
Identification and Characterization of Small RNA Markers of Age in the Blow Fly Cochliomyia macellaria (Fabricius) (Diptera: Calliphoridae)
 , , , , ,
, , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Insect Maintenance and Collection
2.2. Small RNA Isolation and Sequencing
2.3. Sequencing Data Analysis
2.4. Evaluation and Validation
3. Results
3.1. Sequencing Output and Identification of miRNA
3.2. Summary of Results for All of Immature Development
3.3. Identification of Vertebrate miRNA
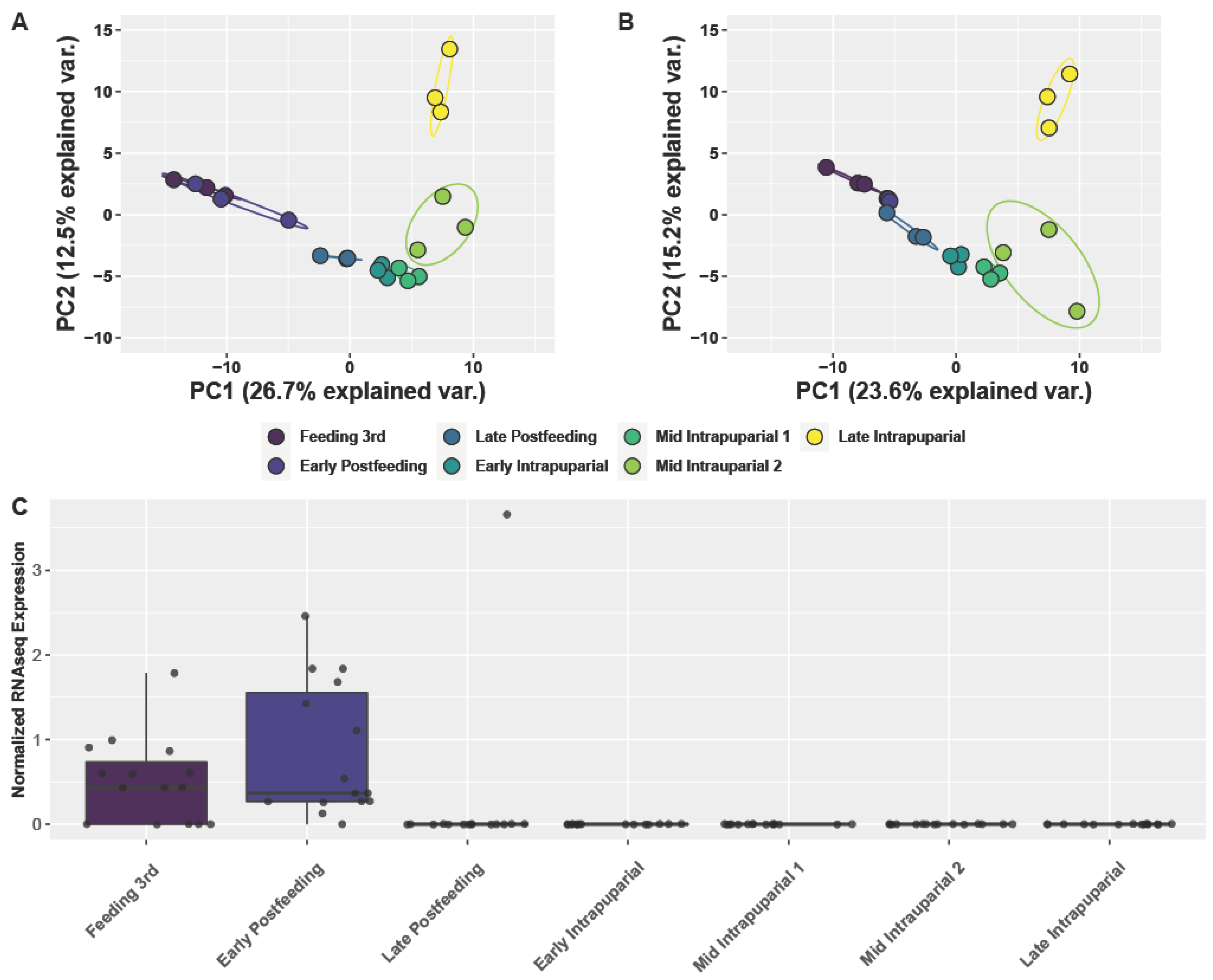
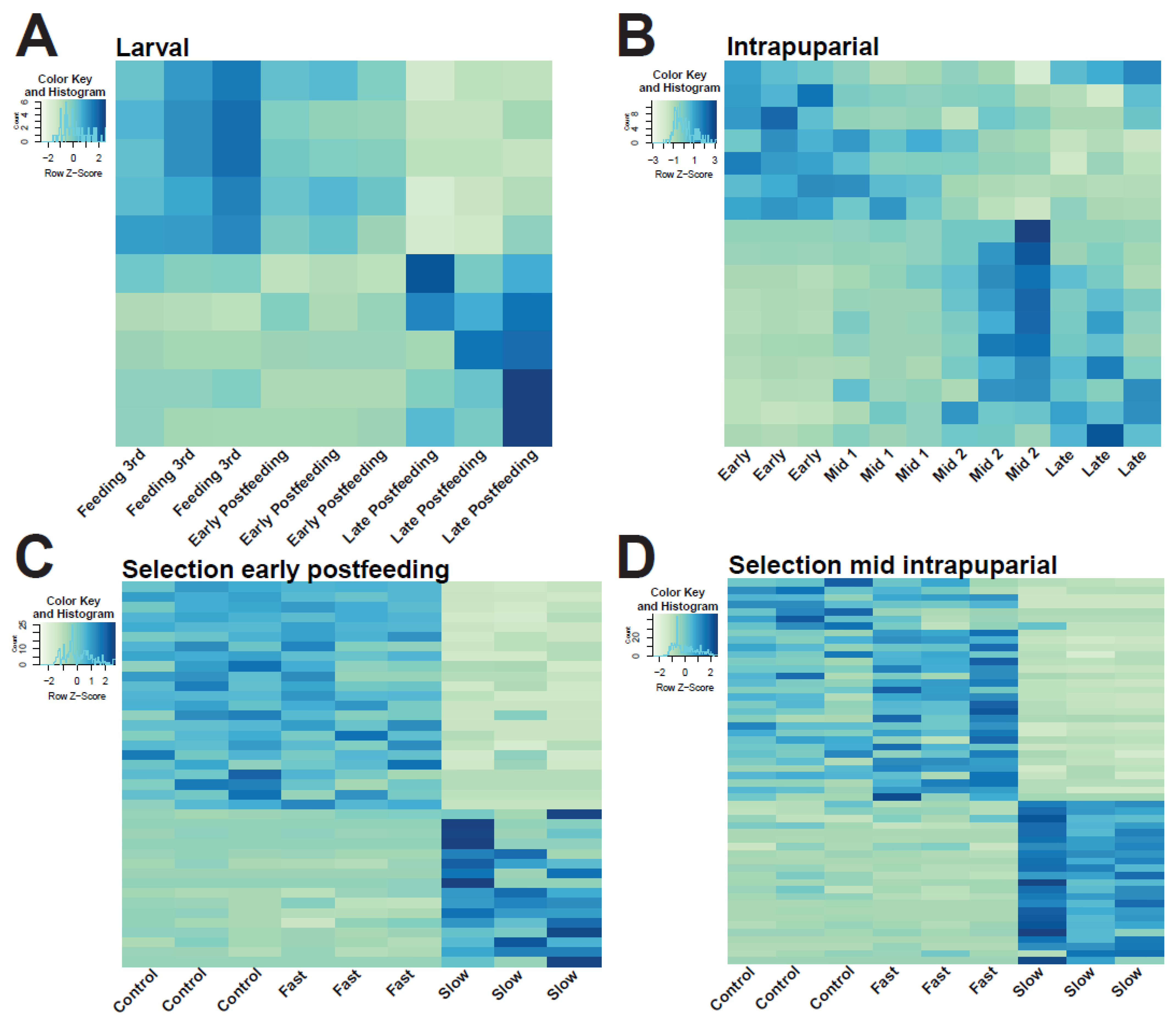
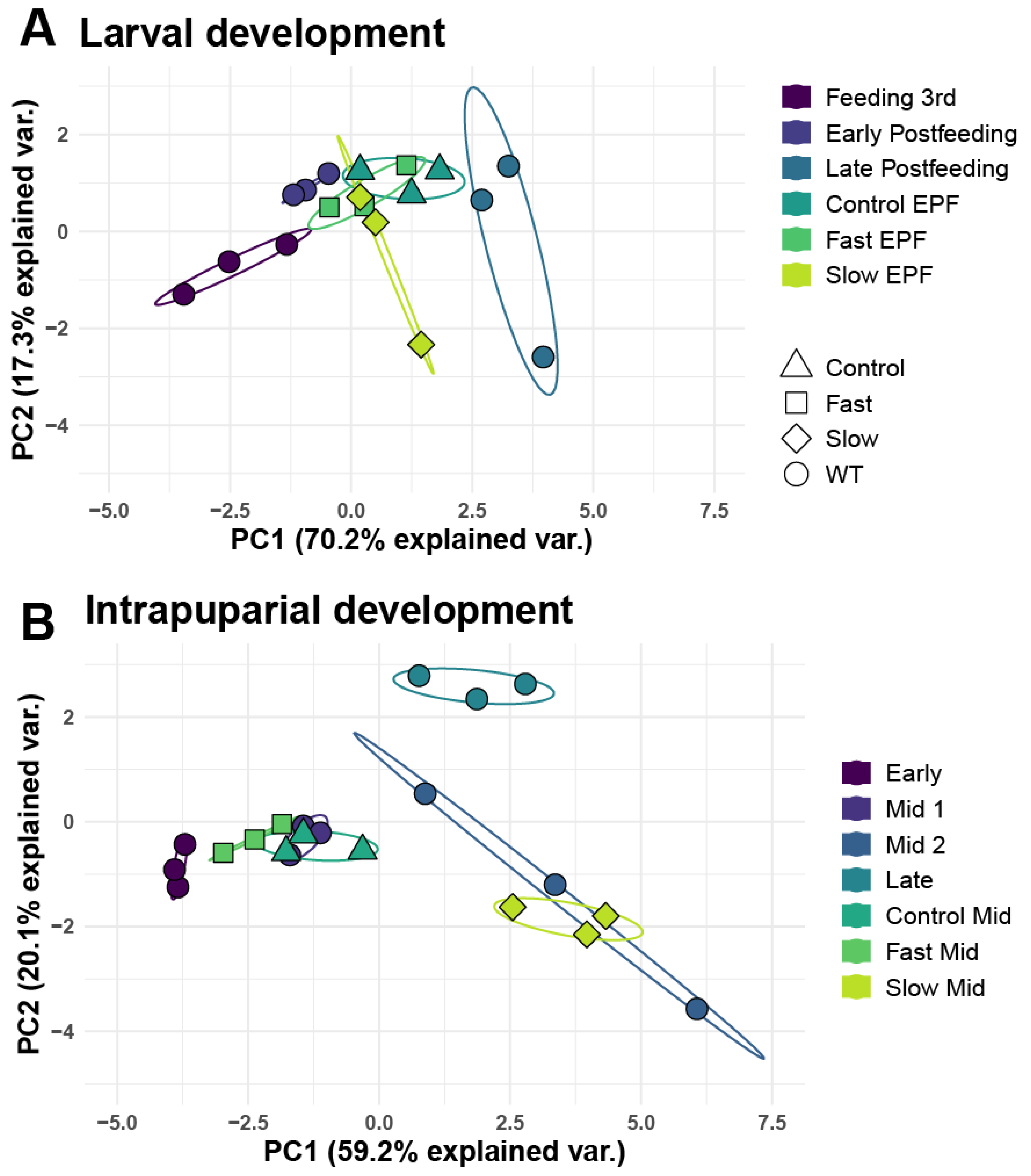
3.4. Analysis of Larval and Intrapuparial Samples with PCA and DESeq2 Methods
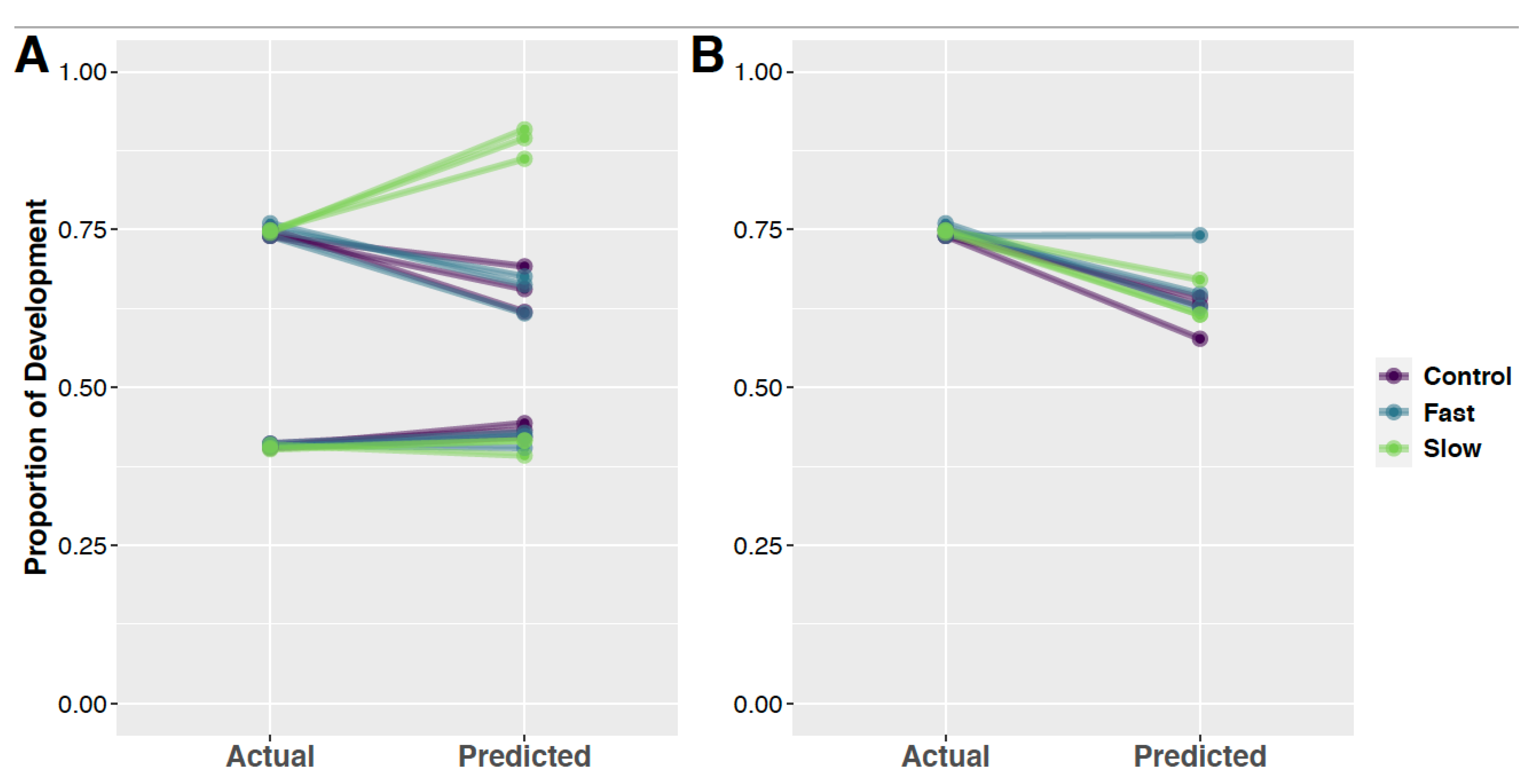
3.5. Random Forest Models and Predication of Development Time
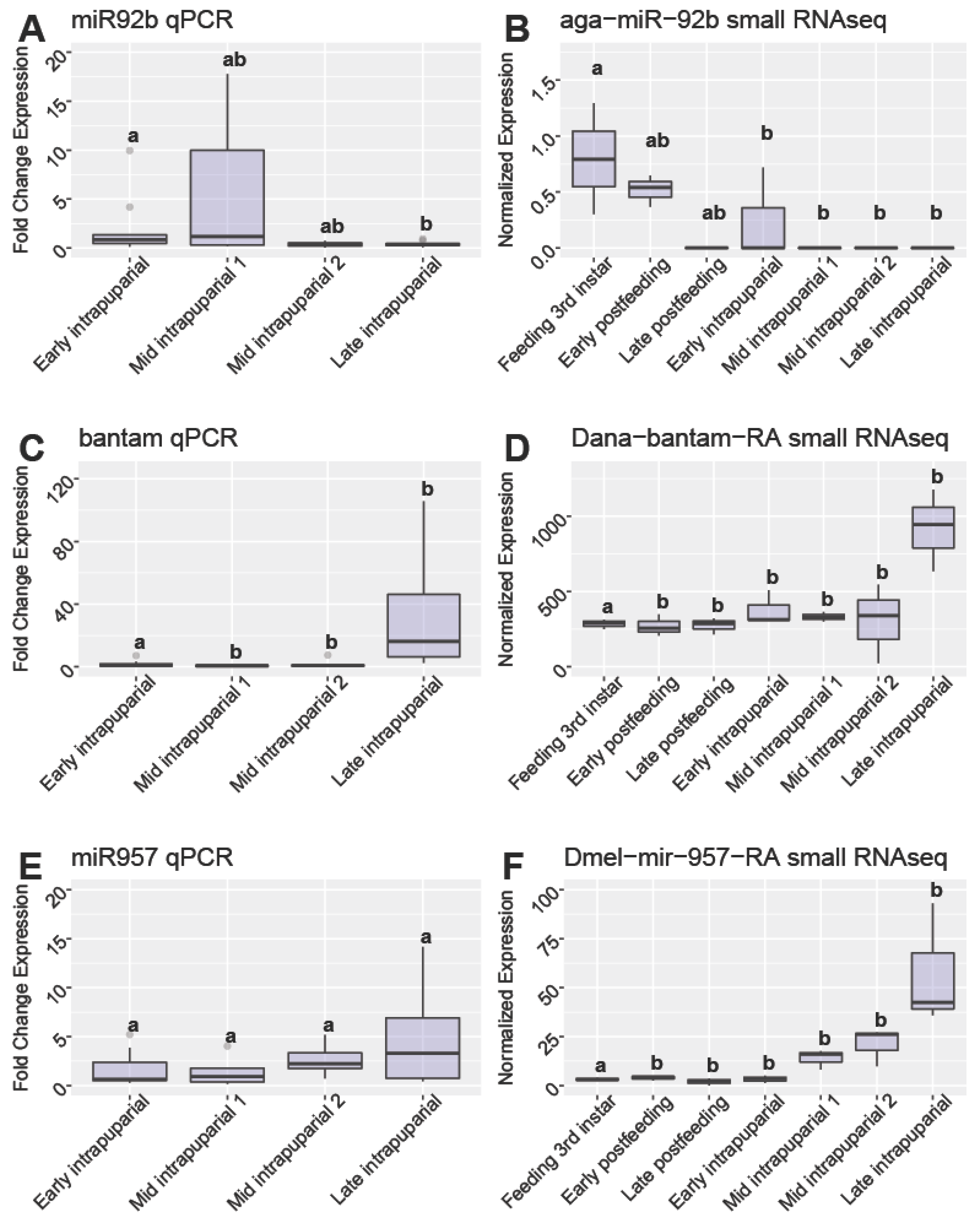
3.6. qPCR Validiation
3.7. Analyses of Sex Biased Expression of miRNA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benbow, M.E.; Tomberlin, J.K.; Tarone, A.M. Introduction to carrion ecology, evolution, and their applications. In Carrion Ecology, Evolution, and Their Applications; CRC Press: Boca Raton, FL, USA, 2015; pp. 3–12. [Google Scholar] [CrossRef]
- Benbow, M.E.; Receveur, J.P.; Lamberti, G.A. Death and Decomposition in Aquatic Ecosystems. Front. Ecol. Evol. 2020, 8. [Google Scholar] [CrossRef] [Green Version]
- Pechal, J.; Benbow, M.E.; Crippen, T.; Tarone, A.; Tomberlin, J.K. Delayed insect access alters carrion decomposition and necrophagous insect community assembly. Ecosphere 2014, 5, art45. [Google Scholar] [CrossRef]
- Payne, J.A. A Summer Carrion Study of the Baby Pig Sus Scrofa Linnaeus. Ecology 1965, 46, 592–602. [Google Scholar] [CrossRef]
- Catts, E. Problems in estimating the postmortem interval in death investigations. J. Agric. Entomol. 1992, 9, 245–255. [Google Scholar]
- Amendt, J.; Richards, C.S.; Campobasso, C.P.; Zehner, R.; Hall, M.J.R. Forensic entomology: Applications and limitations. Forensic Sci. Med. Pathol. 2011, 7, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Byrd, J.H.; Tomberlin, J.K. Forensic Entomology: The Utility of Arthropods in Legal Investigations; CRC Press: Boca Raton, FL, USA, 2019. [Google Scholar] [CrossRef]
- Stevens, J.R. The evolution of myiasis in blowflies (Calliphoridae). Int. J. Parasitol. 2003, 33, 1105–1113. [Google Scholar] [CrossRef]
- Rawlins, S.C.; Alexander, F.C.; Moe, V.; Caesar, E.; Moll, K.; Applewhaite, L. Screwworm (Diptera: Calliphoridae) Myiasis in the Southern Caribbean, and Proposals for Its Management. J. Econ. Èntomol. 1983, 76, 1106–1111. [Google Scholar] [CrossRef] [PubMed]
- Junqueira, A.C.M.; Ratan, A.; Acerbi, E.; Drautz-Moses, D.I.; Premkrishnan, B.N.V.; Costea, P.I.; Linz, B.; Purbojati, R.W.; Paulo, D.; Gaultier, N.E.; et al. The microbiomes of blowflies and houseflies as bacterial transmission reservoirs. Sci. Rep. 2017, 7, 16324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deguenon, J.M.; Travanty, N.; Zhu, J.; Carr, A.; Denning, S.; Reiskind, M.; Watson, D.W.; Roe, R.M.; Ponnusamy, L. Exogenous and endogenous microbiomes of wild-caught Phormia regina (Diptera: Calliphoridae) flies from a suburban farm by 16S rRNA gene sequencing. Sci. Rep. 2019, 9, 20365. [Google Scholar] [CrossRef]
- Tomberlin, J.K.; Crippen, T.L.; Tarone, A.M.; Chaudhury, M.F.B.; Singh, B.; Cammack, J.A.; Meisel, R.P. A Review of Bacterial Interactions With Blow Flies (Diptera: Calliphoridae) of Medical, Veterinary, and Forensic Importance. Ann. Èntomol. Soc. Am. 2016, 110, 19–36. [Google Scholar] [CrossRef]
- Sherman, R.A.; Hall, M.J.R.; Thomas, S. Medicinal Maggots: An Ancient Remedy for Some Contemporary Afflictions. Annu. Rev. Èntomol. 2000, 45, 55–81. [Google Scholar] [CrossRef] [PubMed]
- Benbow, M. Debridement of non-healing wounds. Pract. Nurse 2006, 31, 26–30. [Google Scholar] [CrossRef]
- Clement, S.L.; Hellier, B.C.; Elberson, L.R.; Staska, R.T.; Evans, M.A. Flies (Diptera: Muscidae: Calliphoridae) Are Efficient Pollinators of Allium ampeloprasum L. (Alliaceae) in Field Cages. J. Econ. Èntomol. 2007, 100, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Howlett, B.G. Hybrid carrot seed crop pollination by the fly Calliphora vicina (Diptera: Calliphoridae). J. Appl. Èntomol. 2011, 136, 421–430. [Google Scholar] [CrossRef]
- Saeed, S.; Naqqash, M.N.; Jaleel, W.; Saeed, Q.; Ghouri, F. The effect of blow flies (Diptera: Calliphoridae) on the size and weight of mangos (Mangifera indica L.). PeerJ 2016, 4, e2076. [Google Scholar] [CrossRef] [Green Version]
- Tomberlin, J.; Mohr, R.; Benbow, M.; Tarone, A.; VanLaerhoven, S. A Roadmap for Bridging Basic and Applied Research in Forensic Entomology. Annu. Rev. Èntomol. 2011, 56, 401–421. [Google Scholar] [CrossRef] [Green Version]
- Tarone, A.M.; Sanford, M.R. Is PMI the Hypothesis or the Null Hypothesis? J. Med. Èntomol. 2017, 54, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Tarone, A.M.; Foran, D.R. Generalized Additive Models and Lucilia sericata Growth: Assessing Confidence Intervals and Error Rates in Forensic Entomology. J. Forensic Sci. 2008, 53, 942–948. [Google Scholar] [CrossRef]
- Boatright, S.A.; Tomberlin, J.K. Effects of Temperature and Tissue Type on the Development of Cochliomyia macellaria (Diptera: Calliphoridae). J. Med. Èntomol. 2010, 47, 917–923. [Google Scholar] [CrossRef]
- Anderson, G.S. Minimum and Maximum Development Rates of Some Forensically Important Calliphoridae (Diptera). J. Forensic Sci. 2000, 45, 824–832. [Google Scholar] [CrossRef]
- Anderson, G.S.; Warren, J.-A. Establishing Lower Developmental Thresholds for a Common BlowFly; Defence Research and Development Canada, Centre for Security Science: Dartmouth, NS, Canada, 2011; p. 18. [Google Scholar]
- Tarone, A.M.; Foran, D.R. Components of Developmental Plasticity in a Michigan Population of Lucilia sericata (Diptera: Calliphoridae). J. Med. Èntomol. 2006, 43, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.; Harvey, M. Optical coherence tomography: Age estimation of Calliphora vicina pupae in vivo? Forensic Sci. Int. 2014, 242, 157–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, K.; Thorne, A.; Harvey, M. Calliphora vicina (Diptera: Calliphoridae) pupae: A timeline of external morphological development and a new age and PMI estimation tool. Int. J. Leg. Med. 2014, 129, 835–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, K.; Harvey, M.L. Internal Morphological Analysis for Age Estimation of Blow Fly Pupae (Diptera: Calliphoridae) in Postmortem Interval Estimation*. J. Forensic Sci. 2012, 58, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Voss, S.C.; Magni, P.; Dadour, I.; Nansen, C. Reflectance-based determination of age and species of blowfly puparia. Int. J. Leg. Med. 2016, 131, 263–274. [Google Scholar] [CrossRef]
- Zehner, R.; Amendt, J.; Boehme, P. Gene expression analysis as a tool for age estimation of blowfly pupae. Forensic Sci. Int. Genet. Suppl. Ser. 2009, 2, 292–293. [Google Scholar] [CrossRef]
- Richards, C.S.; Simonsen, T.J.; Abel, R.L.; Hall, M.J.; Schwyn, D.A.; Wicklein, M. Virtual forensic entomology: Improving estimates of minimum post-mortem interval with 3D micro-computed tomography. Forensic Sci. Int. 2012, 220, 251–264. [Google Scholar] [CrossRef] [Green Version]
- Davidson, E.H.; Erwin, D.H. Gene Regulatory Networks and the Evolution of Animal Body Plans. Science 2006, 311, 796–800. [Google Scholar] [CrossRef] [Green Version]
- Carroll, S.B. Evo-Devo and an Expanding Evolutionary Synthesis: A Genetic Theory of Morphological Evolution. Cell 2008, 134, 25–36. [Google Scholar] [CrossRef]
- A Levine, M.; Tjian, R. Transcription regulation and animal diversity. Nature 2003, 424, 147–151. [Google Scholar] [CrossRef]
- Peter, I.S.; Davidson, E.H. Evolution of Gene Regulatory Networks Controlling Body Plan Development. Cell 2011, 144, 970–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rebeiz, M.; Patel, N.H.; Hinman, V.F. Unraveling the Tangled Skein: The Evolution of Transcriptional Regulatory Networks in Development. Annu. Rev. Genom. Hum. Genet. 2015, 16, 103–131. [Google Scholar] [CrossRef] [PubMed]
- Skaer, N.; Pistillo, D.; Simpson, P. Transcriptional Heterochrony of scute and Changes in Bristle Pattern between Two Closely Related Species of Blowfly. Dev. Biol. 2002, 252, 31–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Géminard, C.; Arquier, N.; Layalle, S.; Bourouis, M.; Slaidina, M.; Delanoue, R.; Bjordal, M.; Ohanna, M.; Ma, M.; Colombani, J.; et al. Control of Metabolism and Growth Through Insulin-Like Peptides in Drosophila. Diabetes 2006, 55, S5–S8. [Google Scholar] [CrossRef] [Green Version]
- Feany, M.B.; Bender, W.W. A Drosophila model of Parkinson′s disease. Nature 2000, 404, 394–398. [Google Scholar] [CrossRef]
- Tarone, A.M.; Foran, D.R. Gene Expression During Blow Fly Development: Improving the Precision of Age Estimates in Forensic Entomology*,†. J. Forensic Sci. 2010, 56, S112–S122. [Google Scholar] [CrossRef]
- Tarone, A.; Jennings, K.C.; Foran, D.R. Aging Blow Fly Eggs Using Gene Expression: A Feasibility Study. J. Forensic Sci. 2007, 52, 1350–1354. [Google Scholar] [CrossRef]
- Boehme, P.; Spahn, P.; Amendt, J.; Zehner, R. Differential gene expression during metamorphosis: A promising approach for age estimation of forensically important Calliphora vicina pupae (Diptera: Calliphoridae). Int. J. Leg. Med. 2012, 127, 243–249. [Google Scholar] [CrossRef]
- Sze, S.-H.; Dunham, J.P.; Carey, B.; Chang, P.L.; Li, F.; Edman, R.M.; Fjeldsted, C.; Scott, M.J.; Nuzhdin, S.V.; Tarone, A.M. A de novo transcriptome assembly of Lucilia sericata (Diptera: Calliphoridae) with predicted alternative splices, single nucleotide polymorphisms and transcript expression estimates. Insect Mol. Biol. 2012, 21, 205–221. [Google Scholar] [CrossRef]
- Tarone, A.M.; Singh, B.; Picard, C.J. Molecular Biology in Forensic Entomology. In Forensic Entomology: International Dimensions and Frontiers; CRC Press: Boca Raton, FL, USA, 2015; pp. 297–316. [Google Scholar] [CrossRef]
- Parker, R.; Herrick, D.; Peltz, S.W.; Jacobson, A. Measurement of mRNA decay rates in Saccharomyces cerevisiae. Methods Enzymol. 1991, 194, 415–423. [Google Scholar] [CrossRef]
- Peltz, S.W.; Brewer, G.; Bernstein, P.; A Hart, P.; Ross, J. Regulation of mRNA turnover in eukaryotic cells. Crit. Rev. Eukaryot. Gene Expr. 1991, 194, 99. [Google Scholar]
- Régnier, P.; Arraiano, C.M. Degradation of mRNA in bacteria: Emergence of ubiquitous features. Bioessays 2000, 22, 235–244. [Google Scholar] [CrossRef]
- Wu, Q.; Wang, C.; Guo, L.; Ge, Q.; Lu, Z. Identification and characterization of novel microRNA candidates from deep sequencing. Clin. Chim. Acta 2013, 415, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Aryani, A.; Denecke, B. In vitro application of ribonucleases: Comparison of the effects on mRNA and miRNA stability. BMC Res. Notes 2015, 8, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courts, C.; Madea, B. Micro-RNA–A potential for forensic science? Forensic Sci. Int. 2010, 203, 106–111. [Google Scholar] [CrossRef]
- Courts, C.; Madea, B. Specific Micro-RNA Signatures for the Detection of Saliva and Blood in Forensic Body-fluid Identification. J. Forensic Sci. 2011, 56, 1464–1470. [Google Scholar] [CrossRef]
- Hutvagner, G.; Zamore, P.D. A microRNA in a Multiple-Turnover RNAi Enzyme Complex. Science 2002, 297, 2056–2060. [Google Scholar] [CrossRef] [Green Version]
- Jung, M.; Schaefer, A.; Steiner, I.; Kempkensteffen, C.; Stephan, C.; Erbersdobler, A.; Jung, K. Robust MicroRNA Stability in Degraded RNA Preparations from Human Tissue and Cell Samples. Clin. Chem. 2010, 56, 998–1006. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Ba, Y.; Ma, L.; Cai, X.; Yin, Y.; Wang, K.; Guo, J.; Zhang, Y.; Chen, J.; Guo, X.; et al. Characterization of microRNAs in serum: A novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008, 18, 997–1006. [Google Scholar] [CrossRef]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O′Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [Green Version]
- Blenkiron, C.; Tsai, P.; Brown, L.A.; Tintinger, V.; Askelund, K.J.; Windsor, J.A.; Phillips, A.R. Characterisation of the Small RNAs in the Biomedically Important Green-Bottle Blowfly Lucilia sericata. PLoS ONE 2015, 10, e0122203. [Google Scholar] [CrossRef] [PubMed]
- Martín-Vega, D.; Hall, M.J.R.; Simonsen, T.J. Resolving Confusion in the Use of Concepts and Terminology in Intrapuparial Development Studies of Cyclorrhaphous Diptera. J. Med. Èntomol. 2016, 53, 1249–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarone, A.M. Ecological Genetics. In Carrion Ecology, Evolution, and Their Applications; CRC Press: Boca Raton, FL, USA, 2015; pp. 308–347. [Google Scholar] [CrossRef]
- Hjelmen, C.E.; Parrott, J.J.; Srivastav, S.P.; McGuane, A.S.; Ellis, L.L.; Stewart, A.D.; Johnston, J.S.; Tarone, A.M. Effect of Phenotype Selection on Genome Size Variation in Two Species of Diptera. Genes 2020, 11, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attrill, H.; Falls, K.; Goodman, J.L.; Millburn, G.H.; Antonazzo, G.; Rey, A.J.; Marygold, S.J.; the FlyBase consortium. FlyBase: Establishing a Gene Group resource for Drosophila melanogaster. Nucleic Acids Res. 2015, 44, D786–D792. [Google Scholar] [CrossRef] [Green Version]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2013, 42, D68–D73. [Google Scholar] [CrossRef] [Green Version]
- Jonika, M.M.; Hjelmen, C.; Faris, A.M.; McGuane, A.S.; Tarone, A.M. An Evaluation of Differentially Spliced Genes as Markers of Sex for Forensic Entomology. J. Forensic Sci. 2020, 65, 1579–1587. [Google Scholar] [CrossRef]
- Thurmond, J.; Goodman, J.L.; Strelets, V.B.; Attrill, H.; Gramates, L.S.; Marygold, S.J.; Matthews, B.B.; Millburn, G.; Antonazzo, G.; Trovisco, V.; et al. FlyBase 2.0: The next generation. Nucleic Acids Res. 2019, 47, D759–D765. [Google Scholar] [CrossRef] [Green Version]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res. 2010, 39, D152–D157. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Saini, H.K.; van Dongen, S.; Enright, A.J. miRBase: Tools for microRNA genomics. Nucleic Acids Res. 2008, 36, D154–D158. [Google Scholar] [CrossRef] [Green Version]
- Griffiths-Jones, S. The microRNA Registry. Nucleic Acids Res. 2004, 32, D109–D111. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Differential analysis of count data–the DESeq2 package. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022; Available online: https://www.R-project.org/ (accessed on 10 October 2022).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. Available online: https://ggplot2.tidyverse.org (accessed on 10 October 2022).
- Vu, V. A ggplot2 Based Biplot, R Package Version 0.55. 2011. Available online: http://github.com/vqv/ggbiplot (accessed on 10 October 2022).
- Liaw, A.; Wiener, M. Classification and regression by randomForest. R News 2002, 2, 18–22. Available online: https://CRAN.R-project.org/doc/Rnews/ (accessed on 8 September 2022).
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- Paulo, D.; Azeredo-Espin, A.M.L.; Canesin, L.E.C.; Vicentini, R.; Junqueira, A.C.M. Identification and characterization of microRNAs in the screwworm flies Cochliomyia hominivorax and Cochliomyia macellaria (Diptera: Calliphoridae). Insect Mol. Biol. 2016, 26, 46–57. [Google Scholar] [CrossRef]
- Tuckow, A.P.; Temeyer, K.B.; Olafson, P.U.; De León, A.A.P. Discovery of microRNAs of the stable fly (Diptera: Muscidae) by High-throughput sequencing. J. Med. Èntomol. 2013, 50, 925–930. [Google Scholar] [CrossRef]
- Campobasso, C.P.; Linville, J.G.; Wells, J.D.; Introna, F. Forensic Genetic Analysis of Insect Gut Contents. Am. J. Forensic Med. Pathol. 2005, 26, 161–165. [Google Scholar] [CrossRef]
- Linville, J.G.; Wells, J.D. Surface Sterilization of a Maggot Using Bleach Does Not Interfere with Mitochondrial DNA Analysis of Crop Contents. J. Forensic Sci. 2002, 47, 1–5. [Google Scholar] [CrossRef]
- Monthatong, T.T.M. Analysis of Swine (Sus scrofa) DNA from Blowfly (Chrysomya megacephala) Larval Guts after Post-Feeding Periods by PCR Technique. Walailak J. Sci. Technol. 2016, 13, 117–122. [Google Scholar] [CrossRef]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA Translation and Stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esslinger, S.M.; Schwalb, B.; Helfer, S.; Michalik, K.M.; Witte, H.; Maier, K.C.; Martin, D.; Michalke, B.; Tresch, A.; Cramer, P.; et al. Drosophila miR-277 controls branched-chain amino acid catabolism and affects lifespan. RNA Biol. 2013, 10, 1042–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, P.; Vernooy, S.Y.; Guo, M.; Hay, B.A. The Drosophila MicroRNA Mir-14 Suppresses Cell Death and Is Required for Normal Fat Metabolism. Curr. Biol. 2003, 13, 790–795. [Google Scholar] [CrossRef] [Green Version]
- Aboobaker, A.A.; Tomancak, P.; Patel, N.; Rubin, G.M.; Lai, E.C. Drosophila microRNAs exhibit diverse spatial expression patterns during embryonic development. Proc. Natl. Acad. Sci. USA 2005, 102, 18017–18022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuzin, A.; Kundu, M.; Brody, T.; Odenwald, W.F. The Drosophila nerfin-1 mRNA requires multiple microRNAs to regulate its spatial and temporal translation dynamics in the developing nervous system. Dev. Biol. 2007, 310, 35–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karres, J.S.; Hilgers, V.; Carrera, I.; Treisman, J.; Cohen, S.M. The Conserved microRNA MiR-8 Tunes Atrophin Levels to Prevent Neurodegeneration in Drosophila. Cell 2007, 131, 136–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D.S. MicroRNA targets in Drosophila. Genome Biol. 2003, 5, R1. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Lee, J.-E.; Padgett, R.W.; Edery, I. Circadian regulation of a limited set of conserved microRNAs in Drosophila. BMC Genom. 2008, 9, 83. [Google Scholar] [CrossRef] [Green Version]
- Owings, C.G.; Spiegelman, C.; Tarone, A.; Tomberlin, J.K. Developmental variation among Cochliomyia macellaria Fabricius (Diptera: Calliphoridae) populations from three ecoregions of Texas, USA. Int. J. Leg. Med. 2014, 128, 709–717. [Google Scholar] [CrossRef]
- Barrio, L.; Dekanty, A.; Milán, M. MicroRNA-Mediated Regulation of Dp53 in the Drosophila Fat Body Contributes to Metabolic Adaptation to Nutrient Deprivation. Cell Rep. 2014, 8, 528–541. [Google Scholar] [CrossRef] [Green Version]
- Foronda, D.; Weng, R.; Verma, P.; Chen, Y.-W.; Cohen, S.M. Coordination of insulin and Notch pathway activities by microRNA miR-305 mediates adaptive homeostasis in the intestinal stem cells of the Drosophila gut. Genes Dev. 2014, 28, 2421–2431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collier, T.J.; Coleman, P.D. Divergence of biological and chronological aging: Evidence from rodent studies. Neurobiol. Aging 1991, 12, 685–693. [Google Scholar] [CrossRef]
- Ligout, S.; Munier, D.; Marquereau, L.; Greenfield, M.D. Chronological vs. Physiological Age as Determinants of Mating Decisions: Studies on Female Choice Over Lifespan in An Acoustic Moth. Ethology 2012, 118, 740–751. [Google Scholar] [CrossRef]
- Jarman, S.N.; Polanowski, A.M.; Faux, C.E.; Robbins, J.; De Paoli-Iseppi, R.; Bravington, M.; Deagle, B.E. Molecular biomarkers for chronological age in animal ecology. Mol. Ecol. 2015, 24, 4826–4847. [Google Scholar] [CrossRef]
- Rando, T.A.; Chang, H.Y. Aging, Rejuvenation, and Epigenetic Reprogramming: Resetting the Aging Clock. Cell 2012, 148, 46–57. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.-H.; Marinotti, O.; Zhong, D.; James, A.; Walker, E.; Guda, T.; Kweka, E.J.; Githure, J.; Yan, G. Gene Expression-Based Biomarkers for Anopheles gambiae Age Grading. PLoS ONE 2013, 8, e69439. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Liang, S.; Zhao, Y.; Han, Z. miR-92b regulates Mef2 levels through a negative-feedback circuit during Drosophila muscle development. Development 2012, 139, 3543–3552. [Google Scholar] [CrossRef] [Green Version]
- Brennecke, J.; Hipfner, D.R.; Stark, A.; Russell, R.B.; Cohen, S.M. bantam Encodes a Developmentally Regulated microRNA that Controls Cell Proliferation and Regulates the Proapoptotic Gene hid in Drosophila. Cell 2003, 113, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Hipfner, D.R.; Weigmann, K.; Cohen, S.M. The bantam Gene Regulates Drosophila Growth. Genetics 2002, 161, 1527–1537. [Google Scholar] [CrossRef]
- Li, F.; Ii, S.P.V.; Belikoff, E.J.; Scott, M.J. Conservation and Sex-Specific Splicing of the transformer Gene in the Calliphorids Cochliomyia hominivorax, Cochliomyia macellaria and Lucilia sericata. PLoS ONE 2013, 8, e56303. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples (Three Bio-Reps, Two Technical Reps) | Hours to Collection (Development % Complete) | Total Reads (All Lengths) | Unique Reads (All lengths) | Total Reads Counts (20–25 nt) | Unique Reads (20–25 nt) | Counts of Reads for Predicted miRNA |
|---|---|---|---|---|---|---|
| Feeding 3rd instar | 76–80 h (29.9%) | 13,210,010 | 2,858,189 | 3,513,336 | 767,584 | 220,921 |
| Early postfeeding | 102–106 h (40.5%) | 14,952,640 | 3,213,766 | 3,657,951 | 873,629 | 218,698 |
| Late postfeeding | 120–124 h (46.9%) | 10,088,276 | 944,756 | 268,403 | 100,713 | 23,902 |
| Early intrapuparial | 136–140 h (52.2%) | 8,887,998 | 697,479 | 237,896 | 74,940 | 32,738 |
| Mid-intrapuparial 1 | 180–184 h (70.0%) | 10,959,838 | 1,467,170 | 578,729 | 152,024 | 36,722 |
| Mid-intrapuparial 2 | 236–240 h (90.5%) | 10,731,968 | 1,134,567 | 348,102 | 93,491 | 15,441 |
| Late intrapuparial | 260–264 h (100.0%) | 10,368,840 | 1,159,427 | 209,517 | 89,137 | 20,930 |
| Control early postfeeding | 115–119 h (40.7%) | 15,604,997 | 3,148,131 | 1,819,746 | 593,846 | 147,927 |
| Control-mid-intrapuparial 2 | 212–216 h (74.4%) | 15,912,519 | 2,551,141 | 1,150,734 | 359,491 | 100,406 |
| Fast early postfeeding | 104–108 h (40.9%) | 16,167,408 | 3,530,944 | 2,426,556 | 719,518 | 190,589 |
| Fast mid-intrapuparial 2 | 192–196 h (75.0%) | 14,619,401 | 3,005,893 | 2,103,762 | 585,688 | 139,318 |
| Slow early postfeeding | 127–131 h (40.5%) | 10,596,116 | 1,221,303 | 449,203 | 144,935 | 9,295 |
| Slow mid-intrapuparial 2 | 233–237 h (74.7%) | 14,027,331 | 1,762,262 | 1,424,884 | 217,608 | 45,952 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hjelmen, C.E.; Yuan, Y.; Parrott, J.J.; McGuane, A.S.; Srivastav, S.P.; Purcell, A.C.; Pimsler, M.L.; Sze, S.-H.; Tarone, A.M. Identification and Characterization of Small RNA Markers of Age in the Blow Fly Cochliomyia macellaria (Fabricius) (Diptera: Calliphoridae). Insects 2022, 13, 948. https://doi.org/10.3390/insects13100948
Hjelmen CE, Yuan Y, Parrott JJ, McGuane AS, Srivastav SP, Purcell AC, Pimsler ML, Sze S-H, Tarone AM. Identification and Characterization of Small RNA Markers of Age in the Blow Fly Cochliomyia macellaria (Fabricius) (Diptera: Calliphoridae). Insects. 2022; 13(10):948. https://doi.org/10.3390/insects13100948
Chicago/Turabian StyleHjelmen, Carl E., Ye Yuan, Jonathan J. Parrott, Alexander S. McGuane, Satyam P. Srivastav, Amanda C. Purcell, Meaghan L. Pimsler, Sing-Hoi Sze, and Aaron M. Tarone. 2022. "Identification and Characterization of Small RNA Markers of Age in the Blow Fly Cochliomyia macellaria (Fabricius) (Diptera: Calliphoridae)" Insects 13, no. 10: 948. https://doi.org/10.3390/insects13100948