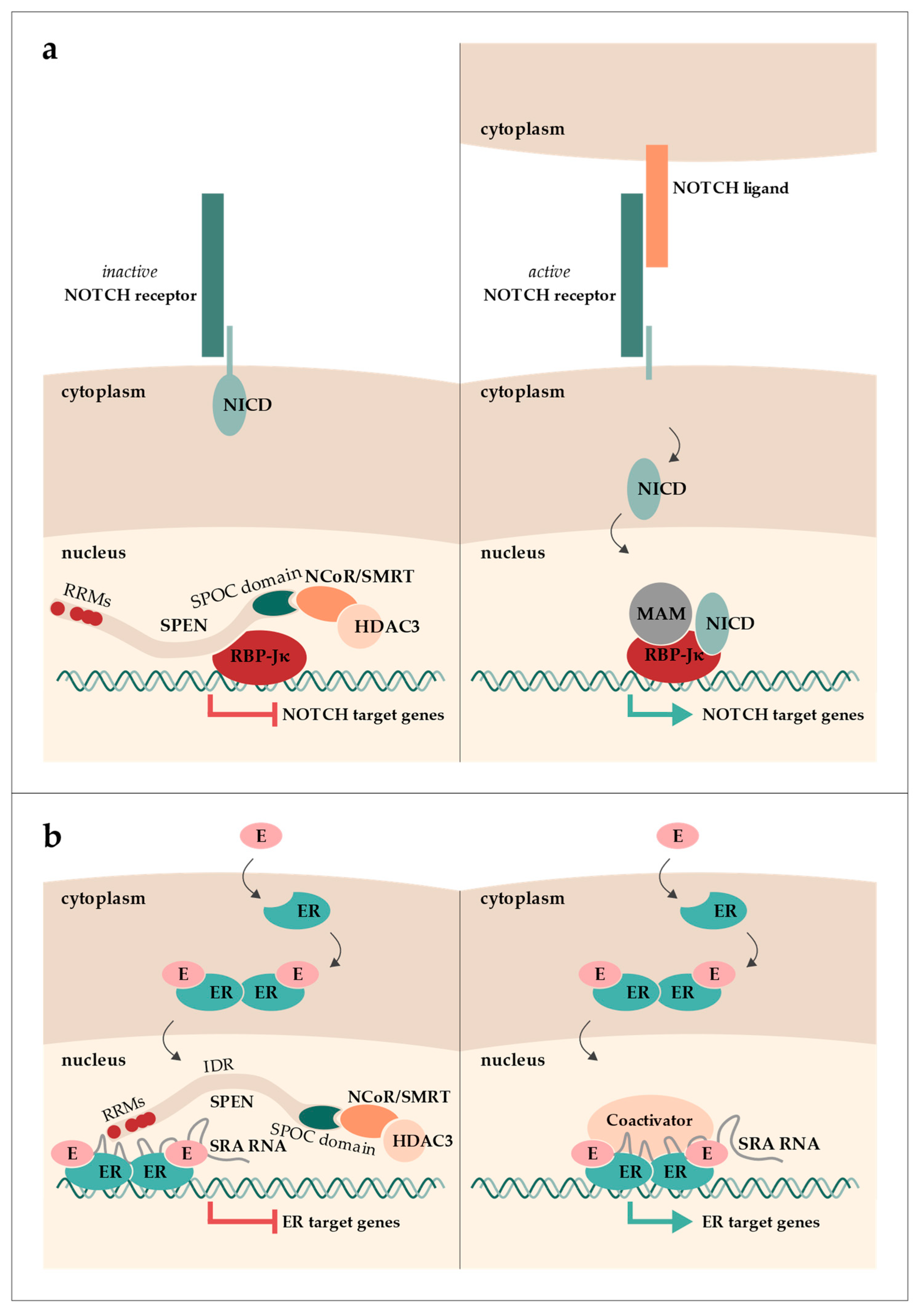
:1. The Discovery of SPEN as a Transcriptional Regulator
2. XIST RNA and X Chromosome Inactivation in Humans and Mice
3. Function of SPEN in Mouse X Chromosome Inactivation
4. Function of SPEN in Human X Chromosome Inactivation
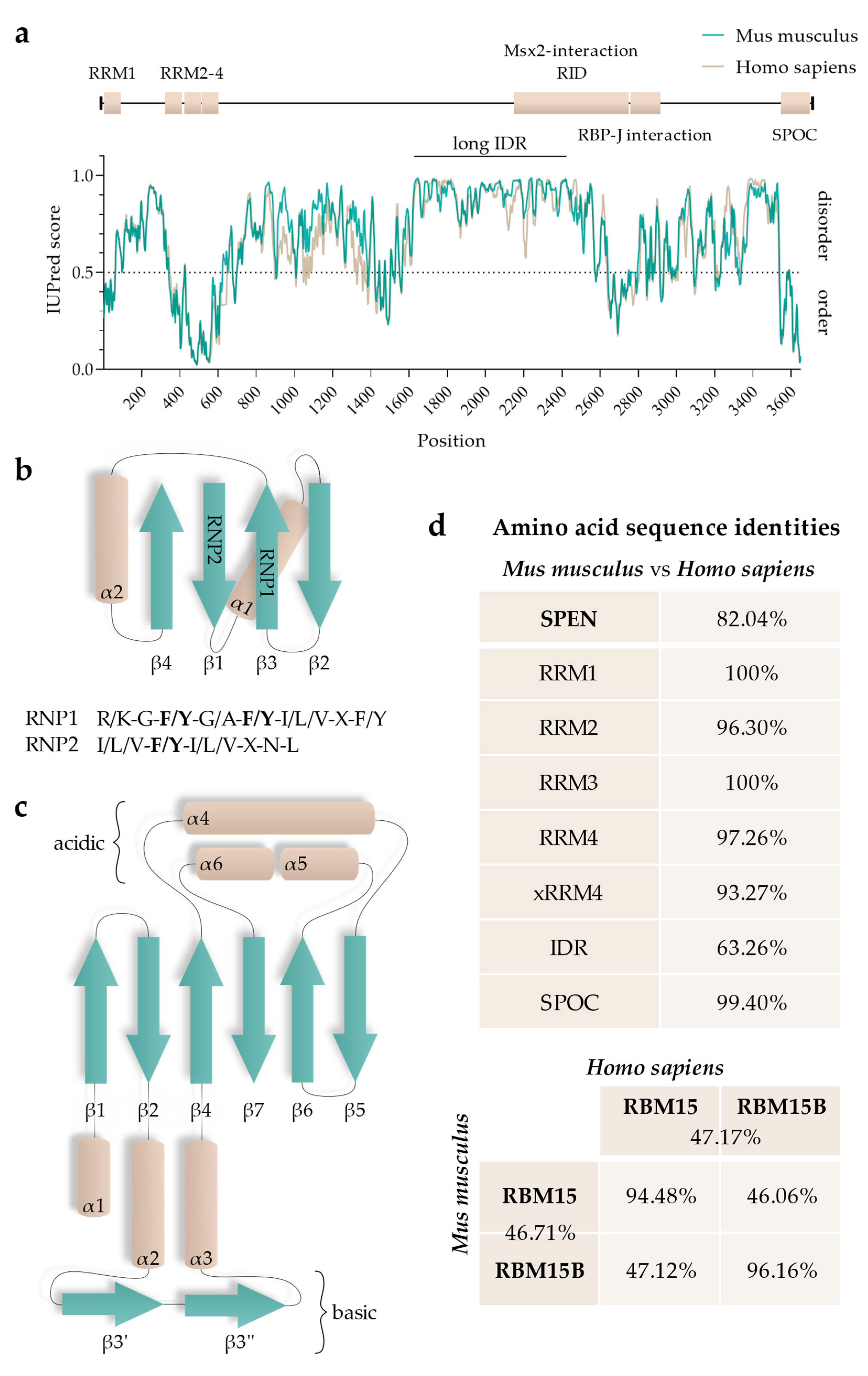
5. Functional Regions of SPEN
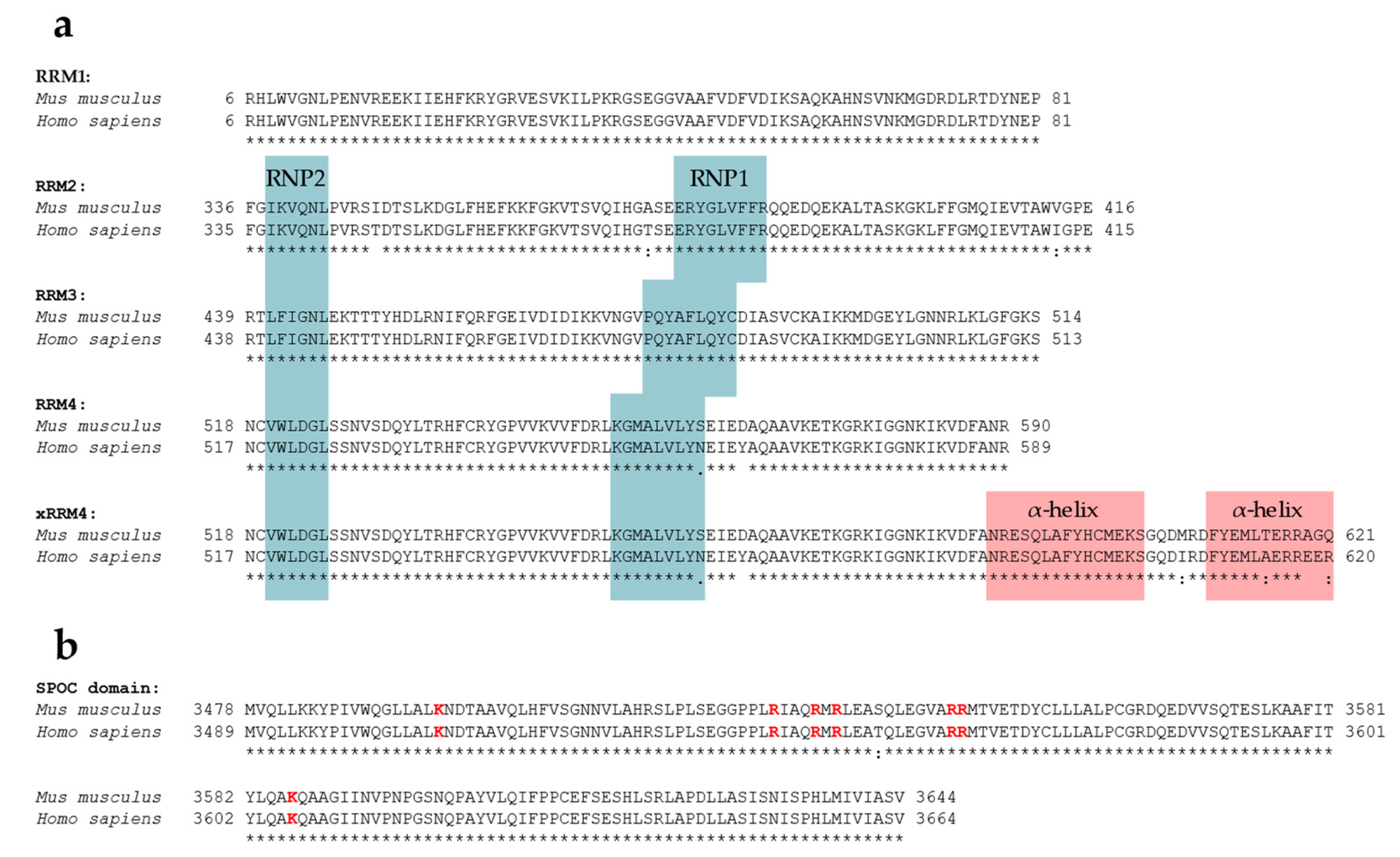
5.1. RNA-Recognition Motifs
5.2. SPOC Domain
5.3. Intrinsically Disordered Regions
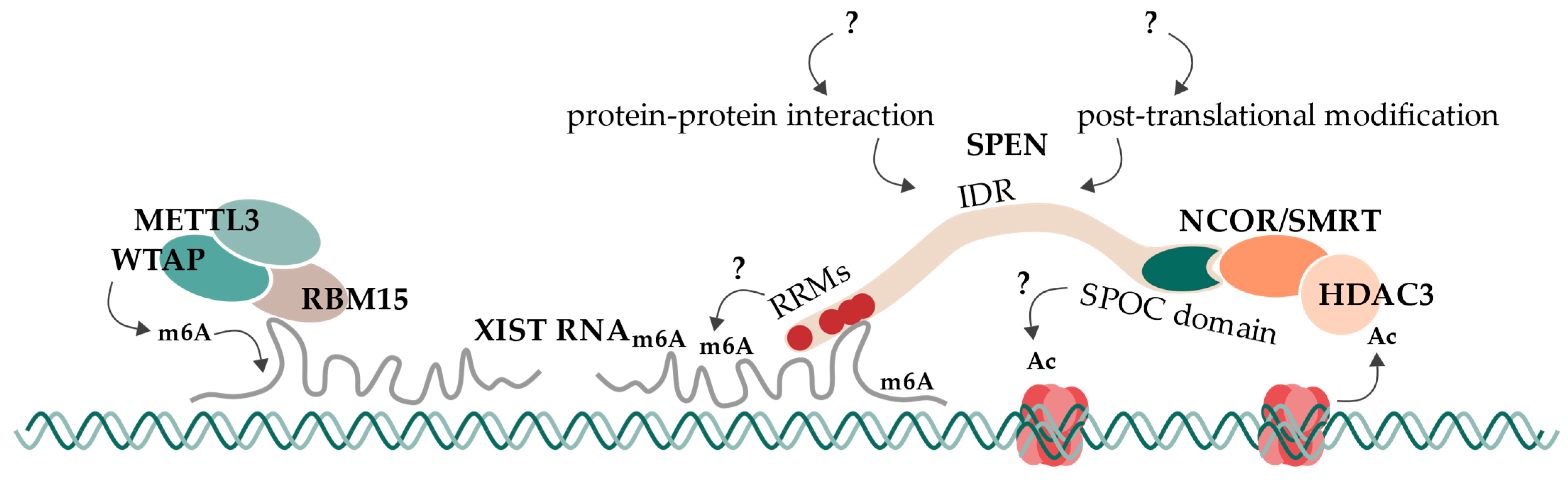
6. Concluding Remarks and Future Directions
6.1. In Vivo and In Vitro Models
6.2. RNA-Recognition Motifs
6.3. SPOC Domain
6.4. Intrinsically Disordered Regions
6.5. Proposed Model and Open Questions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kolodziej, P.A.; Jan, L.Y.; Jan, Y.N. Mutations That Affect the Length, Fasciculation, or Ventral Orientation of Specific Sensory Axons in the Drosophila Embryo. Neuron 1995, 15, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Mace, K.; Tugores, A. The Product of the Split Ends Gene Is Required for the Maintenance of Positional Information during Drosophiladevelopment. BMC Dev. Biol. 2004, 4, 15. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.G.; Dreyfuss, G. Conserved Structures and Diversity of Functions of RNA-Binding Proteins. Science 1994, 265, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Rebay, I.; Chen, F.; Hsiao, F.; Kolodziej, P.A.; Kuang, B.H.; Laverty, T.; Suh, C.; Voas, M.; Williams, A.; Rubin, G.M. A Genetic Screen for Novel Components of the Ras/Mitogen-Activated Protein Kinase Signaling Pathway That Interact with the Yan Gene of Drosophila Identifies Split Ends, a New RNA Recognition Motif-Containing Protein. Genetics 2000, 154, 695–712. [Google Scholar] [CrossRef]
- Kuang, B.; Wu, S.C.; Shin, Y.; Luo, L.; Kolodziej, P. Split Ends Encodes Large Nuclear Proteins That Regulate Neuronal Cell Fate and Axon Extension in the Drosophila Embryo. Development 2000, 127, 1517–1529. [Google Scholar] [CrossRef]
- Newberry, E.P.; Latifi, T.; Towler, D.A. The RRM Domain of MINT, a Novel Msx2 Binding Protein, Recognizes and Regulates the Rat Osteocalcin Promoter. Biochemistry 1999, 38, 10678–10690. [Google Scholar] [CrossRef]
- Shi, Y.; Downes, M.; Xie, W.; Kao, H.Y.; Ordentlich, P.; Tsai, C.C.; Hon, M.; Evans, R.M. Sharp, an Inducible Cofactor That Integrates Nuclear Receptor Repression and Activation. Genes Dev. 2001, 15, 1140–1151. [Google Scholar] [CrossRef]
- Guenther, M.G.; Barak, O.; Lazar, M.A. The SMRT and N-CoR Corepressors Are Activating Cofactors for Histone Deacetylase 3. Mol. Cell. Biol. 2001, 21, 6091–6101. [Google Scholar] [CrossRef]
- You, S.-H.; Lim, H.-W.; Sun, Z.; Broache, M.; Won, K.-J.; Lazar, M.A. Nuclear Receptor Co-Repressors Are Required for the Histone-Deacetylase Activity of HDAC3 in Vivo. Nat. Struct. Mol. Biol. 2013, 20, 182–187. [Google Scholar] [CrossRef]
- Oswald, F.; Kostezka, U.; Astrahantseff, K.; Bourteele, S.; Dillinger, K.; Zechner, U.; Ludwig, L.; Wilda, M.; Hameister, H.; Knöchel, W.; et al. SHARP Is a Novel Component of the Notch/RBP-Jkappa Signalling Pathway. EMBO J. 2002, 21, 5417–5426. [Google Scholar] [CrossRef]
- Oswald, F.; Kovall, R.A. CSL-Associated Corepressor and Coactivator Complexes. In Molecular Mechanisms of Notch Signaling; Borggrefe, T., Giaimo, B.D., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2018; ISBN 978-3-319-89512-3. [Google Scholar]
- Lanz, R.B.; McKenna, N.J.; Onate, S.A.; Albrecht, U.; Wong, J.; Tsai, S.Y.; Tsai, M.J.; O’Malley, B.W. A Steroid Receptor Coactivator, SRA, Functions as an RNA and Is Present in an SRC-1 Complex. Cell 1999, 97, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Lanz, R.B.; Razani, B.; Goldberg, A.D.; O’Malley, B.W. Distinct RNA Motifs Are Important for Coactivation of Steroid Hormone Receptors by Steroid Receptor RNA Activator (SRA). Proc. Natl. Acad. Sci. USA 2002, 99, 16081–16086. [Google Scholar] [CrossRef] [PubMed]
- Lanz, R.B.; Chua, S.S.; Barron, N.; Söder, B.M.; DeMayo, F.; O’Malley, B.W. Steroid Receptor RNA Activator Stimulates Proliferation as Well as Apoptosis in Vivo. Mol. Cell. Biol. 2003, 23, 7163–7176. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Zhang, Q.C.; da Rocha, S.T.; Flynn, R.A.; Bharadwaj, M.; Calabrese, J.M.; Magnuson, T.; Heard, E.; Chang, H.Y. Systematic Discovery of Xist RNA Binding Proteins. Cell 2015, 161, 404–416. [Google Scholar] [CrossRef] [PubMed]
- McHugh, C.A.; Chen, C.-K.; Chow, A.; Surka, C.F.; Tran, C.; McDonel, P.; Pandya-Jones, A.; Blanco, M.; Burghard, C.; Moradian, A.; et al. The Xist LncRNA Interacts Directly with SHARP to Silence Transcription through HDAC3. Nature 2015, 521, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Minajigi, A.; Froberg, J.; Wei, C.; Sunwoo, H.; Kesner, B.; Colognori, D.; Lessing, D.; Payer, B.; Boukhali, M.; Haas, W.; et al. A Comprehensive Xist Interactome Reveals Cohesin Repulsion and an RNA-Directed Chromosome Conformation. Science 2015, 349, aab2276. [Google Scholar] [CrossRef] [PubMed]
- Moindrot, B.; Cerase, A.; Coker, H.; Masui, O.; Grijzenhout, A.; Pintacuda, G.; Schermelleh, L.; Nesterova, T.B.; Brockdorff, N. A Pooled ShRNA Screen Identifies Rbm15, Spen, and Wtap as Factors Required for Xist RNA-Mediated Silencing. Cell Rep. 2015, 12, 562–572. [Google Scholar] [CrossRef]
- Monfort, A.; Di Minin, G.; Postlmayr, A.; Freimann, R.; Arieti, F.; Thore, S.; Wutz, A. Identification of Spen as a Crucial Factor for Xist Function through Forward Genetic Screening in Haploid Embryonic Stem Cells. Cell Rep. 2015, 12, 554–561. [Google Scholar] [CrossRef]
- Lyon, M.F. Gene Action in the X-Chromosome of the Mouse (Mus musculus L.). Nature 1961, 190, 372–373. [Google Scholar] [CrossRef]
- Carrel, L.; Willard, H.F. X-Inactivation Profile Reveals Extensive Variability in X-Linked Gene Expression in Females. Nature 2005, 434, 400–404. [Google Scholar] [CrossRef]
- Penny, G.D.; Kay, G.F.; Sheardown, S.A.; Rastan, S.; Brockdorff, N. Requirement for Xist in X Chromosome Inactivation. Nature 1996, 379, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Chaumeil, J.; Le Baccon, P.; Wutz, A.; Heard, E. A Novel Role for Xist RNA in the Formation of a Repressive Nuclear Compartment into Which Genes Are Recruited When Silenced. Genes Dev. 2006, 20, 2223–2237. [Google Scholar] [CrossRef]
- Żylicz, J.J.; Bousard, A.; Žumer, K.; Dossin, F.; Mohammad, E.; da Rocha, S.T.; Schwalb, B.; Syx, L.; Dingli, F.; Loew, D.; et al. The Implication of Early Chromatin Changes in X Chromosome Inactivation. Cell 2019, 176, 182–197. [Google Scholar] [CrossRef]
- Loda, A.; Collombet, S.; Heard, E. Gene Regulation in Time and Space during X-Chromosome Inactivation. Nat. Rev. Mol. Cell Biol. 2022, 23, 231–249. [Google Scholar] [CrossRef] [PubMed]
- Wutz, A.; Rasmussen, T.P.; Jaenisch, R. Chromosomal Silencing and Localization Are Mediated by Different Domains of Xist RNA. Nat. Genet. 2002, 30, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Kohlmaier, A.; Savarese, F.; Lachner, M.; Martens, J.; Jenuwein, T.; Wutz, A. A Chromosomal Memory Triggered by Xist Regulates Histone Methylation in X Inactivation. PLoS Biol. 2004, 2, E171. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-K.; Blanco, M.; Jackson, C.; Aznauryan, E.; Ollikainen, N.; Surka, C.; Chow, A.; Cerase, A.; McDonel, P.; Guttman, M. Xist Recruits the X Chromosome to the Nuclear Lamina to Enable Chromosome-Wide Silencing. Science 2016, 354, 468–472. [Google Scholar] [CrossRef]
- Patil, D.P.; Chen, C.-K.; Pickering, B.F.; Chow, A.; Jackson, C.; Guttman, M.; Jaffrey, S.R. M(6)A RNA Methylation Promotes XIST-Mediated Transcriptional Repression. Nature 2016, 537, 369–373. [Google Scholar] [CrossRef]
- Dossin, F.; Pinheiro, I.; Żylicz, J.J.; Roensch, J.; Collombet, S.; Le Saux, A.; Chelmicki, T.; Attia, M.; Kapoor, V.; Zhan, Y.; et al. SPEN Integrates Transcriptional and Epigenetic Control of X-Inactivation. Nature 2020, 578, 455–460. [Google Scholar] [CrossRef]
- Wiellette, E.L.; Harding, K.W.; Mace, K.A.; Ronshaugen, M.R.; Wang, F.Y.; McGinnis, W. Spen Encodes an RNP Motif Protein That Interacts with Hox Pathways to Repress the Development of Head-like Sclerites in the Drosophila Trunk. Development 1999, 126, 5373–5385. [Google Scholar] [CrossRef]
- Schoeftner, S.; Sengupta, A.K.; Kubicek, S.; Mechtler, K.; Spahn, L.; Koseki, H.; Jenuwein, T.; Wutz, A. Recruitment of PRC1 Function at the Initiation of X Inactivation Independent of PRC2 and Silencing. EMBO J. 2006, 25, 3110–3122. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.; Pintacuda, G.; Masui, O.; Koseki, Y.; Gdula, M.; Cerase, A.; Brown, D.; Mould, A.; Innocent, C.; Nakayama, M.; et al. PCGF3/5-PRC1 Initiates Polycomb Recruitment in X Chromosome Inactivation. Science 2017, 356, 1081–1084. [Google Scholar] [CrossRef] [PubMed]
- Bousard, A.; Raposo, A.C.; Żylicz, J.J.; Picard, C.; Pires, V.B.; Qi, Y.; Gil, C.; Syx, L.; Chang, H.Y.; Heard, E.; et al. The Role of Xist-Mediated Polycomb Recruitment in the Initiation of X-Chromosome Inactivation. EMBO Rep. 2019, 20, e48019. [Google Scholar] [CrossRef]
- Dixon-McDougall, T.; Brown, C.J. Independent Domains for Recruitment of PRC1 and PRC2 by Human XIST. PLoS Genet. 2021, 17, e1009123. [Google Scholar] [CrossRef]
- Li, J.; Ming, Z.; Yang, L.; Wang, T.; Liu, G.; Ma, Q. Long Noncoding RNA XIST: Mechanisms for X Chromosome Inactivation, Roles in Sex-Biased Diseases, and Therapeutic Opportunities. Genes Dis. 2022, 9, 1478–1492. [Google Scholar] [CrossRef] [PubMed]
- Huynh, K.D.; Lee, J.T. Inheritance of a Pre-Inactivated Paternal X Chromosome in Early Mouse Embryos. Nature 2003, 426, 857–862. [Google Scholar] [CrossRef]
- Okamoto, I.; Otte, A.P.; Allis, C.D.; Reinberg, D.; Heard, E. Epigenetic Dynamics of Imprinted X Inactivation during Early Mouse Development. Science 2004, 303, 644–649. [Google Scholar] [CrossRef]
- Mak, W.; Nesterova, T.B.; de Napoles, M.; Appanah, R.; Yamanaka, S.; Otte, A.P.; Brockdorff, N. Reactivation of the Paternal X Chromosome in Early Mouse Embryos. Science 2004, 303, 666–669. [Google Scholar] [CrossRef]
- Takagi, N.; Sugawara, O.; Sasaki, M. Regional and Temporal Changes in the Pattern of X-Chromosome Replication during the Early Post-Implantation Development of the Female Mouse. Chromosoma 1982, 85, 275–286. [Google Scholar] [CrossRef]
- Van den Berg, I.M.; Laven, J.S.E.; Stevens, M.; Jonkers, I.; Galjaard, R.-J.; Gribnau, J.; van Doorninck, J.H. X Chromosome Inactivation Is Initiated in Human Preimplantation Embryos. Am. J. Hum. Genet. 2009, 84, 771–779. [Google Scholar] [CrossRef]
- Vallot, C.; Patrat, C.; Collier, A.J.; Huret, C.; Casanova, M.; Liyakat Ali, T.M.; Tosolini, M.; Frydman, N.; Heard, E.; Rugg-Gunn, P.J.; et al. XACT Noncoding RNA Competes with XIST in the Control of X Chromosome Activity during Human Early Development. Cell Stem Cell 2017, 20, 102–111. [Google Scholar] [CrossRef]
- Moreira de Mello, J.C.; de Araújo, E.S.S.; Stabellini, R.; Fraga, A.M.; de Souza, J.E.S.; Sumita, D.R.; Camargo, A.A.; Pereira, L.V. Random X Inactivation and Extensive Mosaicism in Human Placenta Revealed by Analysis of Allele-Specific Gene Expression along the X Chromosome. PLoS ONE 2010, 5, e10947. [Google Scholar] [CrossRef]
- Phung, T.N.; Olney, K.C.; Pinto, B.J.; Silasi, M.; Perley, L.; O’Bryan, J.; Kliman, H.J.; Wilson, M.A. X Chromosome Inactivation in the Human Placenta Is Patchy and Distinct from Adult Tissues. Hum. Genet. Genom. Adv. 2022, 3, 100121. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, I.; Patrat, C.; Thépot, D.; Peynot, N.; Fauque, P.; Daniel, N.; Diabangouaya, P.; Wolf, J.-P.; Renard, J.-P.; Duranthon, V.; et al. Eutherian Mammals Use Diverse Strategies to Initiate X-Chromosome Inactivation during Development. Nature 2011, 472, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Petropoulos, S.; Edsgärd, D.; Reinius, B.; Deng, Q.; Panula, S.P.; Codeluppi, S.; Reyes, A.P.; Linnarsson, S.; Sandberg, R.; Lanner, F. Single-Cell RNA-Seq Reveals Lineage and X Chromosome Dynamics in Human Preimplantation Embryos. Cell 2016, 165, 1012–1026. [Google Scholar] [CrossRef]
- Patel, S.; Bonora, G.; Sahakyan, A.; Kim, R.; Chronis, C.; Langerman, J.; Fitz-Gibbon, S.; Rubbi, L.; Skelton, R.J.P.; Ardehali, R.; et al. Human Embryonic Stem Cells Do Not Change Their X Inactivation Status during Differentiation. Cell Rep. 2017, 18, 54–67. [Google Scholar] [CrossRef]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic Stem Cell Lines Derived from Human Blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef]
- Aflatoonian, B.; Ruban, L.; Shamsuddin, S.; Baker, D.; Andrews, P.; Moore, H. Generation of Sheffield (Shef) Human Embryonic Stem Cell Lines Using a Microdrop Culture System. In Vitro Cell. Dev. Biol. Anim. 2010, 46, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Lengner, C.J.; Gimelbrant, A.A.; Erwin, J.A.; Cheng, A.W.; Guenther, M.G.; Welstead, G.G.; Alagappan, R.; Frampton, G.M.; Xu, P.; Muffat, J.; et al. Derivation of Pre-X Inactivation Human Embryonic Stem Cells under Physiological Oxygen Concentrations. Cell 2010, 141, 872–883. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, L.M.; Hall, L.; Batten, J.L.; Young, H.; Pardasani, D.; Baetge, E.E.; Lawrence, J.; Carpenter, M.K. X-Inactivation Status Varies in Human Embryonic Stem Cell Lines. Stem Cells 2005, 23, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Ouyang, Q.; Leng, L.; Hu, L.; Cheng, D.; Tan, Y.; Lu, G.; Lin, G. The Dynamic Changes of X Chromosome Inactivation during Early Culture of Human Embryonic Stem Cells. Stem Cell Res. 2016, 17, 84–92. [Google Scholar] [CrossRef]
- Collier, A.J.; Rugg-Gunn, P.J. Identifying Human Naïve Pluripotent Stem Cells—Evaluating State-Specific Reporter Lines and Cell-Surface Markers. Bioessays 2018, 40, e1700239. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, M.K.; Rosler, E.; Rao, M.S. Characterization and Differentiation of Human Embryonic Stem Cells. Cloning Stem Cells 2003, 5, 79–88. [Google Scholar] [CrossRef]
- Takashima, Y.; Guo, G.; Loos, R.; Nichols, J.; Ficz, G.; Krueger, F.; Oxley, D.; Santos, F.; Clarke, J.; Mansfield, W.; et al. Resetting Transcription Factor Control Circuitry toward Ground-State Pluripotency in Human. Cell 2014, 158, 1254–1269. [Google Scholar] [CrossRef] [PubMed]
- Theunissen, T.W.; Powell, B.E.; Wang, H.; Mitalipova, M.; Faddah, D.A.; Reddy, J.; Fan, Z.P.; Maetzel, D.; Ganz, K.; Shi, L.; et al. Systematic Identification of Culture Conditions for Induction and Maintenance of Naive Human Pluripotency. Cell Stem Cell 2014, 15, 471–487. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; von Meyenn, F.; Rostovskaya, M.; Clarke, J.; Dietmann, S.; Baker, D.; Sahakyan, A.; Myers, S.; Bertone, P.; Reik, W.; et al. Epigenetic Resetting of Human Pluripotency. Development 2017, 144, 2748–2763. [Google Scholar] [CrossRef] [PubMed]
- Theunissen, T.W.; Friedli, M.; He, Y.; Planet, E.; O’Neil, R.C.; Markoulaki, S.; Pontis, J.; Wang, H.; Iouranova, A.; Imbeault, M.; et al. Molecular Criteria for Defining the Naive Human Pluripotent State. Cell Stem Cell 2016, 19, 502–515. [Google Scholar] [CrossRef] [PubMed]
- Bredenkamp, N.; Yang, J.; Clarke, J.; Stirparo, G.G.; von Meyenn, F.; Dietmann, S.; Baker, D.; Drummond, R.; Ren, Y.; Li, D.; et al. Wnt Inhibition Facilitates RNA-Mediated Reprogramming of Human Somatic Cells to Naive Pluripotency. Stem Cell Rep. 2019, 13, 1083–1098. [Google Scholar] [CrossRef]
- Sahakyan, A.; Kim, R.; Chronis, C.; Sabri, S.; Bonora, G.; Theunissen, T.W.; Kuoy, E.; Langerman, J.; Clark, A.T.; Jaenisch, R.; et al. Human Naive Pluripotent Stem Cells Model X Chromosome Dampening and X Inactivation. Cell Stem Cell 2017, 20, 87–101. [Google Scholar] [CrossRef]
- Khan, S.A.; Park, K.-M.; Fischer, L.A.; Dong, C.; Lungjangwa, T.; Jimenez, M.; Casalena, D.; Chew, B.; Dietmann, S.; Auld, D.S.; et al. Probing the Signaling Requirements for Naive Human Pluripotency by High-Throughput Chemical Screening. Cell Rep. 2021, 35, 109233. [Google Scholar] [CrossRef]
- Molè, M.A.; Coorens, T.H.H.; Shahbazi, M.N.; Weberling, A.; Weatherbee, B.A.T.; Gantner, C.W.; Sancho-Serra, C.; Richardson, L.; Drinkwater, A.; Syed, N.; et al. A Single Cell Characterisation of Human Embryogenesis Identifies Pluripotency Transitions and Putative Anterior Hypoblast Centre. Nat. Commun. 2021, 12, 3679. [Google Scholar] [CrossRef] [PubMed]
- Brockdorff, N.; Bowness, J.S.; Wei, G. Progress toward Understanding Chromosome Silencing by Xist RNA. Genes Dev. 2020, 34, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Mikami, S.; Kanaba, T.; Takizawa, N.; Kobayashi, A.; Maesaki, R.; Fujiwara, T.; Ito, Y.; Mishima, M. Structural Insights into the Recruitment of SMRT by the Corepressor SHARP under Phosphorylative Regulation. Structure 2014, 22, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Luikenhuis, S.; Wutz, A.; Jaenisch, R. Antisense Transcription through the Xist Locus Mediates Tsix Function in Embryonic Stem Cells. Mol. Cell. Biol. 2001, 21, 8512–8520. [Google Scholar] [CrossRef]
- Stavropoulos, N.; Lu, N.; Lee, J.T. A Functional Role for Tsix Transcription in Blocking Xist RNA Accumulation but Not in X-Chromosome Choice. Proc. Natl. Acad. Sci. USA 2001, 98, 10232–10237. [Google Scholar] [CrossRef] [PubMed]
- Sado, T.; Hoki, Y.; Sasaki, H. Tsix Silences Xist through Modification of Chromatin Structure. Dev. Cell 2005, 9, 159–165. [Google Scholar] [CrossRef]
- Mutzel, V.; Okamoto, I.; Dunkel, I.; Saitou, M.; Giorgetti, L.; Heard, E.; Schulz, E.G. A Symmetric Toggle Switch Explains the Onset of Random X Inactivation in Different Mammals. Nat. Struct. Mol. Biol. 2019, 26, 350–360. [Google Scholar] [CrossRef]
- Robert-Finestra, T.; Tan, B.F.; Mira-Bontenbal, H.; Timmers, E.; Gontan, C.; Merzouk, S.; Giaimo, B.D.; Dossin, F.; van IJcken, W.F.J.; Martens, J.W.M.; et al. SPEN Is Required for Xist Upregulation during Initiation of X Chromosome Inactivation. Nat. Commun. 2021, 12, 7000. [Google Scholar] [CrossRef]
- Jachowicz, J.W.; Strehle, M.; Banerjee, A.K.; Blanco, M.R.; Thai, J.; Guttman, M. Xist Spatially Amplifies SHARP/SPEN Recruitment to Balance Chromosome-Wide Silencing and Specificity to the X Chromosome. Nat. Struct. Mol. Biol. 2022, 29, 239–249. [Google Scholar] [CrossRef]
- Kuroda, K.; Han, H.; Tani, S.; Tanigaki, K.; Tun, T.; Furukawa, T.; Taniguchi, Y.; Kurooka, H.; Hamada, Y.; Toyokuni, S.; et al. Regulation of Marginal Zone B Cell Development by MINT, a Suppressor of Notch/RBP-J Signaling Pathway. Immunity 2003, 18, 301–312. [Google Scholar] [CrossRef]
- Marahrens, Y.; Panning, B.; Dausman, J.; Strauss, W.; Jaenisch, R. Xist-Deficient Mice Are Defective in Dosage Compensation but Not Spermatogenesis. Genes Dev. 1997, 11, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Sheng, L.; Miao, H.; Saunders, T.L.; MacDougald, O.A.; Koenig, R.J.; Xu, B. SRA Gene Knockout Protects against Diet-Induced Obesity and Improves Glucose Tolerance. J. Biol. Chem. 2014, 289, 13000–13009. [Google Scholar] [CrossRef] [PubMed]
- Satokata, I.; Ma, L.; Ohshima, H.; Bei, M.; Woo, I.; Nishizawa, K.; Maeda, T.; Takano, Y.; Uchiyama, M.; Heaney, S.; et al. Msx2 Deficiency in Mice Causes Pleiotropic Defects in Bone Growth and Ectodermal Organ Formation. Nat. Genet. 2000, 24, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Jepsen, K.; Hermanson, O.; Onami, T.M.; Gleiberman, A.S.; Lunyak, V.; McEvilly, R.J.; Kurokawa, R.; Kumar, V.; Liu, F.; Seto, E.; et al. Combinatorial Roles of the Nuclear Receptor Corepressor in Transcription and Development. Cell 2000, 102, 753–763. [Google Scholar] [CrossRef]
- Jepsen, K.; Gleiberman, A.S.; Shi, C.; Simon, D.I.; Rosenfeld, M.G. Cooperative Regulation in Development by SMRT and FOXP1. Genes Dev. 2008, 22, 740–745. [Google Scholar] [CrossRef]
- Montgomery, R.L.; Potthoff, M.J.; Haberland, M.; Qi, X.; Matsuzaki, S.; Humphries, K.M.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Maintenance of Cardiac Energy Metabolism by Histone Deacetylase 3 in Mice. J. Clin. Investig. 2008, 118, 3588–3597. [Google Scholar] [CrossRef]
- Moumné, L.; Campbell, K.; Howland, D.; Ouyang, Y.; Bates, G.P. Genetic Knock-Down of Hdac3 Does Not Modify Disease-Related Phenotypes in a Mouse Model of Huntington’s Disease. PLoS ONE 2012, 7, e31080. [Google Scholar] [CrossRef]
- Swiatek, P.J.; Gridley, T. Perinatal Lethality and Defects in Hindbrain Development in Mice Homozygous for a Targeted Mutation of the Zinc Finger Gene Krox20. Genes Dev. 1993, 7, 2071–2084. [Google Scholar] [CrossRef]
- Conlon, R.A.; Reaume, A.G.; Rossant, J. Notch1 Is Required for the Coordinate Segmentation of Somites. Development 1995, 121, 1533–1545. [Google Scholar] [CrossRef]
- Krebs, L.T.; Xue, Y.; Norton, C.R.; Shutter, J.R.; Maguire, M.; Sundberg, J.P.; Gallahan, D.; Closson, V.; Kitajewski, J.; Callahan, R.; et al. Notch Signaling Is Essential for Vascular Morphogenesis in Mice. Genes Dev. 2000, 14, 1343–1352. [Google Scholar] [CrossRef]
- Hamada, Y.; Kadokawa, Y.; Okabe, M.; Ikawa, M.; Coleman, J.R.; Tsujimoto, Y. Mutation in Ankyrin Repeats of the Mouse Notch2 Gene Induces Early Embryonic Lethality. Development 1999, 126, 3415–3424. [Google Scholar] [CrossRef] [PubMed]
- Domenga, V.; Fardoux, P.; Lacombe, P.; Monet, M.; Maciazek, J.; Krebs, L.T.; Klonjkowski, B.; Berrou, E.; Mericskay, M.; Li, Z.; et al. Notch3 Is Required for Arterial Identity and Maturation of Vascular Smooth Muscle Cells. Genes Dev. 2004, 18, 2730–2735. [Google Scholar] [CrossRef] [PubMed]
- Migeon, B.R.; Lee, C.H.; Chowdhury, A.K.; Carpenter, H. Species Differences in TSIX/Tsix Reveal the Roles of These Genes in X-Chromosome Inactivation. Am. J. Hum. Genet. 2002, 71, 286–293. [Google Scholar] [CrossRef] [PubMed]
- Boroviak, T.; Stirparo, G.G.; Dietmann, S.; Hernando-Herraez, I.; Mohammed, H.; Reik, W.; Smith, A.; Sasaki, E.; Nichols, J.; Bertone, P. Single Cell Transcriptome Analysis of Human, Marmoset and Mouse Embryos Reveals Common and Divergent Features of Preimplantation Development. Development 2018, 145, dev167833. [Google Scholar] [CrossRef]
- Radio, F.C.; Pang, K.; Ciolfi, A.; Levy, M.A.; Hernández-García, A.; Pedace, L.; Pantaleoni, F.; Liu, Z.; de Boer, E.; Jackson, A.; et al. SPEN Haploinsufficiency Causes a Neurodevelopmental Disorder Overlapping Proximal 1p36 Deletion Syndrome with an Episignature of X Chromosomes in Females. Am. J. Hum. Genet. 2021, 108, 502–516. [Google Scholar] [CrossRef]
- Bandziulis, R.J.; Swanson, M.S.; Dreyfuss, G. RNA-Binding Proteins as Developmental Regulators. Genes Dev. 1989, 3, 431–437. [Google Scholar] [CrossRef]
- Nagai, K.; Oubridge, C.; Jessen, T.H.; Li, J.; Evans, P.R. Crystal Structure of the RNA-Binding Domain of the U1 Small Nuclear Ribonucleoprotein A. Nature 1990, 348, 515–520. [Google Scholar] [CrossRef]
- Swanson, M.S.; Nakagawa, T.Y.; LeVan, K.; Dreyfuss, G. Primary Structure of Human Nuclear Ribonucleoprotein Particle C Proteins: Conservation of Sequence and Domain Structures in Heterogeneous Nuclear RNA, MRNA, and Pre-RRNA-Binding Proteins. Mol. Cell. Biol. 1987, 7, 1731–1739. [Google Scholar] [CrossRef]
- Dreyfuss, G.; Swanson, M.S.; Piñol-Roma, S. Heterogeneous Nuclear Ribonucleoprotein Particles and the Pathway of MRNA Formation. Trends Biochem. Sci. 1988, 13, 86–91. [Google Scholar] [CrossRef]
- Arieti, F.; Gabus, C.; Tambalo, M.; Huet, T.; Round, A.; Thore, S. The Crystal Structure of the Split End Protein SHARP Adds a New Layer of Complexity to Proteins Containing RNA Recognition Motifs. Nucleic Acids Res. 2014, 42, 6742–6752. [Google Scholar] [CrossRef]
- Singh, M.; Wang, Z.; Koo, B.-K.; Patel, A.; Cascio, D.; Collins, K.; Feigon, J. Structural Basis for Telomerase RNA Recognition and RNP Assembly by the Holoenzyme La Family Protein P65. Mol. Cell 2012, 47, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Avis, J.M.; Allain, F.H.; Howe, P.W.; Varani, G.; Nagai, K.; Neuhaus, D. Solution Structure of the N-Terminal RNP Domain of U1A Protein: The Role of C-Terminal Residues in Structure Stability and RNA Binding. J. Mol. Biol. 1996, 257, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Carter, A.C.; Xu, J.; Nakamoto, M.Y.; Wei, Y.; Zarnegar, B.J.; Shi, Q.; Broughton, J.P.; Ransom, R.C.; Salhotra, A.; Nagaraja, S.D.; et al. Spen Links RNA-Mediated Endogenous Retrovirus Silencing and X Chromosome Inactivation. eLife 2020, 9, e54508. [Google Scholar] [CrossRef] [PubMed]
- Bose, E.; Mayes, C.; Ellis, L.; Baker, C.; Tambalotti, S.; Xiong, S.; Sarpong, Y.P.O.; Shalaby, M.; Barry, L.; Lewis, F.; et al. Characterization of RBM15 Protein Binding with Long Noncoding RNAs. bioRxiv 2023. [Google Scholar] [CrossRef]
- Mercher, T.; Coniat, M.B.-L.; Monni, R.; Mauchauffé, M.; Khac, F.N.; Gressin, L.; Mugneret, F.; Leblanc, T.; Dastugue, N.; Berger, R.; et al. Involvement of a Human Gene Related to the Drosophila Spen Gene in the Recurrent t(1;22) Translocation of Acute Megakaryocytic Leukemia. Proc. Natl. Acad. Sci. USA 2001, 98, 5776–5779. [Google Scholar] [CrossRef]
- Lu, Z.; Zhang, Q.C.; Lee, B.; Flynn, R.A.; Smith, M.A.; Robinson, J.T.; Davidovich, C.; Gooding, A.R.; Goodrich, K.J.; Mattick, J.S.; et al. RNA Duplex Map in Living Cells Reveals Higher-Order Transcriptome Structure. Cell 2016, 165, 1267–1279. [Google Scholar] [CrossRef]
- Elisaphenko, E.A.; Kolesnikov, N.N.; Shevchenko, A.I.; Rogozin, I.B.; Nesterova, T.B.; Brockdorff, N.; Zakian, S.M. A Dual Origin of the Xist Gene from a Protein-Coding Gene and a Set of Transposable Elements. PLoS ONE 2008, 3, e2521. [Google Scholar] [CrossRef]
- Nesterova, T.B.; Wei, G.; Coker, H.; Pintacuda, G.; Bowness, J.S.; Zhang, T.; Almeida, M.; Bloechl, B.; Moindrot, B.; Carter, E.J.; et al. Systematic Allelic Analysis Defines the Interplay of Key Pathways in X Chromosome Inactivation. Nat. Commun. 2019, 10, 3129. [Google Scholar] [CrossRef]
- Xiao, W.; Adhikari, S.; Dahal, U.; Chen, Y.-S.; Hao, Y.-J.; Sun, B.-F.; Sun, H.-Y.; Li, A.; Ping, X.-L.; Lai, W.-Y.; et al. Nuclear m(6)A Reader YTHDC1 Regulates MRNA Splicing. Mol. Cell 2016, 61, 507–519. [Google Scholar] [CrossRef]
- Ariyoshi, M.; Schwabe, J.W.R. A Conserved Structural Motif Reveals the Essential Transcriptional Repression Function of Spen Proteins and Their Role in Developmental Signaling. Genes Dev. 2003, 17, 1909–1920. [Google Scholar] [CrossRef]
- Appel, L.-M.; Franke, V.; Benedum, J.; Grishkovskaya, I.; Strobl, X.; Polyansky, A.; Ammann, G.; Platzer, S.; Neudolt, A.; Wunder, A.; et al. The SPOC Domain Is a Phosphoserine Binding Module That Bridges Transcription Machinery with Co- and Post-Transcriptional Regulators. Nat. Commun. 2023, 14, 166. [Google Scholar] [CrossRef] [PubMed]
- Oswald, F.; Rodriguez, P.; Giaimo, B.D.; Antonello, Z.A.; Mira, L.; Mittler, G.; Thiel, V.N.; Collins, K.J.; Tabaja, N.; Cizelsky, W.; et al. A Phospho-Dependent Mechanism Involving NCoR and KMT2D Controls a Permissive Chromatin State at Notch Target Genes. Nucleic Acids Res. 2016, 44, 4703–4720. [Google Scholar] [CrossRef] [PubMed]
- Bornelöv, S.; Reynolds, N.; Xenophontos, M.; Gharbi, S.; Johnstone, E.; Floyd, R.; Ralser, M.; Signolet, J.; Loos, R.; Dietmann, S.; et al. The Nucleosome Remodeling and Deacetylation Complex Modulates Chromatin Structure at Sites of Active Transcription to Fine-Tune Gene Expression. Mol. Cell 2018, 71, 56–72. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Sun, B.K.; Erwin, J.A.; Song, J.-J.; Lee, J.T. Polycomb Proteins Targeted by a Short Repeat RNA to the Mouse X Chromosome. Science 2008, 322, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Siomi, H.; Dreyfuss, G. RNA-Binding Proteins as Regulators of Gene Expression. Curr. Opin. Genet. Dev. 1997, 7, 345–353. [Google Scholar] [CrossRef]
- Uversky, V.N. The Intrinsic Disorder Alphabet. III. Dual Personality of Serine. Intrinsically Disord. Proteins 2015, 3, e1027032. [Google Scholar] [CrossRef]
- Pejaver, V.; Hsu, W.-L.; Xin, F.; Dunker, A.K.; Uversky, V.N.; Radivojac, P. The Structural and Functional Signatures of Proteins That Undergo Multiple Events of Post-Translational Modification. Protein Sci. 2014, 23, 1077–1093. [Google Scholar] [CrossRef]
- Johnson, L.N.; Lewis, R.J. Structural Basis for Control by Phosphorylation. Chem. Rev. 2001, 101, 2209–2242. [Google Scholar] [CrossRef]
- Iakoucheva, L.M.; Radivojac, P.; Brown, C.J.; O’Connor, T.R.; Sikes, J.G.; Obradovic, Z.; Dunker, A.K. The Importance of Intrinsic Disorder for Protein Phosphorylation. Nucleic Acids Res. 2004, 32, 1037–1049. [Google Scholar] [CrossRef]
- Hyman, A.A.; Weber, C.A.; Jülicher, F. Liquid-Liquid Phase Separation in Biology. Annu. Rev. Cell Dev. Biol. 2014, 30, 39–58. [Google Scholar] [CrossRef]
- Lin, Y.; Currie, S.L.; Rosen, M.K. Intrinsically Disordered Sequences Enable Modulation of Protein Phase Separation through Distributed Tyrosine Motifs. J. Biol. Chem. 2017, 292, 19110–19120. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, P.; Salgia, R.; Rangarajan, G. Intrinsically Disordered Proteins and Conformational Noise: The Hypothesis a Decade Later. iScience 2023, 26, 107109. [Google Scholar] [CrossRef] [PubMed]
- Moses, D.; Ginell, G.M.; Holehouse, A.S.; Sukenik, S. Intrinsically Disordered Regions Are Poised to Act as Sensors of Cellular Chemistry. Trends Biochem. Sci. 2023, in press. [Google Scholar] [CrossRef]
- Keul, N.D.; Oruganty, K.; Schaper Bergman, E.T.; Beattie, N.R.; McDonald, W.E.; Kadirvelraj, R.; Gross, M.L.; Phillips, R.S.; Harvey, S.C.; Wood, Z.A. The Entropic Force Generated by Intrinsically Disordered Segments Tunes Protein Function. Nature 2018, 563, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Das, R.K.; Ruff, K.M.; Pappu, R.V. Relating Sequence Encoded Information to Form and Function of Intrinsically Disordered Proteins. Curr. Opin. Struct. Biol. 2015, 32, 102–112. [Google Scholar] [CrossRef]
- Yruela, I.; Oldfield, C.J.; Niklas, K.J.; Dunker, A.K. Evidence for a Strong Correlation Between Transcription Factor Protein Disorder and Organismic Complexity. Genome Biol. Evol. 2017, 9, 1248–1265. [Google Scholar] [CrossRef]
- Yu, F.; Sukenik, S. Structural Preferences Shape the Entropic Force of Disordered Protein Ensembles. J. Phys. Chem. B 2023, 127, 4235–4244. [Google Scholar] [CrossRef]
- Niklas, K.J.; Bondos, S.E.; Dunker, A.K.; Newman, S.A. Rethinking Gene Regulatory Networks in Light of Alternative Splicing, Intrinsically Disordered Protein Domains, and Post-Translational Modifications. Front. Cell Dev. Biol. 2015, 3, 8. [Google Scholar] [CrossRef]
- Markaki, Y.; Chong, J.G.; Wang, Y.; Jacobson, E.C.; Luong, C.; Tan, S.Y.X.; Jachowicz, J.W.; Strehle, M.; Maestrini, D.; Banerjee, A.K.; et al. Xist Nucleates Local Protein Gradients to Propagate Silencing across the X Chromosome. Cell 2021, 184, 6212. [Google Scholar] [CrossRef]
- Pacini, G.; Dunkel, I.; Mages, N.; Mutzel, V.; Timmermann, B.; Marsico, A.; Schulz, E.G. Integrated Analysis of Xist Upregulation and X-Chromosome Inactivation with Single-Cell and Single-Allele Resolution. Nat. Commun. 2021, 12, 3638. [Google Scholar] [CrossRef]
- Ridings-Figueroa, R.; Stewart, E.R.; Nesterova, T.B.; Coker, H.; Pintacuda, G.; Godwin, J.; Wilson, R.; Haslam, A.; Lilley, F.; Ruigrok, R.; et al. The Nuclear Matrix Protein CIZ1 Facilitates Localization of Xist RNA to the Inactive X-Chromosome Territory. Genes Dev. 2017, 31, 876–888. [Google Scholar] [CrossRef] [PubMed]
- Sofi, S.; Williamson, L.; Turvey, G.L.; Scoynes, C.; Hirst, C.; Godwin, J.; Brockdorff, N.; Ainscough, J.; Coverley, D. Prion-like Domains Drive CIZ1 Assembly Formation at the Inactive X Chromosome. J. Cell Biol. 2022, 221, e202103185. [Google Scholar] [CrossRef] [PubMed]
- Pandya-Jones, A.; Markaki, Y.; Serizay, J.; Chitiashvili, T.; Mancia Leon, W.R.; Damianov, A.; Chronis, C.; Papp, B.; Chen, C.-K.; McKee, R.; et al. A Protein Assembly Mediates Xist Localization and Gene Silencing. Nature 2020, 587, 145–151. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaufmann, C.; Wutz, A. IndiSPENsable for X Chromosome Inactivation and Gene Silencing. Epigenomes 2023, 7, 28. https://doi.org/10.3390/epigenomes7040028
Kaufmann C, Wutz A. IndiSPENsable for X Chromosome Inactivation and Gene Silencing. Epigenomes. 2023; 7(4):28. https://doi.org/10.3390/epigenomes7040028
Chicago/Turabian StyleKaufmann, Corinne, and Anton Wutz. 2023. "IndiSPENsable for X Chromosome Inactivation and Gene Silencing" Epigenomes 7, no. 4: 28. https://doi.org/10.3390/epigenomes7040028