
The Mutagenic Consequences of DNA Methylation within and across Generations

{kind=link}
Abstract
:1. Introduction
2. DNA Methylation: Structure, Machinery, Mechanism, and Regulation
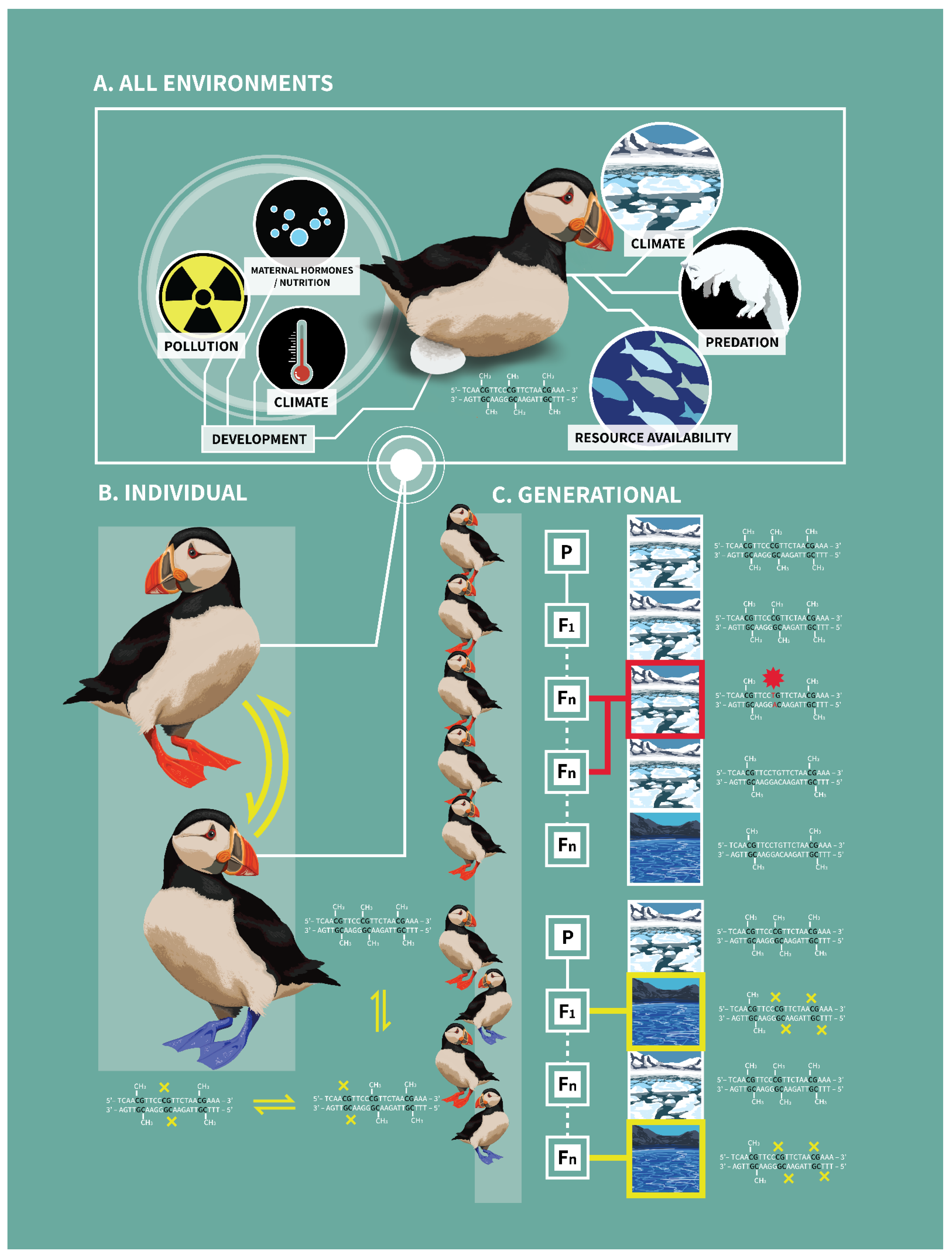
3. Implications of DNA Methylation within and across Generations
4. Mutation of CpG Sites
5. Epigenetic Potential
6. The Evolutionary Consequences of CpG Mutations
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef] [PubMed]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef]
- Zhou, D.; Li, Z.; Yu, D.; Wan, L.; Zhu, Y.; Lai, M.; Zhang, D. Polymorphisms involving gain or loss of CpG sites are significantly enriched in trait-associated SNPs. Oncotarget 2015, 6, 39995–40004. [Google Scholar] [CrossRef] [Green Version]
- Nabel, C.S.; Manning, S.A.; Kohli, R.M. The curious chemical biology of cytosine: Deamination, methylation, and oxidation as modulators of genomic potential. ACS Chem. Biol. 2012, 7, 20–30. [Google Scholar] [CrossRef]
- Blow, M.J.; Clark, T.A.; Daum, C.G.; Deutschbauer, A.M.; Fomenkov, A.; Fries, R.; Froula, J.; Kang, D.D.; Malmstrom, R.R.; Morgan, R.D.; et al. The Epigenomic Landscape of Prokaryotes. PLoS Genet. 2016, 12, e1005854. [Google Scholar] [CrossRef] [Green Version]
- Hoelzer, K.; Shackelton, L.A.; Parrish, C.R. Presence and role of cytosine methylation in DNA viruses of animals. Nucleic Acids Res. 2008, 36, 2825–2837. [Google Scholar] [CrossRef]
- Iyer, L.M.; Abhiman, S.; Aravind, L. Natural History of Eukaryotic DNA Methylation Systems, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2011; Volume 101, ISBN 9780123876850. [Google Scholar]
- Ponger, L.; Li, W.H. Evolutionary diversification of DNA methyltransferases in eukaryotic genomes. Mol. Biol. Evol. 2005, 22, 1119–1128. [Google Scholar] [CrossRef] [Green Version]
- Reik, W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 2007, 447, 425–432. [Google Scholar] [CrossRef]
- Ficz, G. New insights into mechanisms that regulate DNA methylation patterning. J. Exp. Biol. 2015, 218, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lister, R.; Pelizzola, M.; Dowen, R.; Hawkins, R.; Hon, G.; Nery, J.; Lee, L.; Ye, Z.; Ngo, Q.; Edsall, L.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.G.; Wilson, G.A.; Butcher, L.M.; Roos, C.; Walter, L.; Beck, S. Human-specific CpG “beacons” identify loci associated with human-specific traits and disease. Epigenetics 2012, 7, 1188–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Long, H.K.; Sims, D.; Heger, A.; Blackledge, N.P.; Kutter, C.; Wright, M.L.; Grützner, F.; Odom, D.T.; Patient, R.; Ponting, C.P.; et al. Epigenetic conservation at gene regulatory elements revealed by non-methylated DNA profiling in seven vertebrates. Elife 2013, 2013, e00348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, S.; Liu, Z.; Zhang, B.; Zhou, J.; Zhu, B.D.; Ji, J.; Deng, D. Methylation status of individual CpG sites within Alu elements in the human genome and Alu hypomethylation in gastric carcinomas. BMC Cancer 2010, 10, 44. [Google Scholar] [CrossRef] [Green Version]
- Xin, Y.; O’Donnell, A.H.; Ge, Y.; Chanrion, B.; Milekic, M.; Rosoklija, G.; Stankov, A.; Arango, V.; Dwork, A.J.; Gingrich, J.A.; et al. Role of CpG context and content in evolutionary signatures of brain DNA methylation. Epigenetics 2011, 6, 1308–1318. [Google Scholar] [CrossRef] [Green Version]
- Zemach, A.; McDaniel, I.E.; Silva, P.; Zilberman, D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 2010, 328, 916–919. [Google Scholar] [CrossRef] [Green Version]
- Varriale, A. DNA Methylation, Epigenetics, and Evolution in Vertebrates: Facts and Challenges. Int. J. Evol. Biol. 2014, 2014, 1–7. [Google Scholar] [CrossRef]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef]
- Feng, S.; Cokus, S.J.; Zhang, X.; Chen, P.Y.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jain, J.; Strauss, S.H.; Halpern, M.E.; et al. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. USA 2010, 107, 8689–8694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Wang, G.; Qian, J. Transcription factors as readers and effectors of DNA methylation. Nat. Rev. Genet. 2016, 17, 551–565. [Google Scholar] [CrossRef]
- Tate, P.H.; Bird, A.P. Effects of DNA methylation on DNA-binding proteins and gene expression. Curr. Opin. Genet. Dev. 1993, 3, 226–231. [Google Scholar] [CrossRef]
- Yin, Y.; Morgunova, E.; Jolma, A.; Kaasinen, E.; Sahu, B.; Khund-Sayeed, S.; Das, P.K.; Kivioja, T.; Dave, K.; Zhong, F.; et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 2017, 356. [Google Scholar] [CrossRef]
- Rishi, V.; Bhattacharya, P.; Chatterjee, R.; Rozenberg, J.; Zhao, J.; Glass, K.; Fitzgerald, P.; Vinson, C. CpG methylation of half-CRE sequences creates C/EBPα binding sites that activate some tissue-specific genes. Proc. Natl. Acad. Sci. USA 2010, 107, 20311–20316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Wan, J.; Su, Y.; Song, Q.; Zeng, Y.; Nguyen, H.N.; Shin, J.; Cox, E.; Rho, H.S.; Woodard, C.; et al. DNA methylation presents distinct binding sites for human transcription factors. Elife 2013, 2013, e00726. [Google Scholar] [CrossRef]
- Maunakea, A.K.; Nagarajan, R.P.; Bilenky, M.; Ballinger, T.J.; Dsouza, C.; Fouse, S.D.; Johnson, B.E.; Hong, C.; Nielsen, C.; Zhao, Y.; et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 2010, 466, 253–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurent, L.; Wong, E.; Li, G.; Huynh, T.; Tsirigos, A.; Ong, C.T.; Low, H.M.; Sung, K.W.K.; Rigoutsos, I.; Loring, J.; et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010, 20, 320–331. [Google Scholar] [CrossRef] [Green Version]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilvitis, H.J.; Hanson, H.; Schrey, A.W.; Martin, L.B. Epigenetic potential as a mechanism of phenotypic plasticity in vertebrate range expansions. Integr. Comp. Biol. 2017, 57, 385–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bestor, T.H. The DNA methyltransferases of mammals. Hum. Mol. Genet. 2000, 9, 2395–2402. [Google Scholar] [CrossRef] [Green Version]
- Baubec, T.; Colombo, D.F.; Wirbelauer, C.; Schmidt, J.; Burger, L.; Krebs, A.R.; Akalin, A.; Schübeler, D. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature 2015, 520, 243–247. [Google Scholar] [CrossRef]
- Neri, F.; Rapelli, S.; Krepelova, A.; Incarnato, D.; Parlato, C.; Basile, G.; Maldotti, M.; Anselmi, F.; Oliviero, S. Intragenic DNA methylation prevents spurious transcription initiation. Nature 2017, 32, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Bourc’his, D.; Xu, G.L.; Lin, C.S.; Bollman, B.; Bestor, T.H. Dnmt3L and the establishment of maternal genomic imprints. Science 2001, 294, 2536–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aapola, U.; Shibuya, K.; Scott, H.S.; Ollila, J.; Vihinen, M.; Heino, M.; Shintani, A.; Kawasaki, K.; Minoshima, S.; Krohn, K.; et al. Isolation and initial characterization of a novel zinc finger gene, DNMT3L, on 21q22.3, related to the cytosine-5-methyltransferase 3 gene family. Genomics 2000, 65, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Yarychkivska, O.; Shahabuddin, Z.; Comfort, N.; Boulard, M.; Bestor, T.H. BAH domains and a histone-like motif in DNA methyltransferase 1 (DNMT1) regulate de novo and maintenance methylation in vivo. J. Biol. Chem. 2018, 293, 19466–19475. [Google Scholar] [CrossRef] [Green Version]
- Li, J.-Y.; Pu, M.-T.; Hirasawa, R.; Li, B.-Z.; Huang, Y.-N.; Zeng, R.; Jing, N.-H.; Chen, T.; Li, E.; Sasaki, H.; et al. Synergistic Function of DNA Methyltransferases Dnmt3a and Dnmt3b in the Methylation of Oct4 and Nanog. Mol. Cell. Biol. 2007, 27, 8748–8759. [Google Scholar] [CrossRef] [Green Version]
- Li, E.; Zhang, Y. DNA Methylation in Mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a019133. [Google Scholar] [CrossRef] [Green Version]
- Sharif, J.; Muto, M.; Takebayashi, S.I.; Suetake, I.; Iwamatsu, A.; Endo, T.A.; Shinga, J.; Mizutani-Koseki, Y.; Toyoda, T.; Okamura, K.; et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature 2007, 450, 908–912. [Google Scholar] [CrossRef]
- Kagiwada, S.; Kurimoto, K.; Hirota, T.; Yamaji, M.; Saitou, M. Replication-coupled passive DNA demethylation for the erasure of genome imprints in mice. EMBO J. 2013, 32, 340–353. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Saitou, M.; Kagiwada, S.; Kurimoto, K. Epigenetic reprogramming in mouse pre-implantation development and primordial germ cells. Development 2012, 139, 15–31. [Google Scholar] [CrossRef]
- Wu, H.; Zhang, Y. Reversing DNA methylation: Mechanisms, genomics, and biological functions. Cell 2014, 156, 45–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.U.; Su, Y.; Zhong, C.; Ming, G.L.; Song, H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 2011, 145, 423–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabel, C.S.; Jia, H.; Ye, Y.; Shen, L.; Goldschmidt, H.L.; Stivers, J.T.; Zhang, Y.; Kohli, R.M. AID/APOBEC deaminases disfavor modified cytosines implicated in DNA demethylation. Nat. Chem. Biol. 2012, 8, 751–758. [Google Scholar] [CrossRef] [Green Version]
- Kohli, R.M.; Zhang, Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Affinito, O.; Palumbo, D.; Fierro, A.; Cuomo, M.; De Riso, G.; Monticelli, A.; Miele, G.; Chiariotti, L.; Cocozza, S. Nucleotide distance influences co-methylation between nearby CpG sites. Genomics 2020, 112, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, D.N.; Rosenbaum, P.; Chen, X.; Barrows, D.; Horth, C.; Marunde, M.R.; Popova, I.K.; Gillespie, Z.B.; Keogh, M.C.; Lu, C.; et al. Two competing mechanisms of DNMT3A recruitment regulate the dynamics of de novo DNA methylation at PRC1-targeted CpG islands. Nat. Genet. 2021, 53, 794–800. [Google Scholar] [CrossRef]
- Blattler, A.; Farnham, P.J. Cross-talk between site-specific transcription factors and DNA methylation states. J. Biol. Chem. 2013, 288, 34287–34294. [Google Scholar] [CrossRef] [Green Version]
- Ravichandran, M.; Jurkowska, R.Z.; Jurkowski, T.P. Target specificity of mammalian DNA methylation and demethylation machinery. Org. Biomol. Chem. 2018, 16, 1419–1435. [Google Scholar] [CrossRef]
- Laisné, M.; Gupta, N.; Kirsh, O.; Pradhan, S.; Defossez, P.A. Mechanisms of DNA methyltransferase recruitment in mammals. Genes 2018, 9, 617. [Google Scholar] [CrossRef] [Green Version]
- Johnsson, P.; Ackley, A.; Vidarsdottir, L.; Lui, W.O.; Corcoran, M.; Grandér, D.; Morris, K.V. A pseudogene long-noncoding-RNA network regulates PTEN transcription and translation in human cells. Nat. Struct. Mol. Biol. 2013, 20, 440–446. [Google Scholar] [CrossRef]
- Stathopoulou, A.; Chhetri, J.B.; Ambrose, J.C.; Estève, P.O.; Ji, L.; Erdjument-Bromage, H.; Zhang, G.; Neubert, T.A.; Pradhan, S.; Herrero, J.; et al. A novel requirement for DROSHA in maintenance of mammalian CG methylation. Nucleic Acids Res. 2017, 45, 9398–9412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozata, D.M.; Gainetdinov, I.; Zoch, A.; O’Carroll, D.; Zamore, P.D. PIWI-interacting RNAs: Small RNAs with big functions. Nat. Rev. Genet. 2019, 20, 89–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, R.J.; Weiner, M.M.; Lin, H. PIWI proteins and PIWI-interacting RNAs in the soma. Nature 2014, 505, 353–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Zhu, J.K. Active DNA demethylation in plants and animals. Cold Spring Harb. Symp. Quant. Biol. 2012, 77, 161–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanata, C.M.; Chung, S.A.; Criswell, L.A. DNA methylation 101: What is important to know about DNA methylation and its role in SLE risk and disease heterogeneity. Lupus Sci. Med. 2018, 5, e000285. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Zhou, Y.; Lin, N.; Lowdon, R.F.; Hong, C.; Nagarajan, R.P.; Cheng, J.B.; Li, D.; Stevens, M.; Lee, H.J.; et al. Functional DNA methylation differences between tissues, cell types, and across individuals discovered using the M&M algorithm. Genome Res. 2013, 23, 1522–1540. [Google Scholar] [CrossRef] [Green Version]
- Schultz, M.D.; He, Y.; Whitaker, J.W.; Hariharan, M.; Mukamel, E.A.; Leung, D.; Rajagopal, N.; Nery, J.R.; Urich, M.A.; Chen, H.; et al. Human body epigenome maps reveal noncanonical DNA methylation variation. Nature 2015, 523, 212–216. [Google Scholar] [CrossRef] [Green Version]
- Weaver, I.C.G. Reversal of Maternal Programming of Stress Responses in Adult Offspring through Methyl Supplementation: Altering Epigenetic Marking Later in Life. J. Neurosci. 2005, 25, 11045–11054. [Google Scholar] [CrossRef] [Green Version]
- Weaver, I.C.G.; Cervoni, N.; Champagne, F.A.; D’Alessio, A.C.; Sharma, S.; Seckl, J.R.; Dymov, S.; Szyf, M.; Meaney, M.J. Epigenetic programming by maternal behavior. Nat. Neurosci. 2004, 7, 847–854. [Google Scholar] [CrossRef]
- Metzger, D.C.H.; Schulte, P.M. Persistent and plastic effects of temperature on dna methylation across the genome of threespine stickleback (Gasterosteus aculeatus). Proc. R. Soc. B Biol. Sci. 2017, 284, 20171667. [Google Scholar] [CrossRef]
- Morán, P.; Marco-Rius, F.; Megías, M.; Covelo-Soto, L.; Pérez-Figueroa, A. Environmental induced methylation changes associated with seawater adaptation in brown trout. Aquaculture 2013, 392–395, 77–83. [Google Scholar] [CrossRef]
- Lea, A.J.; Altmann, J.; Alberts, S.C.; Tung, J. Resource base influences genome-wide DNA methylation levels in wild baboons (Papio cynocephalus). Mol. Ecol. 2016, 25, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Herb, B.R.; Shook, M.S.; Fields, C.J.; Robinson, G.E. Defense against territorial intrusion is associated with DNA methylation changes in the honey bee brain. BMC Genom. 2018, 19, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDade, T.W.; Ryan, C.; Jones, M.J.; MacIsaac, J.L.; Morin, A.M.; Meyer, J.M.; Borja, J.B.; Miller, G.E.; Kobor, M.S.; Kuzawa, C.W. Social and physical environments early in development predict DNA methylation of inflammatory genes in young adulthood. Proc. Natl. Acad. Sci. USA 2017, 114, 7611–7616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubenstein, D.R.; Skolnik, H.; Berrio, A.; Champagne, F.A.; Phelps, S.; Solomon, J. Sex-specific fitness effects of unpredictable early life conditions are associated with DNA methylation in the avian glucocorticoid receptor. Mol. Ecol. 2016, 25, 1714–1728. [Google Scholar] [CrossRef] [PubMed]
- Head, J.A. Patterns of DNA methylation in animals: An ecotoxicological perspective. J. Environ. Law 2014, 54, 77–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angers, B.; Castonguay, E.; Massicotte, R. Environmentally induced phenotypes and DNA methylation: How to deal with unpredictable conditions until the next generation and after. Mol. Ecol. 2010, 19, 1283–1295. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Barrett, R.D.H. Epigenetics in natural animal populations. J. Evol. Biol. 2017, 30, 1612–1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laine, V.N.; Sepers, B.; Lindner, M.; Gawehns, F.; Ruuskanen, S.; van Oers, K. An ecologist’s guide for studying DNA methylation variation in wild vertebrates. Mol. Ecol. Resour. 2022, 1–21. [Google Scholar] [CrossRef]
- Vogt, G. Paradigm shifts in animal epigenetics: Research on non-model species leads to new insights into dependencies, functions and inheritance of DNA methylation. BioEssays 2022, 44, 2200040. [Google Scholar] [CrossRef] [PubMed]
- Barrès, R.; Yan, J.; Egan, B.; Treebak, J.T.; Rasmussen, M.; Fritz, T.; Caidahl, K.; Krook, A.; O’Gorman, D.J.; Zierath, J.R. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 2012, 15, 405–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiechmann, T.; Röh, S.; Sauer, S.; Czamara, D.; Arloth, J.; Ködel, M.; Beintner, M.; Knop, L.; Menke, A.; Binder, E.B.; et al. Identification of dynamic glucocorticoid-induced methylation changes at the FKBP5 locus. Clin. Epigenetics 2019, 11, 1–14. [Google Scholar] [CrossRef]
- Métivier, R.; Gallais, R.; Tiffoche, C.; Le Péron, C.; Jurkowska, R.Z.; Carmouche, R.P.; Ibberson, D.; Barath, P.; Demay, F.; Reid, G.; et al. Cyclical DNA methylation of a transcriptionally active promoter. Nature 2008, 452, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Kangaspeska, S.; Stride, B.; Métivier, R.; Polycarpou-Schwarz, M.; Ibberson, D.; Carmouche, R.P.; Benes, V.; Gannon, F.; Reid, G. Transient cyclical methylation of promoter DNA. Nature 2008, 452, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Bruniquel, D.; Schwartz, R.H. Selective, stable demethylation of the interleukin-2 gene enhances transcription by an active process. Nat. Immunol. 2003, 4, 235–240. [Google Scholar] [CrossRef]
- Bonduriansky, R.; Crean, A.J.; Day, T. The implications of nongenetic inheritance for evolution in changing environments. Evol. Appl. 2012, 5, 192–201. [Google Scholar] [CrossRef]
- West-Eberhard, M.J. Developmental Plasticity and Evolution; Oxford University Press: Oxford, NY, USA, 2003; ISBN 978-0195122350. [Google Scholar]
- Pigliucci, M. Evolution of phenotypic plasticity: Where are we going now? Trends Ecol. Evol. 2005, 20, 481–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevenson, T.J.; Prendergast, B.J. Reversible DNA methylation regulates seasonal photoperiodic time measurement. Proc. Natl. Acad. Sci. USA 2013, 110, 16651–16656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, G.; Koncevičius, K.; Ebrahimi, S.; Carlucci, M.; Groot, D.E.; Nair, A.; Zhang, A.; Kriščiūnas, A.; Oh, E.S.; Labrie, V.; et al. Circadian oscillations of cytosine modification in humans contribute to epigenetic variability, aging, and complex disease 06 Biological Sciences 0604 Genetics. Genome Biol. 2019, 20, 2. [Google Scholar] [CrossRef] [PubMed]
- Oh, G.; Ebrahimi, S.; Carlucci, M.; Zhang, A.; Nair, A.; Groot, D.E.; Labrie, V.; Jia, P.; Oh, E.S.; Jeremian, R.H.; et al. Cytosine modifications exhibit circadian oscillations that are involved in epigenetic diversity and aging. Nat. Commun. 2018, 9, 644. [Google Scholar] [CrossRef]
- Biggar, Y.; Storey, K.B. Global DNA modifications suppress transcription in brown adipose tissue during hibernation. Cryobiology 2014, 69, 333–338. [Google Scholar] [CrossRef]
- Alvarado, S.; Mak, T.; Liu, S.; Storey, K.B.; Szyf, M. Dynamic changes in global and gene-specific DNA methylation during hibernation in adult thirteen-lined ground squirrels, Ictidomys tridecemlineatus. J. Exp. Biol. 2015, 218, 1787–1795. [Google Scholar] [CrossRef] [Green Version]
- Viitaniemi, H.M.; Verhagen, I.; Visser, M.E.; Honkela, A.; Van Oers, K.; Husby, A.; Meyer, M. Seasonal Variation in Genome-Wide DNA Methylation Patterns and the Onset of Seasonal Timing of Reproduction in Great Tits. Genome Biol. Evol. 2019, 11, 970–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsh, A.G.; Pasqualone, A.A. DNA methylation and temperature stress in an Antarctic polychaete, Spiophanes tcherniai. Front. Physiol. 2014, 5, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsaprouni, L.G.; Yang, T.P.; Bell, J.; Dick, K.J.; Kanoni, S.; Nisbet, J.; Viñuela, A.; Grundberg, E.; Nelson, C.P.; Meduri, E.; et al. Cigarette smoking reduces DNA methylation levels at multiple genomic loci but the effect is partially reversible upon cessation. Epigenetics 2014, 9, 1382–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.C.; Wang, M.Y.; Feng, J.R.; Chang, Y.; Ji, S.R.; Wu, Y. Reversible promoter methylation determines fluctuating expression of acute phase proteins. Elife 2020, 9, e51317. [Google Scholar] [CrossRef] [PubMed]
- Dugué, P.A.; Jung, C.H.; Joo, J.E.; Wang, X.; Wong, E.M.; Makalic, E.; Schmidt, D.F.; Baglietto, L.; Severi, G.; Southey, M.C.; et al. Smoking and blood DNA methylation: An epigenome-wide association study and assessment of reversibility. Epigenetics 2020, 15, 358–368. [Google Scholar] [CrossRef] [Green Version]
- Massicotte, R.; Whitelaw, E.; Angers, B. DNA methylation: A source of random variation in natural populations. Epigenetics 2011, 6, 422–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souren, N.Y.; Gerdes, L.A.; Lutsik, P.; Gasparoni, G.; Beltrán, E.; Salhab, A.; Kümpfel, T.; Weichenhan, D.; Plass, C.; Hohlfeld, R.; et al. DNA methylation signatures of monozygotic twins clinically discordant for multiple sclerosis. Nat. Commun. 2019, 10, 2094. [Google Scholar] [CrossRef] [Green Version]
- Navarro-Martín, L.; Viñas, J.; Ribas, L.; Díaz, N.; Gutiérrez, A.; Di Croce, L.; Piferrer, F. DNA methylation of the gonadal aromatase (cyp19a) promoter is involved in temperature-dependent sex ratio shifts in the European sea bass. PLoS Genet. 2011, 7, e1002447. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Buemio, A.; Chu, R.; Vafaee, M.; Crews, D. Epigenetic Control of Gonadal Aromatase (cyp19a1) in Temperature-Dependent Sex Determination of Red-Eared Slider Turtles. PLoS ONE 2013, 8, e63599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrott, B.B.; Kohno, S.; Cloy-McCoy, J.A.; Guillette, L.J. Differential incubation temperatures result in dimorphic DNA methylation patterning of the SOX9 and aromatase promoters in gonads of alligator (Alligator mississippiensis) embryos. Biol. Reprod. 2014, 90, 2. [Google Scholar] [CrossRef]
- Szyf, M.; Weaver, I.; Meaney, M. Maternal care, the epigenome and phenotypic differences in behavior. Reprod. Toxicol. 2007, 24, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Danchin, E.; Pocheville, A.; Rey, O.; Pujol, B.; Blanchet, S. Epigenetically facilitated mutational assimilation: Epigenetics as a hub within the inclusive evolutionary synthesis. Biol. Rev. 2019, 94, 259–282. [Google Scholar] [CrossRef] [Green Version]
- Jablonka, E.V.A.; Raz, G.A.L. Transgenerational epigenetic inheritance: Prevalence, mechanisms, and implications for the study of heredity and evolution. Q. Rev. Biol. 2009, 84, 131–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Liu, H.; Sun, Z. Lamarck rises from his grave: Parental environment-induced epigenetic inheritance in model organisms and humans. Biol. Rev. 2017, 92, 2084–2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quadrana, L.; Colot, V. Plant Transgenerational Epigenetics. Annu. Rev. Genet. 2016, 50, 467–491. [Google Scholar] [CrossRef] [PubMed]
- Morgan, H.D.; Santos, F.; Green, K.; Dean, W.; Reik, W. Epigenetic reprogramming in mammals. Hum. Mol. Genet. 2005, 14, 47–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.J.; Hore, T.A.; Reik, W. Reprogramming the methylome: Erasing memory and creating diversity. Cell Stem Cell 2014, 14, 710–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daxinger, L.; Whitelaw, E. Understanding transgenerational epigenetic inheritance via the gametes in mammals. Nat. Rev. Genet. 2012, 13, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Heard, E.; Martienssen, R.A. Transgenerational epigenetic inheritance: Myths and mechanisms. Cell 2014, 157, 95–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias, B.G.; Ressler, K.J. Parental olfactory experience influences behavior and neural structure in subsequent generations. Nat. Neurosci. 2014, 17, 89–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weyrich, A.; Benz, S.; Karl, S.; Jeschek, M.; Jewgenow, K.; Fickel, J. Paternal heat exposure causes DNA methylation and gene expression changes of Stat3 in Wild guinea pig sons. Ecol. Evol. 2016, 6, 2657–2666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nätt, D.; Rubin, C.J.; Wright, D.; Johnsson, M.; Beltéky, J.; Andersson, L.; Jensen, P. Heritable genome-wide variation of gene expression and promoter methylation between wild and domesticated chickens. BMC Genom. 2012, 13, 59. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Wuitchik, S.J.S.; Barry, T.N.; Jamniczky, H.A.; Rogers, S.M.; Barrett, R.D.H. Heritability of DNA methylation in threespine stickleback (Gasterosteus aculeatus). Genetics 2021, 217, iyab001. [Google Scholar] [CrossRef]
- Heckwolf, M.J.; Meyer, B.S.; Häsler, R.; Höppner, M.P.; Eizaguirre, C.; Reusch, T.B.H. Two different epigenetic information channels in wild three-spined sticklebacks are involved in salinity adaptation. Sci. Adv. 2020, 6, eaaz1138. [Google Scholar] [CrossRef] [Green Version]
- Walser, J.C.; Furano, A. V The mutational spectrum of non-CpG DNA varies with CpG content. Genome Res. 2010, 20, 875–882. [Google Scholar] [CrossRef] [Green Version]
- Hwang, D.G.; Green, P. Bayesian Markov chain Monte Carlo sequence analysis reveals varying neutral substitution patterns in mammalian evolution. Proc. Natl. Acad. Sci. USA 2004, 101, 13994–14001. [Google Scholar] [CrossRef] [Green Version]
- Bird, A.P. DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. 1980, 8, 1499–1504. [Google Scholar] [CrossRef] [Green Version]
- Gorelick, R. Evolution of dioecy and sex chromosomes via methylation driving Muller’s ratchet. Biol. J. Linn. Soc. 2003, 80, 353–368. [Google Scholar] [CrossRef]
- Panchin, A.Y.; Makeev, V.J.; Medvedeva, Y.A. Preservation of methylated CpG dinucleotides in human CpG islands. Biol. Direct 2016, 11, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, A.; Frigge, M.L.; Masson, G.; Besenbacher, S.; Sulem, P.; Magnusson, G.; Gudjonsson, S.A.; Sigurdsson, A.; Jonasdottir, A.; Jonasdottir, A.; et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature 2012, 488, 471–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amster, G.; Sella, G. Life history effects on the molecular clock of autosomes and sex chromosomes. Proc. Natl. Acad. Sci. USA 2016, 113, 1588–1593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmutte, C.; Yang, A.S.; Beart, R.W.; Jones, P.A. Base Excision Repair of U:G Mismatches at a Mutational Hotspot in the p53 Gene Is More Efficient Than Base Excision Repair of T:G Mismatches in Extracts of Human Colon Tumors. Cancer Res. 1995, 55, 3742–3746. [Google Scholar] [PubMed]
- Bellacosa, A.; Drohat, A.C. Role of base excision repair in maintaining the genetic and epigenetic integrity of CpG sites. DNA Repair 2015, 32, 33–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Z.; Wyman, M.J.; Sella, G.; Przeworski, M. Interpreting the Dependence of Mutation Rates on Age and Time. PLoS Biol. 2016, 14, e1002355. [Google Scholar] [CrossRef]
- MacRae, S.L.; Croken, M.M.K.; Calder, R.B.; Aliper, A.; Milholland, B.; White, R.R.; Zhavoronkov, A.; Gladyshev, V.N.; Seluanov, A.; Gorbunova, V.; et al. DNA repair in species with extreme lifespan differences. Aging 2015, 7, 1171–1184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeifer, G.P. Mutagenesis at methylated CpG sequences. Curr. Top. Microbiol. Immunol. 2006, 301, 259–281. [Google Scholar] [CrossRef] [PubMed]
- Poulos, R.C.; Olivier, J.; Wong, J.W.H. The interaction between cytosine methylation and processes of DNA replication and repair shape the mutational landscape of cancer genomes. Nucleic Acids Res. 2017, 45, 7786–7795. [Google Scholar] [CrossRef]
- Shen, J.C.; Rideout, W.M.; Jones, P.A. High frequency mutagenesis by a DNA methyltransferase. Cell 1992, 71, 1073–1080. [Google Scholar] [CrossRef]
- Tomkova, M.; McClellan, M.; Kriaucionis, S.; Schuster-Böckler, B. DNA Replication and associated repair pathways are involved in the mutagenesis of methylated cytosine. DNA Repair 2018, 62, 1–7. [Google Scholar] [CrossRef]
- Ollila, J.; Lappalainen, I.; Vihinen, M. Sequence specificity in CpG mutation hotspots. FEBS Lett. 1996, 396, 119–122. [Google Scholar] [CrossRef] [Green Version]
- Branciamore, S.; Chen, Z.-X.; Riggs, A.D.; Rodin, S.N. CpG island clusters and pro-epigenetic selection for CpGs in protein-coding exons of HOX and other transcription factors. Proc. Natl. Acad. Sci. USA 2010, 107, 15485–15490. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Elango, N.; Warden, C.; Vigoda, E.; Yi, S.V. Heterogeneous genomic molecular clocks in primates. PLoS Genet. 2006, 2, 1527–1534. [Google Scholar] [CrossRef] [Green Version]
- Moorjani, P.; Amorim, C.E.G.; Arndt, P.F.; Przeworski, M. Variation in the molecular clock of primates. Proc. Natl. Acad. Sci. USA 2016, 113, 10607–10612. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.; Moorjani, P.; Sasani, T.A.; Pedersen, B.S.; Quinlan, A.R.; Jorde, L.B.; Amster, G.; Przeworski, M. Overlooked roles of DNA damage and maternal age in generating human germline mutations. Proc. Natl. Acad. Sci. USA 2019, 116, 9491–9500. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.S. Cancer mutation signatures, DNA damage mechanisms, and potential clinical implications. Genome Med. 2013, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Qu, J.; Hodges, E.; Molaro, A.; Gagneux, P.; Dean, M.D.; Hannon, G.J.; Smith, A.D. Evolutionary expansion of DNA hypomethylation in the mammalian germline genome. Genome Res. 2018, 28, 145–158. [Google Scholar] [CrossRef]
- Saxonov, S.; Berg, P.; Brutlag, D.L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. USA 2006, 103, 1412–1417. [Google Scholar] [CrossRef] [PubMed]
- Fryxell, K.J.; Moon, W.J. CpG mutation rates in the human genome are highly dependent on local GC content. Mol. Biol. Evol. 2005, 22, 650–658. [Google Scholar] [CrossRef]
- Molaro, A.; Hodges, E.; Fang, F.; Song, Q.; McCombie, W.R.; Hannon, G.J.; Smith, A.D. Sperm methylation profiles reveal features of epigenetic inheritance and evolution in primates. Cell 2011, 146, 1029–1041. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.; Han, L.; Zhao, Z. Investigating the relationship of DNA methylation with mutation rate and allele frequency in the human genome. BMC Genom. 2012, 13 (Suppl. 8), S7. [Google Scholar] [CrossRef] [Green Version]
- Zhi, D.; Aslibekyan, S.; Irvin, M.R.; Claas, S.A.; Borecki, I.B.; Ordovas, J.M.; Absher, D.M.; Arnett, D.K. SNPs located at CpG sites modulate genome-epigenome interaction. Epigenetics 2013, 8, 802–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanson, H.E.; Wang, C.; Schrey, A.W.; Liebl, A.L.; Ravinet, M.; Jiang, R.H.Y.; Martin, L.B. Epigenetic Potential and DNA Methylation in an Ongoing House Sparrow (Passer domesticus) Range Expansion. Am. Nat. 2022, 200. [Google Scholar] [CrossRef]
- Shi, Y.; Xu, L.; Feng, Q.; Li, A.; Jia, J.; Xu, Y.; Yang, D.; Zhang, Y.; Zhang, X.; Zhao, H.; et al. Allele-specific methylation contributed by CpG-SNP is associated with regulation of ALOX5AP gene expression in ischemic stroke. Neurol. Sci. 2018, 39, 1717–1724. [Google Scholar] [CrossRef]
- Izzi, B.; Pistoni, M.; Cludts, K.; Akkor, P.; Lambrechts, D.; Verfaillie, C.; Verhamme, P.; Freson, K.; Hoylaerts, M.F. Allele-specific DNA methylation reinforces PEAR1 enhancer activity. Blood 2016, 128, 1003–1012. [Google Scholar] [CrossRef] [Green Version]
- Shoemaker, R.; Deng, J.; Wang, W.; Zhang, K. Allele-specific methylation is prevalent and is contributed by CpG-SNPs in the human genome. Genome Res. 2010, 20, 883–889. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Lou, D.; Wang, Z. Crosstalk of genetic variants, allele-specific DNA methylation, and environmental factors for complex disease risk. Front. Genet. 2019, 10, 695. [Google Scholar] [CrossRef] [PubMed]
- Hanson, H.E.; Zimmer, C.; Koussayer, B.; Schrey, A.W.; Maddox, J.D.; Martin, L.B. Epigenetic Potential Affects Immune Gene Expression in House Sparrows. J. Exp. Biol. 2021, 224, jeb238451. [Google Scholar] [CrossRef]
- Agrawal, A.A. A scale-dependent framework for trade-offs, syndromes, and specialization in organismal biology. Ecology 2020, 101, e02924. [Google Scholar] [CrossRef] [PubMed]
- Lande, R. Evolution of phenotypic plasticity in colonizing species. Mol. Ecol. 2015, 24, 2038–2045. [Google Scholar] [CrossRef] [PubMed]
- Hanson, H.E.; Koussayer, B.; Kilvitis, H.J.; Schrey, A.W.; Maddox, J.D.; Martin, L.B. Epigenetic Potential in Native and Introduced Populations of House Sparrows (Passer domesticus). Integr. Comp. Biol. 2020, 60, 1458–1468. [Google Scholar] [CrossRef]
- Liebl, A.L.; Schrey, A.W.; Richards, C.L.; Martin, L.B. Patterns of DNA methylation throughout a range expansion of an introduced songbird. Integr. Comp. Biol. 2013, 53, 351–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrey, A.W.; Coon, C.A.C.; Grispo, M.T.; Awad, M.; Imboma, T.; McCoy, E.D.; Mushinsky, H.R.; Richards, C.L.; Martin, L.B. Epigenetic Variation May Compensate for Decreased Genetic Variation with Introductions: A Case Study Using House Sparrows (Passer domesticus) on Two Continents. Genet. Res. Int. 2012, 2012, 979751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, L.B.; Liebl, A.L. Physiological flexibility in an avian range expansion. Gen. Comp. Endocrinol. 2014, 206, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Liebl, A.L.; Martin, L.B. Exploratory behaviour and stressor hyper responsiveness facilitate range expansion of an introduced songbird. Proc. R. Soc. B Biol. Sci. 2012, 279, 4375–4381. [Google Scholar] [CrossRef]
- Liebl, A.L.; Martin, L.B. Living on the edge: Range edge birds consume novel foods sooner than established ones. Behav. Ecol. 2014, 25, 1089–1096. [Google Scholar] [CrossRef]
- Storz, J.F.; Natarajan, C.; Signore, A.V.; Witt, C.C.; McCandlish, D.M.; Stoltzfus, A. The role of mutation bias in adaptive molecular evolution: Insights from convergent changes in protein function. Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 979751. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Bosagna, C. From epigenotype to new genotypes: Relevance of epigenetic mechanisms in the emergence of genomic evolutionary novelty. Semin. Cell Dev. Biol. 2019, 97, 86–92. [Google Scholar] [CrossRef]
- Guerrero-Bosagna, C. Finalism in Darwinian and Lamarckian Evolution: Lessons from Epigenetics and Developmental Biology. Evol. Biol. 2012, 39, 283–300. [Google Scholar] [CrossRef]
- Guerrero-Bosagna, C.; Sabat, P.; Valladares, L. Environmental signaling and evolutionary change: Can exposure of pregnant mammals to environmental estrogens lead to epigenetically induced evolutionary changes in embryos? Evol. Dev. 2005, 7, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Flores, K.B.; Wolschin, F.; Amdam, G.V. The role of methylation of DNA in environmental adaptation. Integr. Comp. Biol. 2013, 53, 359–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bateson, P.; Gluckman, P. Plasticity, Robustness, Development and Evolution; The University of Chicago Press: Chicago, IL, USA, 2011; ISBN 9780511842382. [Google Scholar]
- Flores, K.B.; Amdam, G.V. Deciphering a methylome: What can we read into patterns of DNA methylation? J. Exp. Biol. 2011, 214, 3155–3163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mugal, C.F.; Arndt, P.F.; Holm, L.; Ellegren, H. Evolutionary consequences of DNA methylation on the GC content in vertebrate genomes. G3 Genes Genomes Genet. 2015, 5, 441–447. [Google Scholar] [CrossRef]
- Simmen, M.W. Genome-scale relationships between cytosine methylation and dinucleotide abundances in animals. Genomics 2008, 92, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.H.; Zheng, J.B.; Gu, X.; Saunders, G.F.; Alfred Yung, W.K. Novel PAX6 binding sites in the human genome and the role of repetitive elements in the evolution of gene regulation. Genome Res. 2002, 12, 1716–1722. [Google Scholar] [CrossRef] [Green Version]
- Zemojtel, T.; Kiebasa, S.M.; Arndt, P.F.; Behrens, S.; Bourque, G.; Vingron, M. CpG deamination creates transcription factor-binding sites with high efficiency. Genome Biol. Evol. 2011, 3, 1304–1311. [Google Scholar] [CrossRef]
- Zemojtel, T.; Kielbasa, S.M.; Arndt, P.F.; Chung, H.R.; Vingron, M. Methylation and deamination of CpGs generate p53-binding sites on a genomic scale. Trends Genet. 2009, 25, 63–66. [Google Scholar] [CrossRef]
- He, X.; Tillo, D.; Vierstra, J.; Syed, K.S.; Deng, C.; Ray, G.J.; Stamatoyannopoulos, J.; FitzGerald, P.C.; Vinson, C. Methylated Cytosines Mutate to Transcription Factor Binding Sites that Drive Tetrapod Evolution. Genome Biol. Evol. 2015, 7, 3155–3169. [Google Scholar] [CrossRef]
- Galen, S.C.; Natarajan, C.; Moriyama, H.; Weber, R.E.; Fago, A.; Benham, P.M.; Chavez, A.N.; Cheviron, Z.A.; Storz, J.F.; Witt, C.C. Contribution of a mutational hot spot to hemoglobin adaptation in high-Altitude Andean house wrens. Proc. Natl. Acad. Sci. USA 2015, 112, 13958–13963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, H.; Huttley, G. Exploiting CpG hypermutability to identify phenotypically significant variation within human protein-coding genes. Genome Biol. Evol. 2011, 3, 938–949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikkelsen, T.S.; Hillier, L.W.; Eichler, E.E.; Zody, M.C.; Jaffe, D.B.; Yang, S.P.; Enard, W.; Hellmann, I.; Lindblad-Toh, K.; Altheide, T.K.; et al. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 2005, 437, 69–87. [Google Scholar] [CrossRef] [Green Version]
- Pértille, F.; Da Silva, V.H.; Johansson, A.M.; Lindström, T.; Wright, D.; Coutinho, L.L.; Jensen, P.; Guerrero-Bosagna, C. Mutation dynamics of CpG dinucleotides during a recent event of vertebrate diversification. Epigenetics 2019, 14, 685–707. [Google Scholar] [CrossRef] [Green Version]
- Cooper, D.N.; Youssoufian, H. The CpG dinucleotide and human genetic disease. Hum. Genet. 1988, 78, 151–155. [Google Scholar] [CrossRef]
- Cooper, D.N.; Mort, M.; Stenson, P.D.; Ball, E.V.; Chuzhanova, N.A. Methylation-mediated deamination of 5-methylcytosine appears to give rise to mutations causing human inherited disease in CpNpG trinucleotides, as well as in CpG dinucleotides. Hum. Genomics 2010, 4, 406–410. [Google Scholar] [CrossRef] [Green Version]
- Chae, H.; Lee, S.; Nephew, K.P.; Kim, S. Subtype-specific CpG island shore methylation and mutation patterns in 30 breast cancer cell lines. BMC Syst. Biol. 2016, 10, 433–443. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hanson, H.E.; Liebl, A.L. The Mutagenic Consequences of DNA Methylation within and across Generations. Epigenomes 2022, 6, 33. https://doi.org/10.3390/epigenomes6040033
Hanson HE, Liebl AL. The Mutagenic Consequences of DNA Methylation within and across Generations. Epigenomes. 2022; 6(4):33. https://doi.org/10.3390/epigenomes6040033
Chicago/Turabian StyleHanson, Haley E., and Andrea L. Liebl. 2022. "The Mutagenic Consequences of DNA Methylation within and across Generations" Epigenomes 6, no. 4: 33. https://doi.org/10.3390/epigenomes6040033