Diet Breadth Affects Bacterial Identity but Not Diversity in the Pollen Provisions of Closely Related Polylectic and Oligolectic Bees

 , ,
, ,
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Locations
2.2. DNA Extraction and Library Preparation
2.3. Bioinformatics and Statistics
3. Results
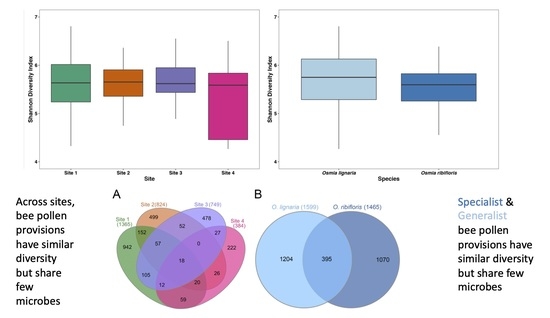
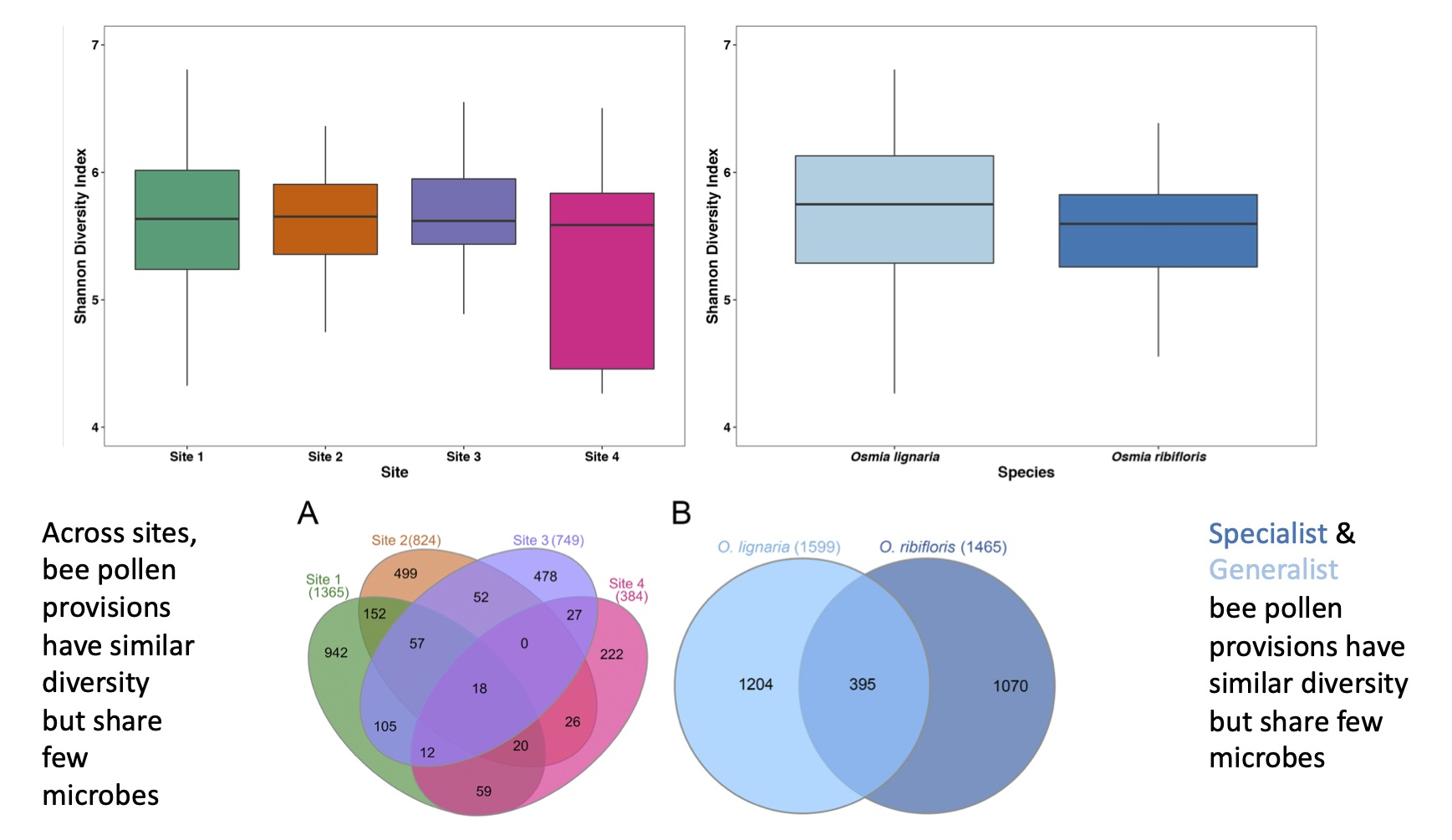
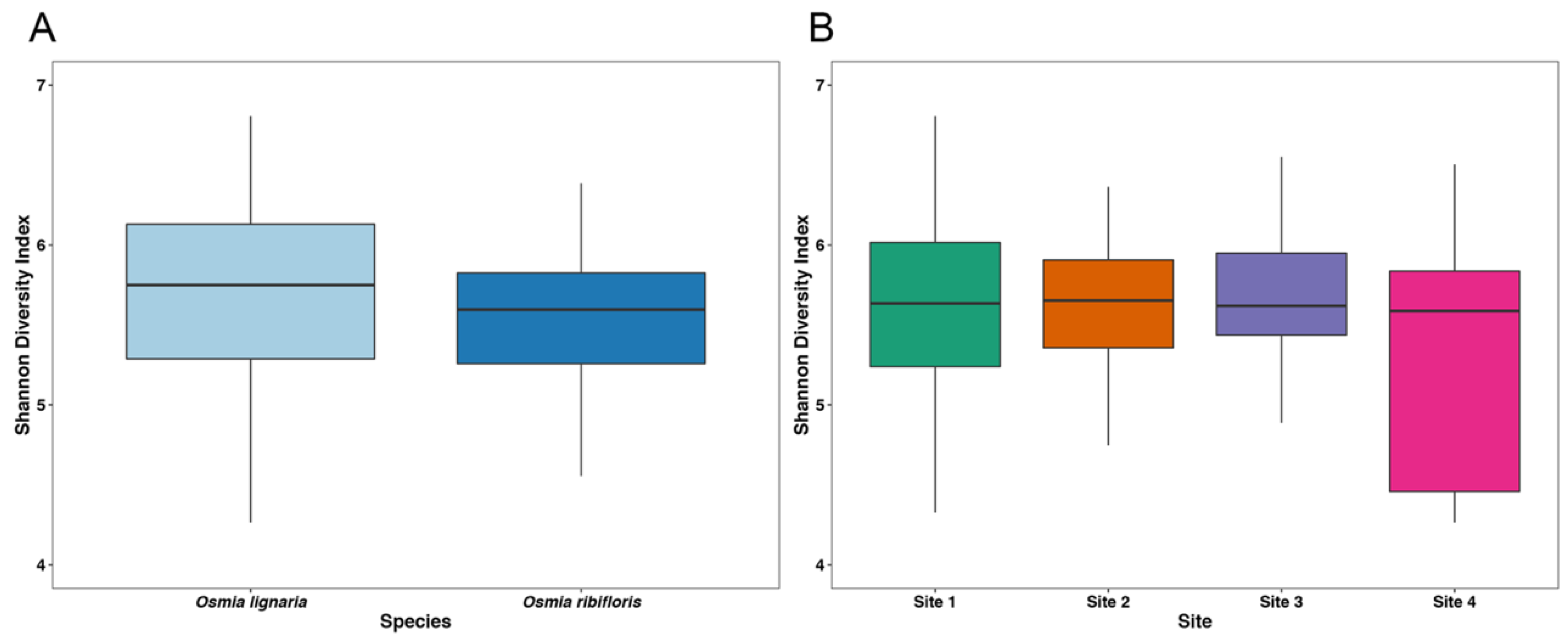
3.1. Alpha Diversity and Library Statistics
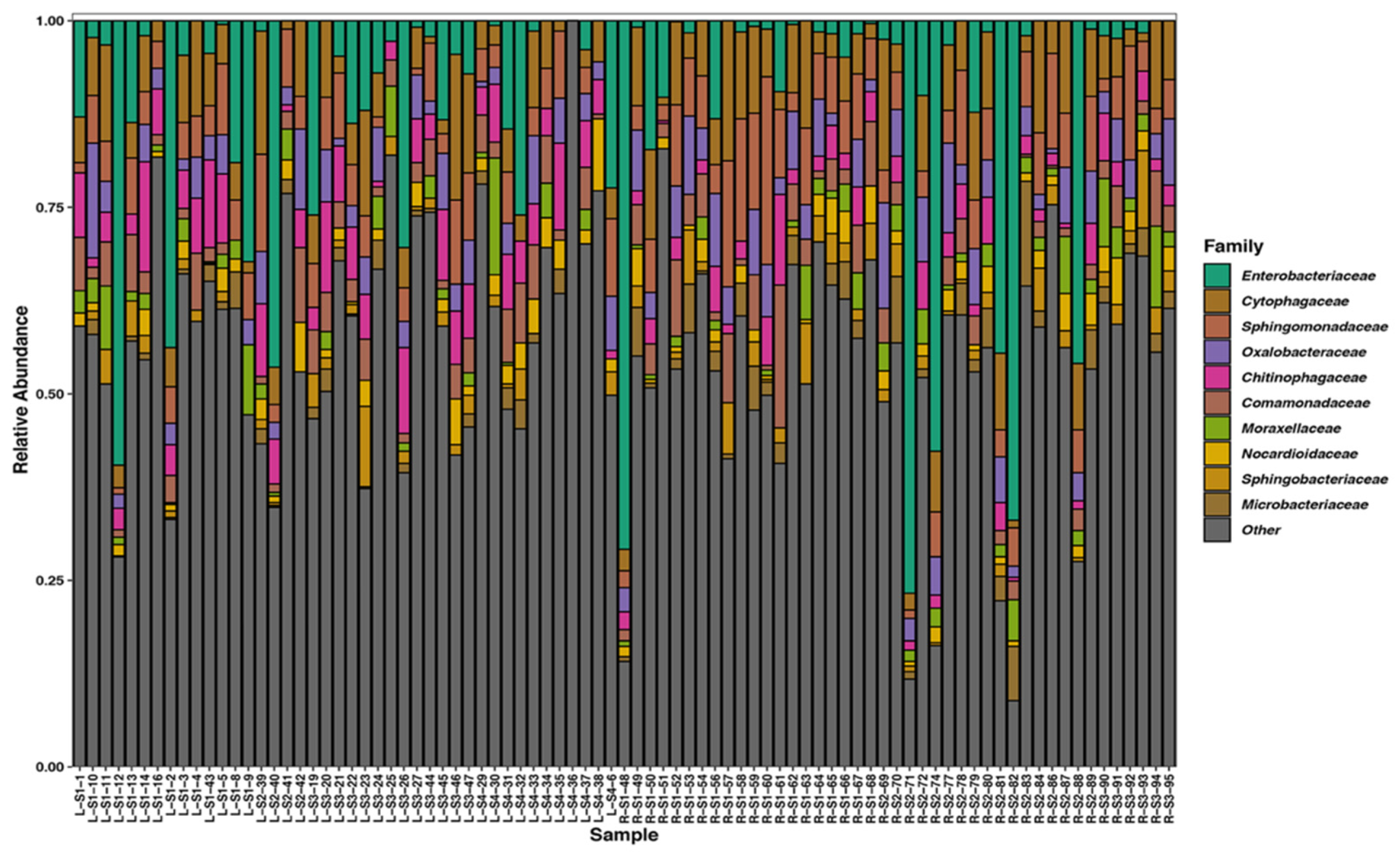
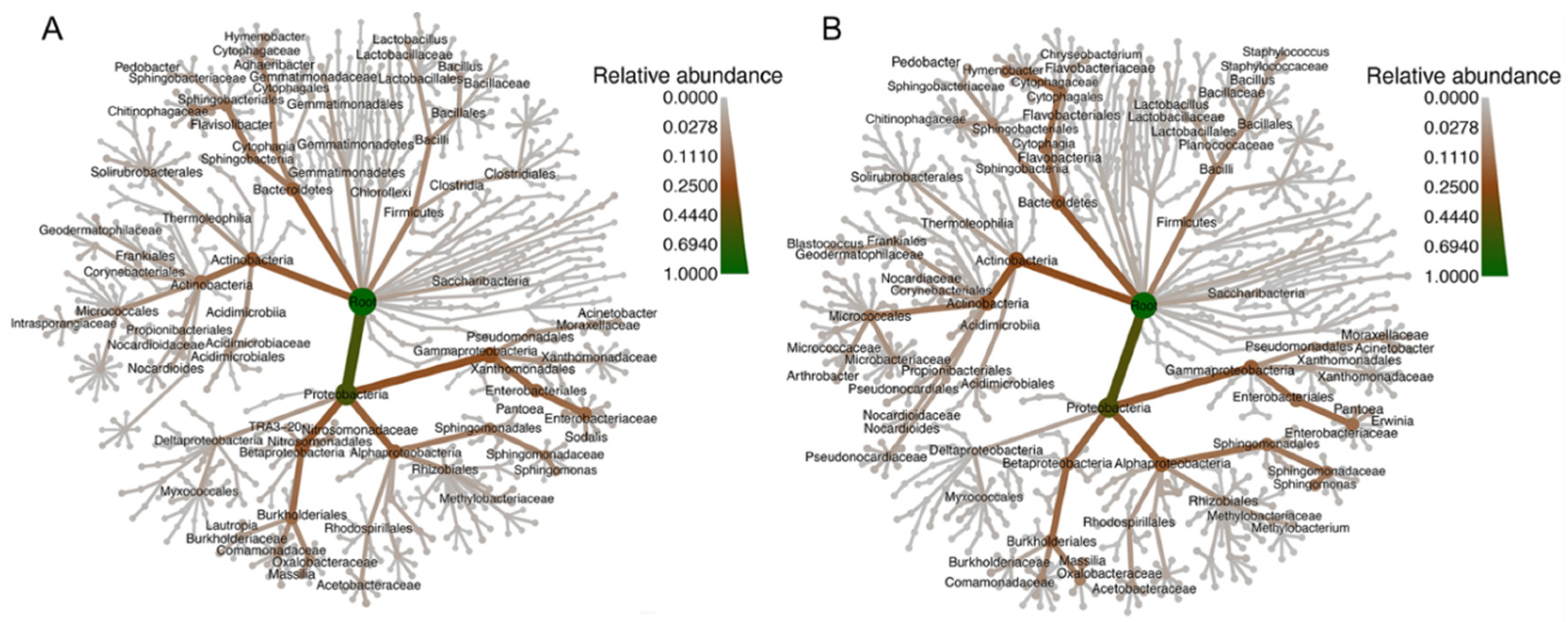
3.2. Taxonomic Description of the Data
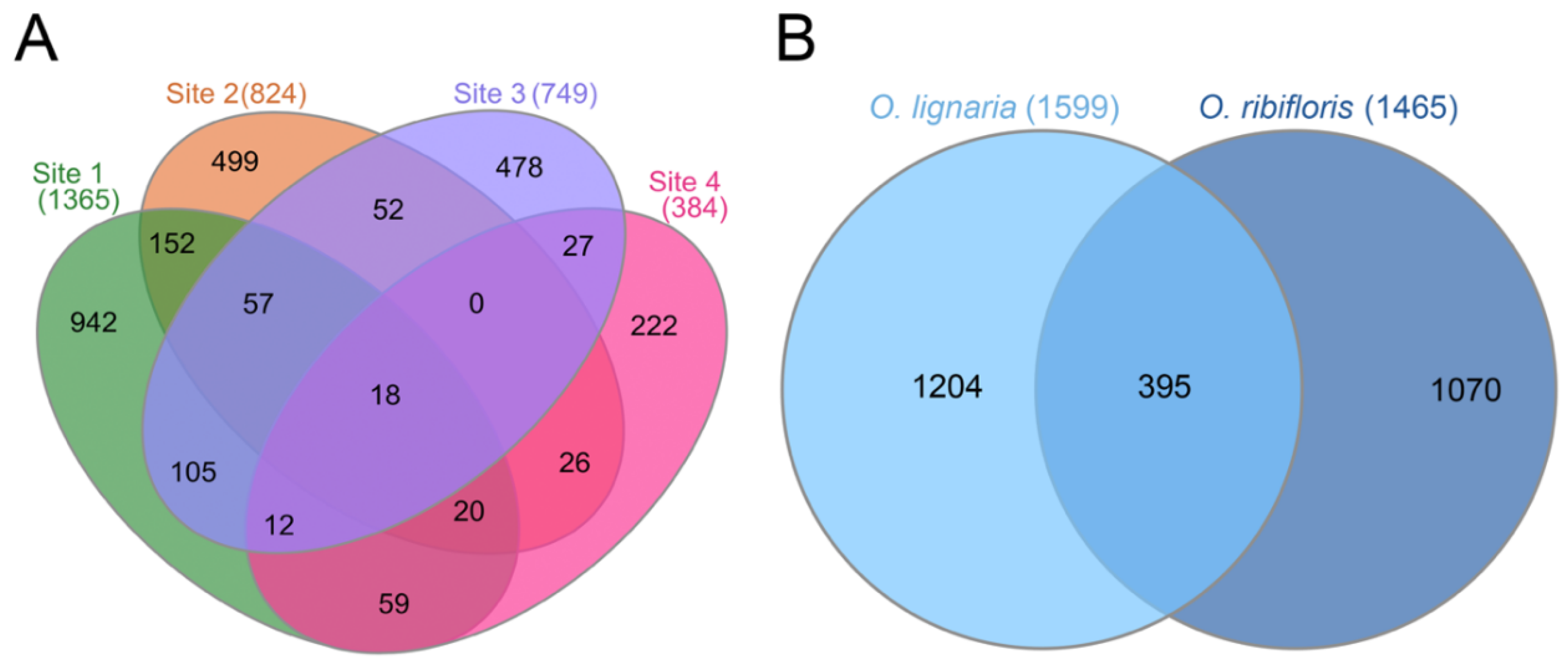
3.3. Shared and Unique Amplicon Sequence Variants by Bee Species and Site
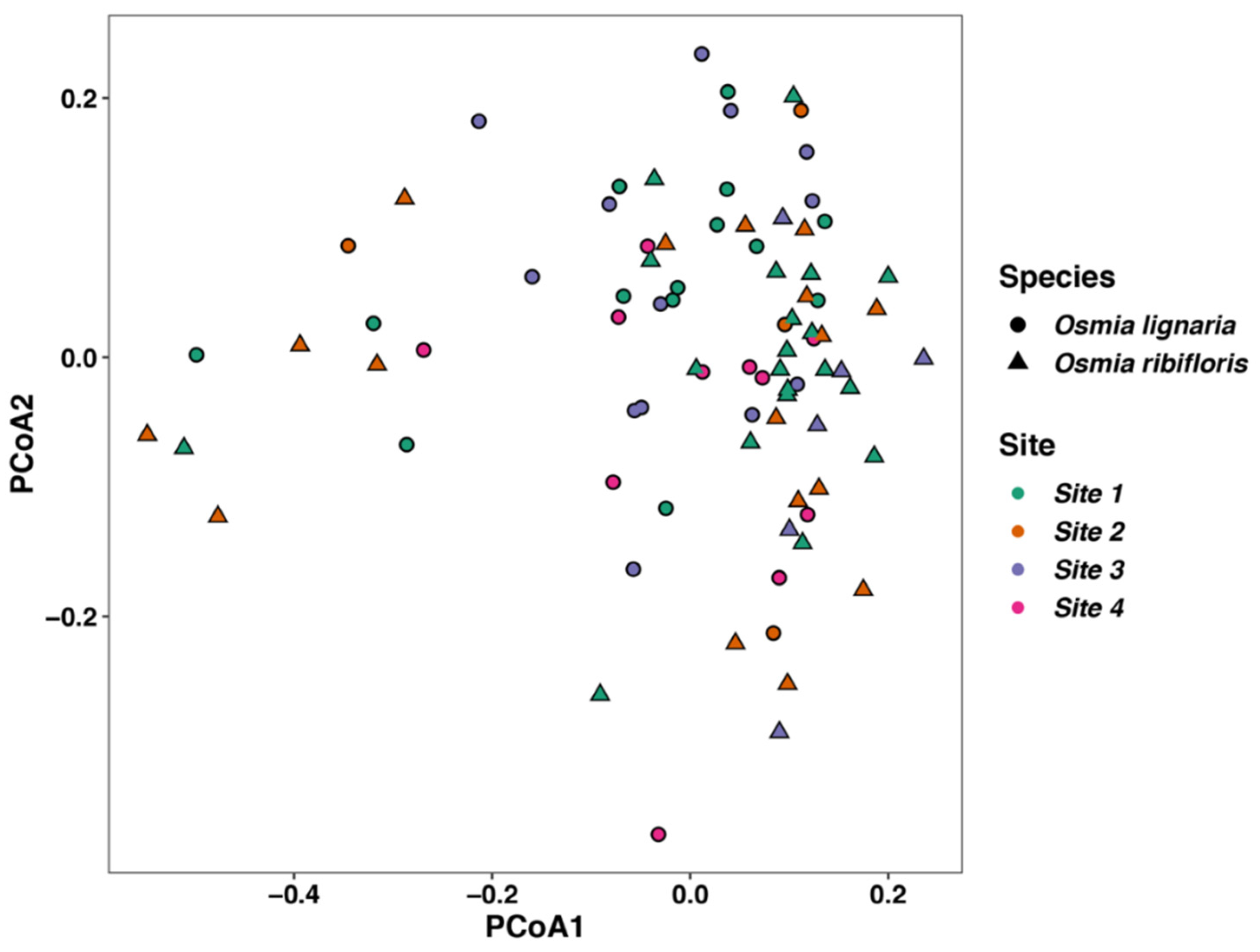
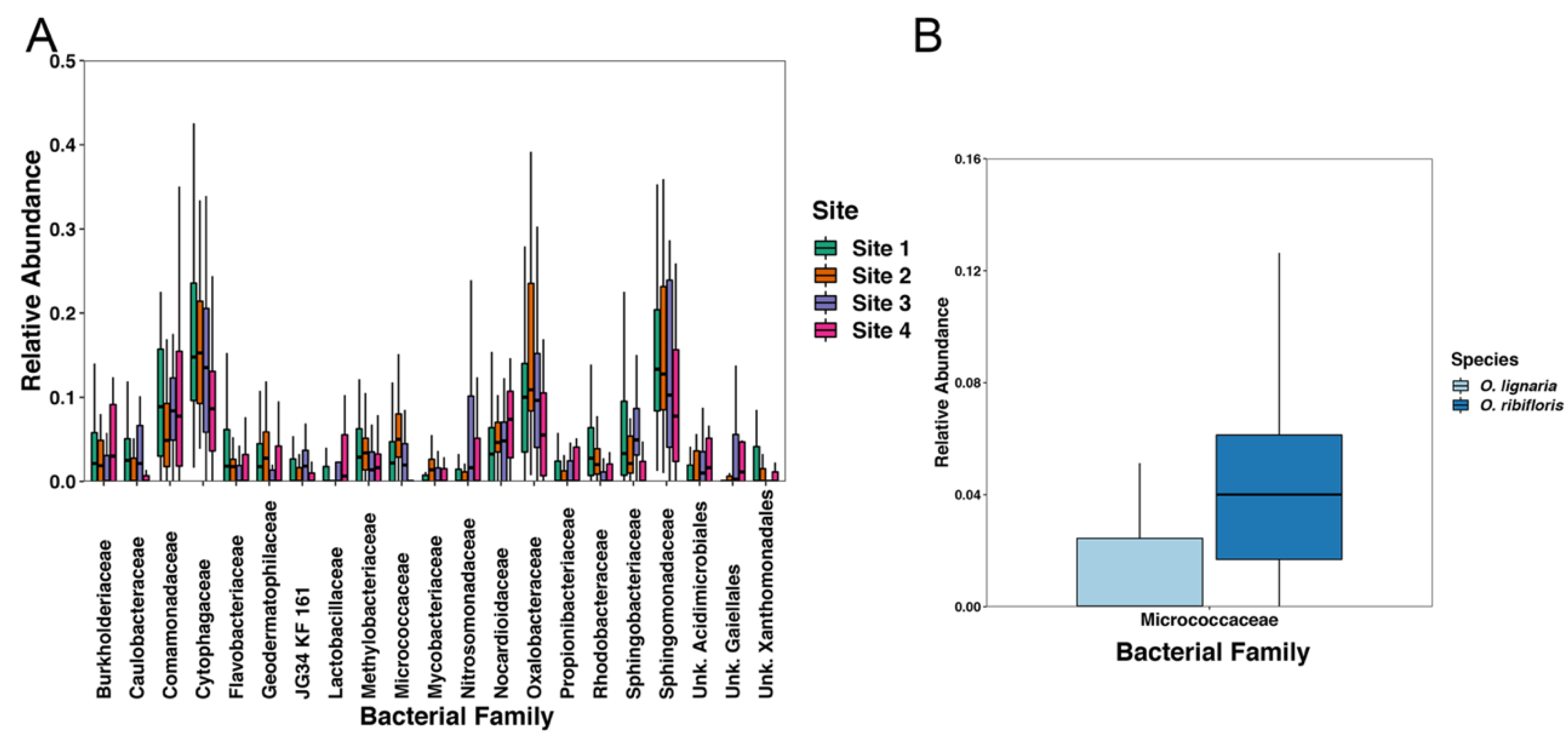
3.4. Beta Diversity of Microbes within Osmia Species and Site Location, and Differential Abundance of Bacterial Families
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Michener, C.D. The Bees of the World; John Hopkins University Press: Baltimore, MD, USA, 2007. [Google Scholar]
- McFrederick, Q.S.; Roulston, T.H.; Taylor, D.R. Evolution of conflict and cooperation of nematodes associated with solitary and social sweat bees. Insectes Sociaux 2013, 60, 309–317. [Google Scholar] [CrossRef]
- Biani, N.B.; Mueller, U.G.; Wcislo, W.T. Cleaner mites: Sanitary mutualism in the miniature ecosystem of Neotropical bee nests. Am. Nat. 2009, 173, 841–847. [Google Scholar] [CrossRef] [Green Version]
- Mari Mutt, J.A. New species of melittophilous Pseudocyphoderijs from Costa Rica and remarks on taxonomy. Entomol. News 1977, 88, 229–234. [Google Scholar]
- Gilliam, M.; Buchmann, S.; Lorenz, B. Microbial flora of the larval provisions of the solitary bees, Centris pallida and Anthophora sp. Apidologie 1984, 15, 1–10. [Google Scholar] [CrossRef]
- Gilliam, M. Microbiology of pollen and bee bread—The yeasts. Apidologie 1979, 10, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Voulgari-Kokota, A.; McFrederick, Q.S.; Steffan-Dewenter, I.; Keller, A. Drivers, diversity, and functions of the solitary-bee microbiota. Trends Microbiol. 2019, 27, 1034–1044. [Google Scholar] [CrossRef]
- McFrederick, Q.S.; Wcislo, W.T.; Taylor, D.R.; Ishak, H.D.; Dowd, S.E.; Mueller, U.G. Environment or kin: Whence do bees obtain acidophilic bacteria? Mol. Ecol. 2012, 21, 1754–1768. [Google Scholar] [CrossRef]
- Keller, A.; Grimmer, G.; Steffan-Dewenter, I. Diverse microbiota Identified in whole intact nest chambers of the red mason bee Osmia bicornis (Linnaeus 1758). PLoS ONE 2013, 8, 78296. [Google Scholar] [CrossRef] [Green Version]
- McFrederick, Q.S.; Thomas, J.M.; Neff, J.L.; Vuong, H.Q.; Russell, K.A.; Hale, A.R.; Mueller, U.G. Flowers and wild megachilid bees share microbes. Microb. Ecol. 2017, 73, 188–200. [Google Scholar] [CrossRef]
- Voulgari-Kokota, A.; Ankenbrand, M.J.; Grimmer, G.; Steffan-Dewenter, I.; Keller, A. Linking pollen foraging of megachilid bees to their nest bacterial microbiota. Ecol. Evol. 2019, 9, 10788–10800. [Google Scholar] [CrossRef] [Green Version]
- McFrederick, Q.S.; Rehan, S.M. Characterization of pollen and bacterial community composition in brood provisions of a small carpenter bee. Mol. Ecol. 2016, 25, 2302–2311. [Google Scholar] [CrossRef] [PubMed]
- McFrederick, Q.S.; Rehan, S.M. Wild bee pollen usage and microbial communities co-vary across landscapes. Microb. Ecol. 2019, 77, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Vuong, H.Q.; McFrederick, Q.S. Comparative genomics of wild bee and flower isolated Lactobacillus reveals potential adaptation to the bee host. Genome Biol. Evol. 2019, 11, 2151–2161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, R.F.; Knorr, R.; Müller, M.R.A.; Steudel, U.; Gänzle, M.G.; Ehrmann, M.A. Non-dairy lactic fermentations: The cereal world. Antonie Van Leeuwenhoek 1999, 76, 403–411. [Google Scholar] [CrossRef]
- Steffan, S.A.; Dharampal, P.S.; Danforth, B.N.; Gaines-Day, H.R.; Takizawa, Y.; Chikaraishi, Y. Omnivory in bees: Elevated trophic positions among all major bee families. Am. Nat. 2019, 194, 414–421. [Google Scholar] [CrossRef]
- Dharampal, P.S.; Carlson, C.; Currie, C.R.; Steffan, S.A. Pollen-borne microbes shape bee fitness. Proc. R. Soc. B Biol. Sci. 2019, 286, 20182894. [Google Scholar] [CrossRef] [Green Version]
- Dharampal, P.; Hetherington, M.; Steffan, S. Microbes make the meal: Oligolectic bees require microbes within their host pollen to thrive. Ecol. Entomol. 2020, in press. [Google Scholar] [CrossRef]
- Cane, J.; Sipes, S. Characterizing floral specialization by bees: Analytical methods and a revised lexicon for oligolecty. In Plant-Pollinator Interactions: From Specialization to Generalization; University of Chicago Press: Chicago, IL, USA, 2006; pp. 99–122. [Google Scholar]
- Zemenick, A.T.; Vannette, R.L.; Rosenheim, J.A. Linked networks reveal dual roles of insect dispersal and species sorting for bacterial communities in flowers. bioRxiv 2019, 847376. [Google Scholar] [CrossRef]
- Bosch, J.; Kemp, W.P. Development and emergence of the orchard pollinator Osmia lignaria (Hymenoptera: Megachilidae). Environ. Entomol. 2000, 29, 8–13. [Google Scholar] [CrossRef]
- Torchio, P.F. Osmia ribifloris, a native bee species developed as a commercially managed pollinator of highbush blueberry (Hymenoptera: Megachilidae). J. Kans. Entomol. Soc. 1990, 63, 427–436. [Google Scholar]
- Pinilla-Gallego, M.S.; Isaacs, R. Pollen use by Osmia lignaria (Hymenoptera: Megachilidae) in highbush blueberry fields. Ann. Entomol. Soc. Am. 2018, 111, 335–340. [Google Scholar] [CrossRef]
- Engel, P.; James, R.R.; Koga, R.; Kwong, W.K.; Mcfrederick, Q.S.; Moran, N.A. Standard methods for research on Apis mellifera gut symbionts. J. Apic. Res. 2013, 524, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Rothman, J.A.; Carroll, M.J.; Meikle, W.G.; Anderson, K.E.; McFrederick, Q.S. Longitudinal effects of supplemental forage on the honey bee (Apis mellifera) microbiota and inter- and intra-colony variability. Microb. Ecol. 2018, 76, 814–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennington, M.J.; Rothman, J.A.; Jones, M.B.; McFrederick, Q.S.; Gan, J.; Trumble, J.T. Effects of contaminants of emerging concern on Myzus persicae (Sulzer, Hemiptera: Aphididae) biology and on their host plant, Capsicum annuum. Environ. Monit. Assess. 2018, 190, 125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothman, J.A.; Leger, L.; Graystock, P.; Russell, K.; McFrederick, Q.S. The bumble bee microbiome increases survival of bees exposed to selenate toxicity. Environ. Microbiol. 2019, 21, 3417–3429. [Google Scholar] [CrossRef] [Green Version]
- Pennington, M.J.; Rothman, J.A.; Jones, M.B.; McFrederick, Q.S.; Gan, J.; Trumble, J.T. Effects of contaminants of emerging concern on Megaselia scalaris (Lowe, Diptera: Phoridae) and its microbial community. Sci. Rep. 2017, 7, 8165. [Google Scholar] [CrossRef] [Green Version]
- Hanshew, A.S.; Mason, C.J.; Raffa, K.F.; Currie, C.R. Minimization of chloroplast contamination in 16S rRNA gene pyrosequencing of insect herbivore bacterial communities. J. Microbiol. Methods 2013, 95, 149–155. [Google Scholar] [CrossRef] [Green Version]
- Kembel, S.W.; O’Connor, T.K.; Arnold, H.K.; Hubbell, S.P.; Wright, S.J.; Green, J.L. Relationships between phyllosphere bacterial communities and plant functional traits in a neotropical forest. Proc. Natl. Acad. Sci. USA 2014, 111, 13715–13720. [Google Scholar] [CrossRef] [Green Version]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, 590–596. [Google Scholar] [CrossRef]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2-approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, 9490. [Google Scholar] [CrossRef] [PubMed]
- R Core Team R. A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. 2017. Available online: https://CRAN.R-project.org/package=vegan (accessed on 20 September 2020).
- Mandal, S.; Van Treuren, W.; White, R.A.; Eggesbø, M.; Knight, R.; Peddada, S.D. Analysis of composition of microbiomes: A novel method for studying microbial composition. Microb. Ecol. Health Dis. 2015, 26, 27663. [Google Scholar] [CrossRef] [Green Version]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin, Germany, 2009. [Google Scholar]
- Foster, Z.S.L.; Sharpton, T.J.; Grünwald, N.J. Metacoder: An R package for visualization and manipulation of community taxonomic diversity data. PLoS Comput. Biol. 2017, 13, 1005404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothman, J.A.; Andrikopoulos, C.; Cox-Foster, D.; McFrederick, Q.S. Floral and foliar source affect the bee nest microbial community. Microb. Ecol. 2019, 78, 506–516. [Google Scholar] [CrossRef] [Green Version]
- Al Atrouni, A.; Joly-Guillou, M.-L.; Hamze, M.; Kempf, M. Reservoirs of non-baumannii Acinetobacter species. Front. Microbiol. 2016, 7, 49. [Google Scholar] [CrossRef] [Green Version]
- McFrederick, Q.S.; Wcislo, W.T.; Hout, M.C.; Mueller, U.G. Host species and developmental stage, but not host social structure, affects bacterial community structure in socially polymorphic bees. FEMS Microbiol. Ecol. 2014, 88, 398–406. [Google Scholar] [CrossRef] [Green Version]
- McFrederick, Q.S.; Vuong, H.Q.; Rothman, J.A. Lactobacillus micheneri sp. nov., Lactobacillus timberlakei sp. nov. and Lactobacillus quenuiae sp. nov., lactic acid bacteria isolated from wild bees and flowers. Int. J. Syst. Evol. Microbiol. 2018, 68, 1879–1884. [Google Scholar] [CrossRef] [PubMed]
- Voulgari-Kokota, A.; Grimmer, G.; Steffan-Dewenter, I.; Keller, A. Bacterial community structure and succession in nests of two megachilid bee genera. FEMS Microbiol. Ecol. 2019, 95, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, H.; McFrederick, Q.S.; Philpott, S.M. Environment shapes the microbiome of the blue orchard bee, Osmia lignaria: RRH: Environmental drivers of bee microbiome. Microb. Ecol. 2020. [Google Scholar] [CrossRef]
- Ofek, M.; Hadar, Y.; Minz, D. Ecology of root colonizing Massilia (Oxalobacteraceae). PLoS ONE 2012, 7, 40117. [Google Scholar] [CrossRef] [PubMed]
- Bosch, J.; Kemp, W.P. How to Manage the Blue Orchard Bee; Suistainable Agriculture Network: Beltsville, MD, USA, 2001. [Google Scholar]
- Lozo, J.; Berić, T.; Terzić-Vidojević, A.; Stanković, S.; Fira, D.; Stanisavljević, L. Microbiota associated with pollen, bee bread, larvae and adults of solitary bee Osmia cornuta (Hymenoptera: Megachilidae). Bull. Entomol. Res. 2015, 105, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Loncaric, I.; Heigl, H.; Licek, E.; Moosbeckhofer, R.; Busse, H.-J.; Rosengarten, R. Typing of Pantoea agglomerans isolated from colonies of honey bees (Apis mellifera) and culturability of selected strains from honey. Apidologie 2009, 40, 40–54. [Google Scholar] [CrossRef]
- Smith; Neafie; Yeager; Skelton Micrococcus folliculitis in HIV-1 disease. Br. J. Dermatol. 1999, 141, 558–561. [CrossRef]
- Lourenço, A.P.; Guidugli-Lazzarini, K.R.; Freitas, F.C.P.; Bitondi, M.M.G.; Simões, Z.L.P. Bacterial infection activates the immune system response and dysregulates microRNA expression in honey bees. Insect Biochem. Mol. Biol. 2013, 43, 474–482. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rothman, J.A.; Cox-Foster, D.L.; Andrikopoulos, C.; McFrederick, Q.S. Diet Breadth Affects Bacterial Identity but Not Diversity in the Pollen Provisions of Closely Related Polylectic and Oligolectic Bees. Insects 2020, 11, 645. https://doi.org/10.3390/insects11090645
Rothman JA, Cox-Foster DL, Andrikopoulos C, McFrederick QS. Diet Breadth Affects Bacterial Identity but Not Diversity in the Pollen Provisions of Closely Related Polylectic and Oligolectic Bees. Insects. 2020; 11(9):645. https://doi.org/10.3390/insects11090645
Chicago/Turabian StyleRothman, Jason A., Diana L. Cox-Foster, Corey Andrikopoulos, and Quinn S. McFrederick. 2020. "Diet Breadth Affects Bacterial Identity but Not Diversity in the Pollen Provisions of Closely Related Polylectic and Oligolectic Bees" Insects 11, no. 9: 645. https://doi.org/10.3390/insects11090645