Diverse Diets with Consistent Core Microbiome in Wild Bee Pollen Provisions


Abstract
:1. Introduction
2. Methods
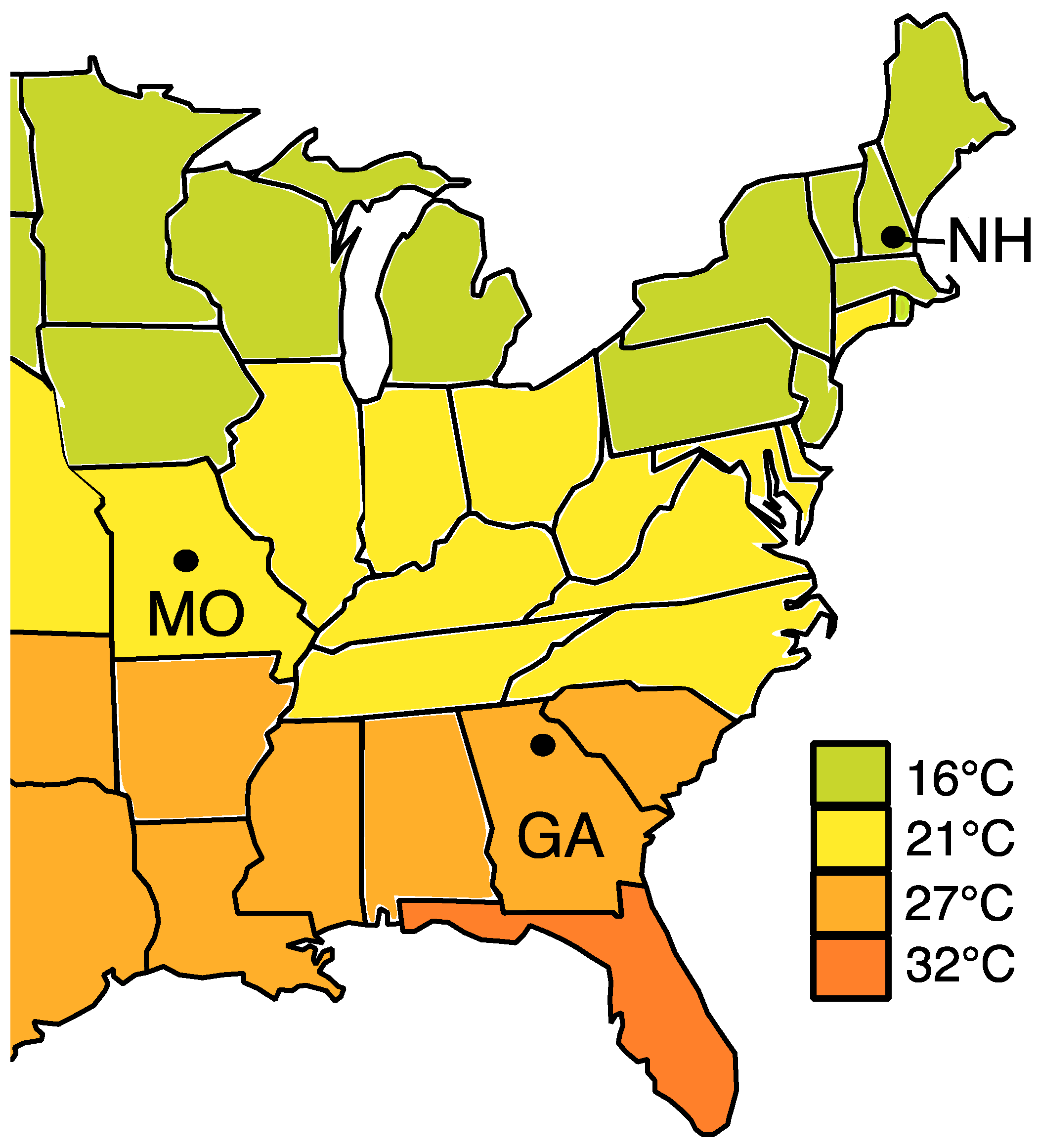
2.1. Brood Provision Collection
2.2. DNA Extraction and Sequencing
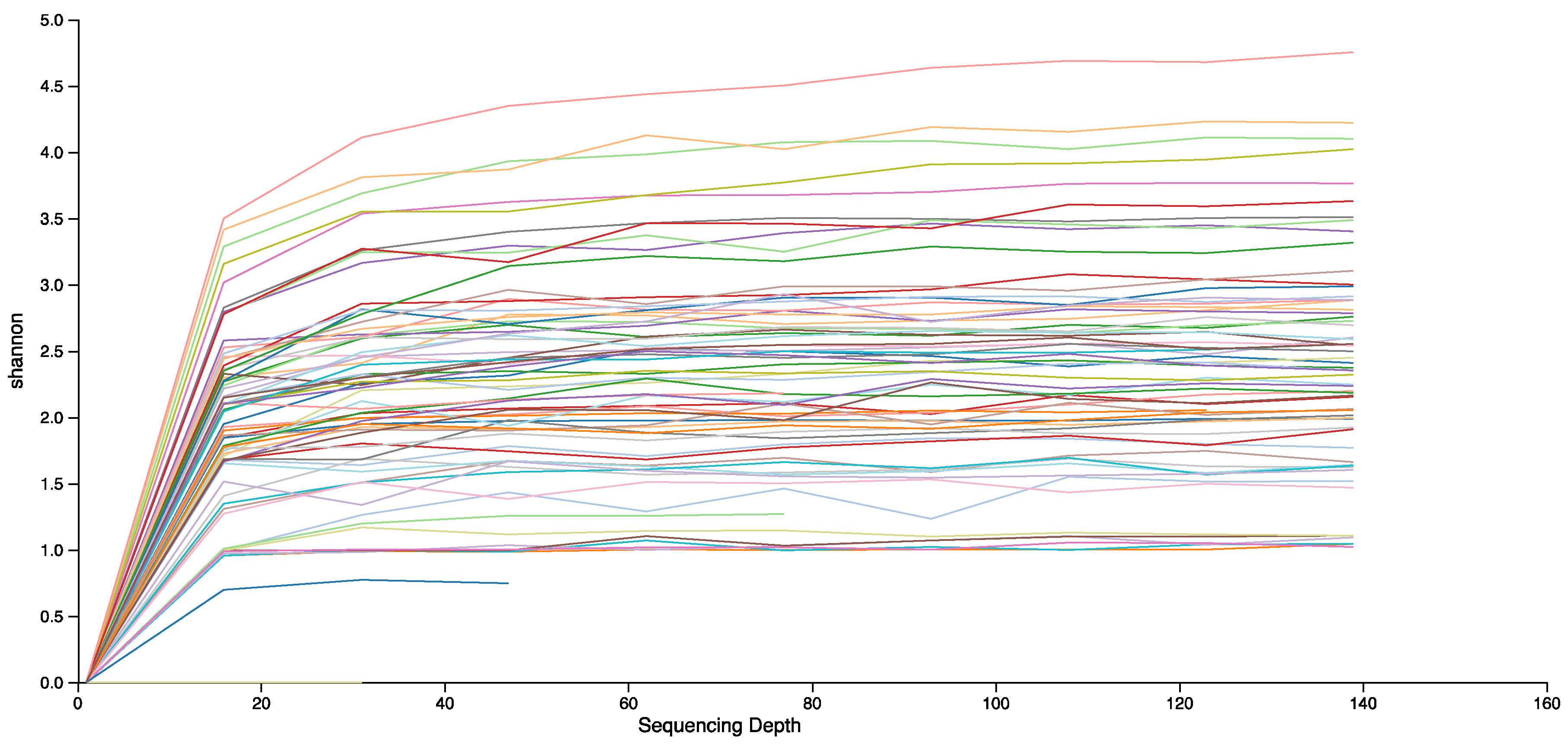
3. Data Analysis
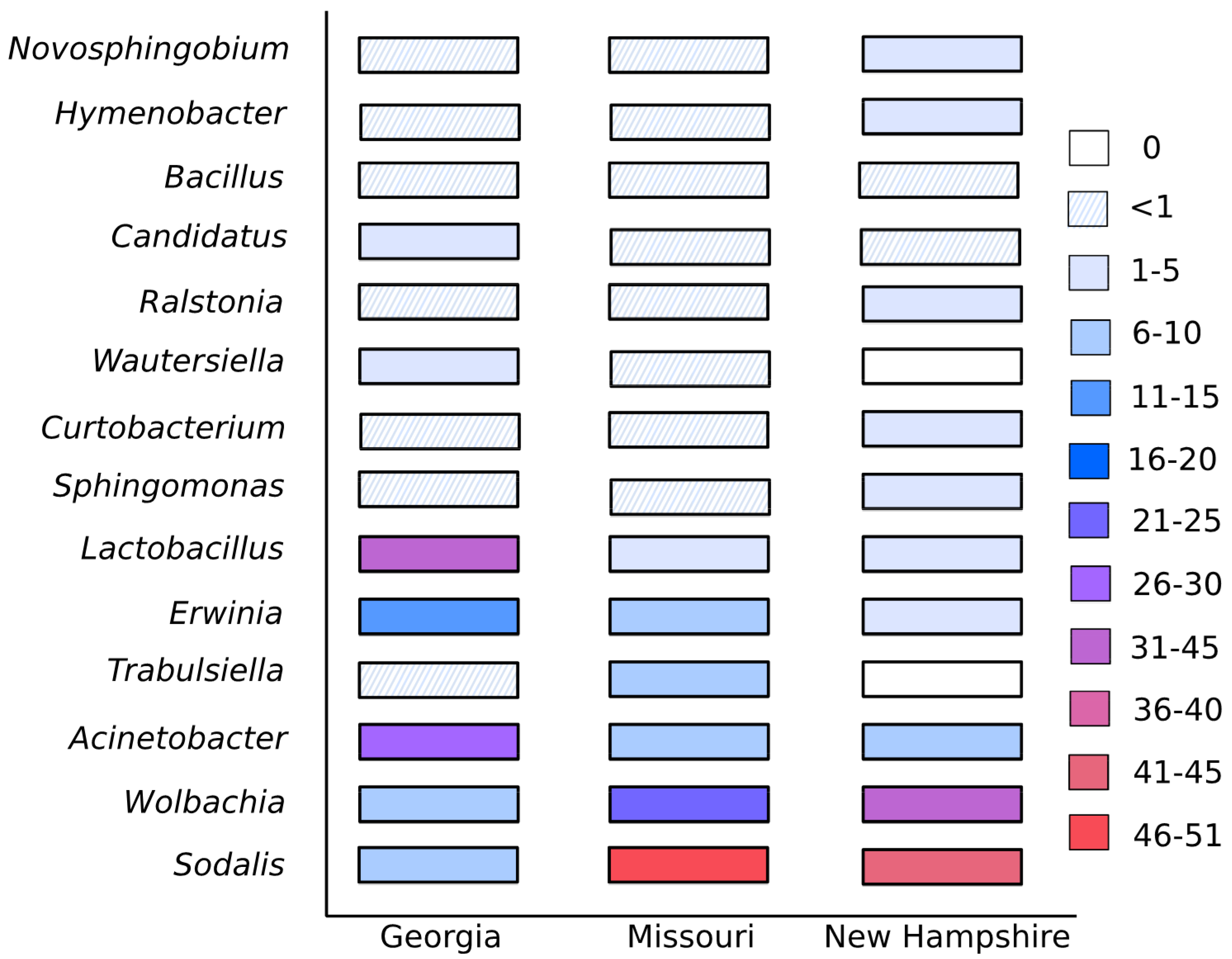
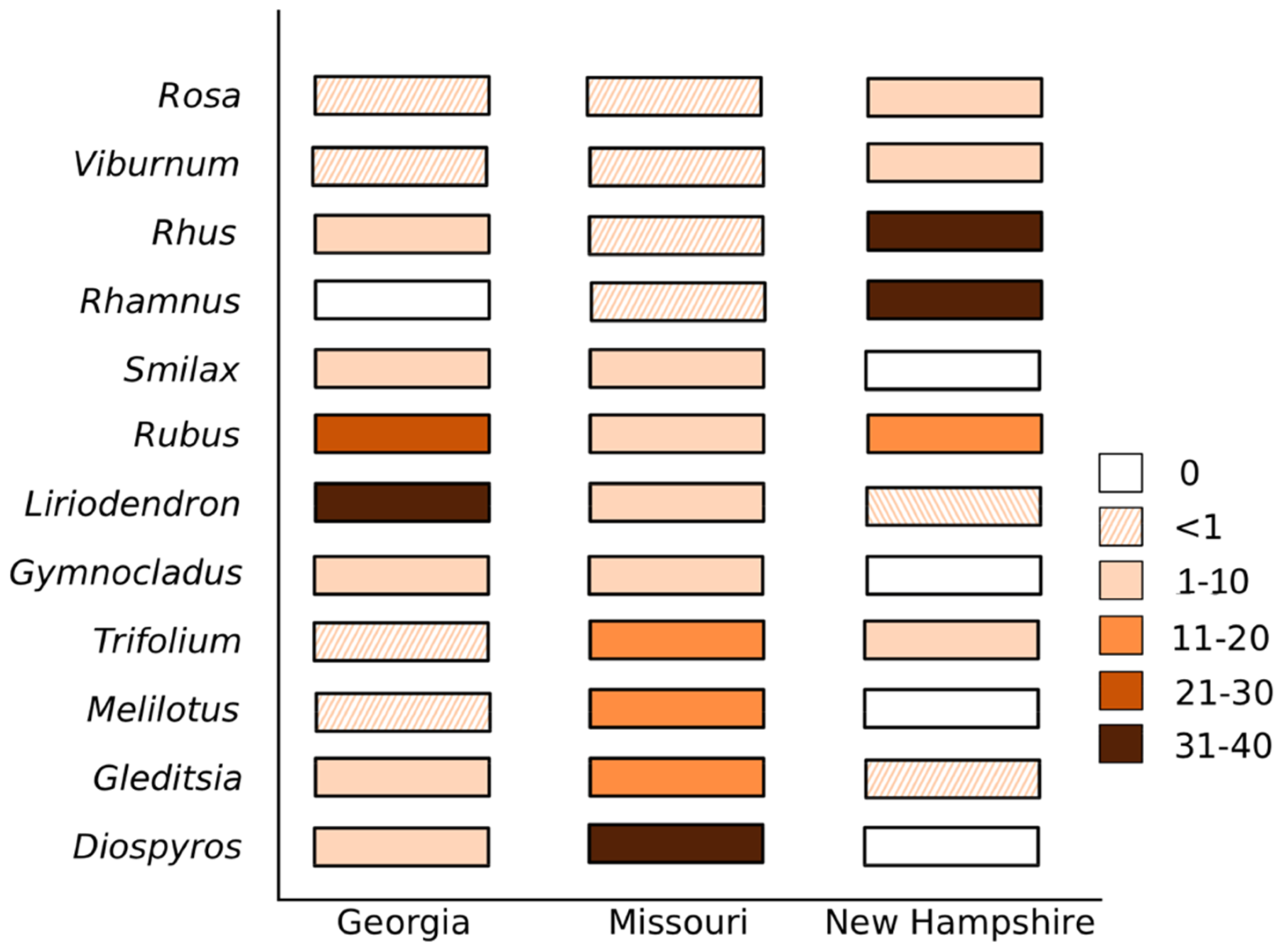
4. Results
5. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Accessibility
References
- Alberoni, D.; Gaggìa, F.; Baffoni, L.; Di Gioia, D. Beneficial microorganisms for honey bees: Problems and progresses. Appl. Microbiol. Biotechnol. 2016, 100, 9469–9482. [Google Scholar] [CrossRef]
- Engel, P.; Moran, N.A. The gut microbiota of insects—Diversity in structure and function. FEMS Microbiol. Rev. 2013, 37, 699–735. [Google Scholar] [CrossRef]
- Arredondo, D.; Castelli, L.; Porrini, M.P.; Garrido, P.M.; Eguaras, M.J.; Zunino, P.; Antúnez, K. Lactobacillus kunkeei strains decreased the infection by honey bee pathogens Paenibacillus larvae and Nosema ceranae. Benef. Microbes 2018, 9, 279–290. [Google Scholar] [CrossRef]
- Mockler, B.K.; Kwong, W.K.; Moran, N.A.; Koch, H. Microbiome structure influences infection by the parasite Crithidia bombi in bumble bees. Appl. Environ. Microbiol. 2018, 84, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Dharampal, P.S.; Carlson, C.; Currie, C.R.; Steffan, S. Pollen-borne microbes shape bee fitness. Proc. R. Soc. B Biol. Sci. 2019, 286, 20182894-99. [Google Scholar] [CrossRef] [Green Version]
- Koch, H.; Schmid-Hempel, P. Socially transmitted gut microbiota protect bumble bees against an intestinal parasite. Proc. Natl. Acad. Sci. USA 2011, 108, 19288–19292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwong, W.K.; Medina, L.A.; Koch, H.; Sing, K.W.; Soh, E.J.Y.; Ascher, J.S.; Moran, N.A. Dynamic microbiome evolution in social bees. Sci. Adv. 2017, 3, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Kwong, W.K.; Moran, N.A. Gut microbial communities of social bees. Nat. Rev. Microbiol. 2016, 14, 374–384. [Google Scholar] [CrossRef]
- Zheng, J.; Wittouck, S.; Salvetti, E.; Franz, C.M.; Harris, H.M.; Mattarelli, P.; O’Toole, P.W.; Pot, B.; Vandamme, P.; Walter, J.; et al. A taxonomic note on the genus Lactobacillus: Description of 23 novel genera, emended description of the genus Lactobacillus Beijerinck 1901, and union of Lactobacillaceae and Leuconostocaceae. Int. J. Syst. Evol. Microbiol. 2020, 70, 2782–2858. [Google Scholar] [CrossRef]
- Graystock, P.; Rehan, S.M.; McFrederick, Q.S. Hunting for healthy microbiomes: Determining the core microbiomes of Ceratina, Megalopta, and Apis bees and how they associate with microbes in bee collected pollen. Conserv. Genet. 2017, 18, 701–711. [Google Scholar] [CrossRef]
- Lozo, J.; Berić, T.; Terzić-Vidojević, A.; Stanković, S.; Fira, D.; Stanisavljević, L. Microbiota associated with pollen, bee bread, larvae and adults of solitary bee Osmia cornuta (Hymenoptera: Megachilidae). Bull. Entomol. Res. 2015, 105, 470–476. [Google Scholar] [CrossRef]
- Martinson, V.G.; Danforth, B.N.; Minckley, R.L.; Rueppell, O.; Tingek, S.; Moran, N.A. A simple and distinctive microbiota associated with honey bees and bumble bees. Mol. Ecol. 2011, 20, 619–628. [Google Scholar] [CrossRef]
- McFrederick, Q.S.; Thomas, J.M.; Neff, J.L.; Vuong, H.Q.; Russell, K.A.; Hale, A.R.; Mueller, U.G. Flowers and wild megachilid bees share microbes. Microb. Ecol. 2017, 73, 188–200. [Google Scholar] [CrossRef]
- McFrederick, Q.S.; Wcislo, W.T.; Taylor, D.R.; Ishak, H.D.; Dowd, S.E.; Mueller, U.G. Environment or kin: Whence do bees obtain acidophilic bacteria? Mol. Ecol. 2012, 21, 1754–1768. [Google Scholar] [CrossRef]
- McFrederick, Q.S.; Rehan, S.M. Wild bee pollen usage and microbial communities co-vary across landscapes. Microb. Ecol. 2019, 77, 513–522. [Google Scholar] [CrossRef]
- Koch, H.; Abrol, D.P.; Li, J.; Schmid-Hempel, P. Diversity and evolutionary patterns of bacterial gut associates of corbiculate bees. Mol. Ecol. 2013, 22, 2028–2044. [Google Scholar] [CrossRef]
- Martinson, V.G.; Moy, J.; Moran, N.A. Establishment of characteristic gut bacteria during development of the honeybee worker. Appl. Environ. Microbiol. 2012, 78, 2830–2840. [Google Scholar] [CrossRef] [Green Version]
- Parmentier, A.; Meeus, I.; Van Nieuwerburgh, F.; Deforce, D.; Vandamme, P.; Smagghe, G. A different gut microbial community between larvae and adults of a wild bumblebee nest (Bombus pascuorum). Insect Sci. 2018, 25, 66–74. [Google Scholar] [CrossRef]
- Michener, C.D. The Social Behavior of the Bees; Belknap Press of Harvard University Press: Cambridge, MA, USA, 1974. [Google Scholar]
- McFrederick, Q.S.; Rehan, S.M. Characterization of pollen and bacterial community composition in brood provisions of a small carpenter bee. Mol. Ecol. 2016, 25, 2302–2311. [Google Scholar] [CrossRef]
- McFrederick, Q.S.; Wcislo, W.T.; Hout, M.C.; Mueller, U.G. Host species and developmental stage, but not host social structure, affects bacterial community structure in socially polymorphic bees. FEMS Microbiol. Ecol. 2014, 88, 398–406. [Google Scholar] [CrossRef] [Green Version]
- Rothman, J.A.; Andrikopoulos, C.; Cox-Foster, D.Q.S. McFrederick. Floral and foliar source affect the bee nest microbial community. Microb. Ecol. 2019, 78, 506–516. [Google Scholar] [CrossRef] [Green Version]
- Durrer, S.; Schmid-hempel, P. Shared use of flowers leads to horizontal pathogen transmission. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1994, 258, 299–302. [Google Scholar] [CrossRef]
- Evison, S.E.F.; Roberts, K.E.; Laurenson, L.; Pietravalle, S.; Hui, J.; Biesmeijer, J.C.; Hughes, W.O.H. Pervasiveness of parasites in pollinators. PLoS ONE 2012, 7, e30641. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Levitt, A.L.; Rajotte, E.G.; Holmes, E.C.; Ostiguy, N.; VanEngelsdorp, D.; Cox-Foster, D.L. RNA viruses in hymenopteran pollinators: Evidence of inter-taxa virus transmission via pollen and potential impact on non-Apis hymenopteran species. PLoS ONE 2010, 5, e14357. [Google Scholar] [CrossRef]
- McArt, S.H.; Koch, H.; Irwin, R.E.; Adler, L.S. Arranging the bouquet of disease: Floral traits and the transmission of plant and animal pathogens. Ecol. Lett. 2014, 17, 624–636. [Google Scholar] [CrossRef]
- Richardson, R.T.; Lin, C.-H.; Quijia, J.O.; Riusech, N.S.; Goodell, K.; Johnson, R.M. Rank-based characterization of pollen assemblages collected by honey bees using a multi-locus metabarcoding approach. Appl. Plant Sci. 2015, 3, 1500043. [Google Scholar] [CrossRef]
- Gardiner, M.A.; Tuell, J.K.; Isaacs, R.; Gibbs, J.; Ascher, J.S.; Landis, D.A. Implications of three biofuel crops for beneficial arthropods in agricultural landscapes. Bioenergy Res. 2010, 3, 6–19. [Google Scholar] [CrossRef] [Green Version]
- Tucker, E.M.; Rehan, S.M. Farming for bees: Annual variation in pollinator populations across agricultural landscapes. Agric. For. Entomol. 2018, 20, 541–548. [Google Scholar] [CrossRef]
- Tuell, J.K.; Ascher, J.S.; Isaacs, R. Wild bees (Hymenoptera: Apoidea: Anthophila) of the Michigan highbush blueberry agroecosystem. Ann. Entomol. Soc. Am. 2009, 102, 275–287. [Google Scholar] [CrossRef] [Green Version]
- Rehan, S.M.; Richards, M.H. Nesting biology and subsociality in Ceratina calcarata (Hymenoptera: Apidae). Can. Entomol. 2010, 142, 65–74. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Knight, R. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [Green Version]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Friedman, J.; Alm, E.J. Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 2012, 8, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Faust, K.; Sathirapongsasuti, J.F.; Izard, J.; Segata, N.; Gevers, D.; Raes, J.; Huttenhower, C. Microbial co-occurrence relationships in the human microbiome. PLoS Comput. Biol. 2012, 8, e1002606. [Google Scholar] [CrossRef]
- Layeghifard, M.; Hwang, D.M.; Guttman, D.S. Disentangling interactions in the microbiome: A network Perspective. Trends Microbiol. 2017, 25, 217–228. [Google Scholar] [CrossRef]
- Dalet, C.; Maudlin, I. Sodalis gen. nov. and Sodalis glossinidius sp. nov. a microaerophilic secondary endosymbiont of the tsetse fly GIossina morsitans morsitans. Int. J. Syst. Bacteriol. 1999, 49, 267–275. [Google Scholar] [CrossRef]
- Saeed, A.; White, J.A. Surveys for maternally-inherited endosymbionts reveal novel and variable infections within solitary bee species. J. Invertebr. Pathol. 2015, 132, 111–114. [Google Scholar] [CrossRef]
- Keller, A.; Danner, N.; Grimmer, G.; Ankenbrand, M.; von der Ohe, K.; von der Ohe, W.; Steffan-Dewenter, I. Evaluating multiplexed next-generation sequencing as a method in palynology for mixed pollen samples. Plant Biol. 2015, 17, 558–566. [Google Scholar] [CrossRef] [Green Version]
- Kraaijeveld, K.; de Weger, L.A.; Ventayol García, M.; Buermans, H.; Frank, J.; Hiemstra, P.S.; den Dunnen, J.T. Efficient and sensitive identification and quantification of airborne pollen using next-generation DNA sequencing. Mol. Ecol. Resour. 2015, 15, 8–16. [Google Scholar] [CrossRef]
- Richardson, R.T.; Curtis, H.R.; Matcham, E.G.; Hua Lin, C.; Suresh, S.; Sponsler, D.B.; Johnson, R.M. Quantitative multi-locus metabarcoding and waggle dance interpretation reveal honey bee spring foraging patterns in Midwest agroecosystems. Mol. Ecol. 2019, 28, 686–697. [Google Scholar] [CrossRef]
- Bell, K.L.; Burgess, K.S.; Botsch, J.C.; Dobbs, E.K.; Read, T.D.; Brosi, B.J. Quantitative and qualitative assessment of pollen DNA metabarcoding using constructed species mixtures. Mol. Ecol. 2018, 28, 431–455. [Google Scholar] [CrossRef]
- Roulston, T.H.I.; Cane, J.H. Pollen nutritional content and digestibility for animals. Plant Syst. 2000, 222, 187–209. [Google Scholar] [CrossRef]
- Eckhardt, M.; Haider, M.; Dorn, S.; Müller, A. Pollen mixing in pollen generalist solitary bees: A possible strategy to complement or mitigate unfavourable pollen properties? J. Anim. Ecol. 2014, 83, 588–597. [Google Scholar] [CrossRef]
- Vanderplanck, M.; Decleves, S.; Roger, N.; Decroo, C.; Caulier, G.; Glauser, G.; Michez, D. Is non-host pollen suitable for generalist bumblebees? Insect Sci. 2018, 25, 259–272. [Google Scholar] [CrossRef]
- Weiss, S.; Van Treuren, W.; Lozupone, C.; Faust, K.; Friedman, J.; Deng, Y.; Knight, R. Correlation detection strategies in microbial data sets vary widely in sensitivity and precision. ISME J. 2016, 10, 1669–1681. [Google Scholar] [CrossRef]
- Aydogan, E.L.; Moser, G.; Müller, C.; Kämpfer, P.; Glaeser, S.P. Long-term warming shifts the composition of bacterial communities in the phyllosphere of Galium album in a permanent grassland field-experiment. Front. Microbiol. 2018, 9, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Jia, S.; Zhang, X.; Zhang, G.; Yin, A.; Zhang, S.; Li, F.; Yu, J. Seasonally variable intestinal metagenomes of the red palm weevil (Rhynchophorus ferrugineus). Environ. Microbiol. 2013, 15, 3020–3029. [Google Scholar] [CrossRef] [Green Version]
- Toju, H.; Fukatsu, T. Diversity and infection prevalence of endosymbionts in natural populations of the chestnut weevil: Relevance of local climate and host plants. Mol. Ecol. 2011, 20, 853–868. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-X.; Song, Y.-L.; Zhang, Y.-K.; Hoffmann, A.A.; Zhou, J.-C.; Sun, J.-T.; Hong, X.-Y. Incidence of facultative bacterial endosymbionts in spider mites associated with local environments and host plants. Appl. Environ. Microbiol. 2018, 84, e02546-17. [Google Scholar] [CrossRef] [PubMed]
- Aizenberg-Gershtein, Y.; Izhaki, I.; Halpern, M. Do honeybees shape the bacterial community composition in floral nectar? PLoS ONE 2013, 8, e67556. [Google Scholar] [CrossRef] [Green Version]
- Samuni-Blank, M.; Izhaki, I.; Laviad, S.; Bar-Massada, A.; Gerchman, Y.; Halpern, M. The role of abiotic environmental conditions and herbivory in shaping bacterial community composition in floral nectar. PLoS ONE 2014, 9, e99107. [Google Scholar] [CrossRef] [Green Version]
- Vannette, R.L.; Fukami, T. Dispersal enhances beta diversity in nectar microbes. Ecol. Lett. 2017, 20, 901–910. [Google Scholar] [CrossRef]
- Vannette, R.L.; Bichier, P.; Philpott, S.M. The presence of aggressive ants is associated with fewer insect visits to and altered microbe communities in coffee flowers. Basic Appl. Ecol. 2017, 20, 62–74. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacteria | Plants | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Brunia | Camptotheca | Gleditsia | Gymonocladus | Liriodendron | Rhus | Rosa | Rubus | Smilax | Trifolium | |
| Acinetobacter | 0.287 NH | |||||||||
| Lactobacillus | 0.231 NH | 0.393 | 0.225 NH | |||||||
| Sodalis | 0.249 NH | |||||||||
| Sphingomonas | 0.420 GA | |||||||||
| Trabulsiella | 0.579 MO | 0.578 MO | ||||||||
| Wautersiella | 0.654 MO | |||||||||
| Wolbachia | 0.600 MO | 0.587 MO | 0.574 MO | 0.566 MO | ||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dew, R.M.; McFrederick, Q.S.; Rehan, S.M. Diverse Diets with Consistent Core Microbiome in Wild Bee Pollen Provisions. Insects 2020, 11, 499. https://doi.org/10.3390/insects11080499
Dew RM, McFrederick QS, Rehan SM. Diverse Diets with Consistent Core Microbiome in Wild Bee Pollen Provisions. Insects. 2020; 11(8):499. https://doi.org/10.3390/insects11080499
Chicago/Turabian StyleDew, Rebecca M., Quinn S. McFrederick, and Sandra M. Rehan. 2020. "Diverse Diets with Consistent Core Microbiome in Wild Bee Pollen Provisions" Insects 11, no. 8: 499. https://doi.org/10.3390/insects11080499