
Landscape of Secondary Findings in Chinese Population: A Practice of ACMG SF v3.0 List
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Enrollment
2.2. Sequencing and Variant Calling
2.3. Variant Interpretation
3. Results
3.1. Actionable EP and KP Variants
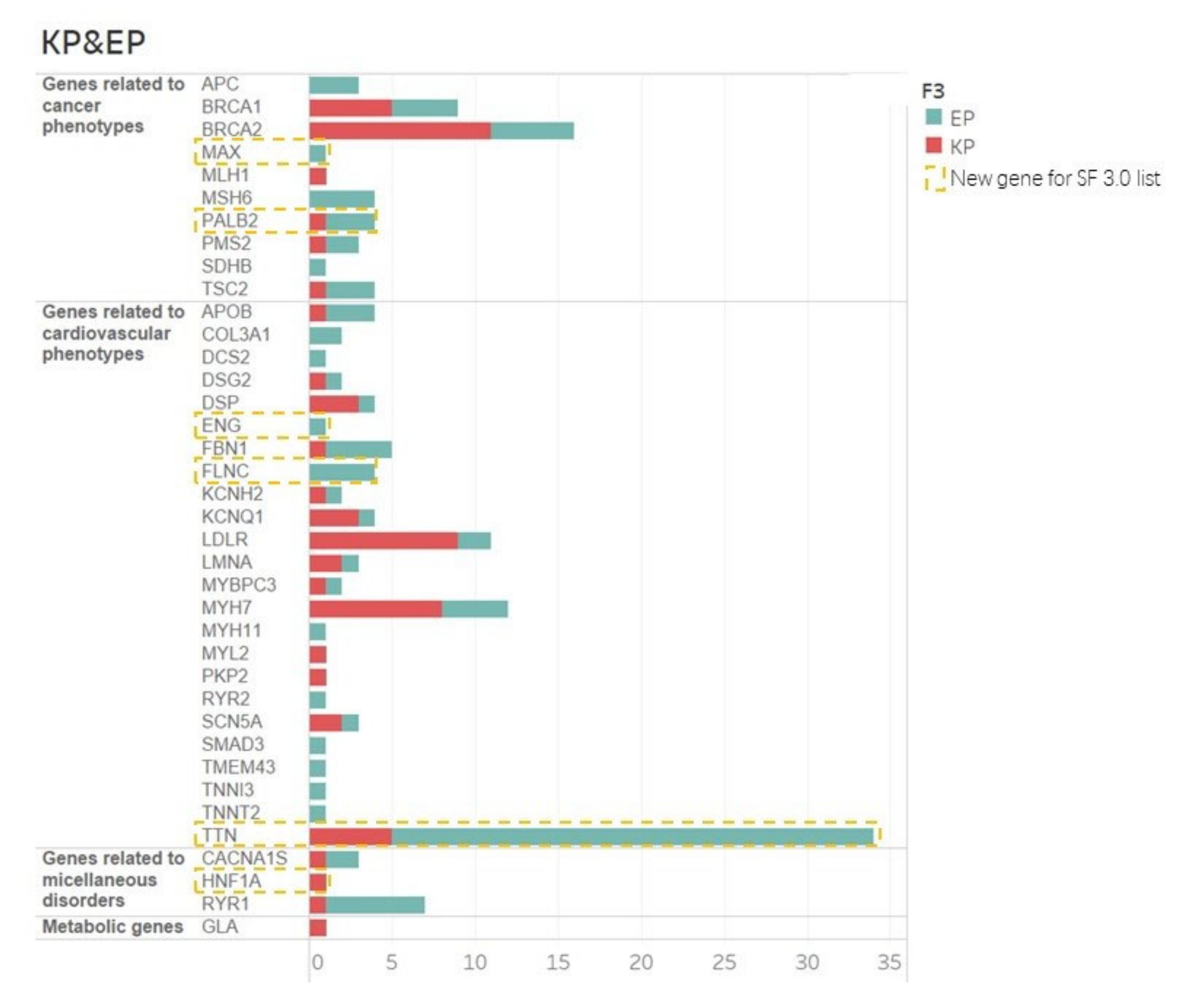
3.2. SF v 3.0 Updated Genes
3.3. Phenotypes of KP Variant Carriers
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koboldt, D.C.; Steinberg, K.M.; Larson, D.E.; Wilson, R.K.; Mardis, E.R. The next-generation sequencing revolution and its impact on genomics. Cell 2013, 155, 27–38. [Google Scholar] [CrossRef]
- Caskey, T. Precision Medicine: Functional Advancements. Annu. Rev. Med. 2018, 69, 1–18. [Google Scholar] [CrossRef]
- Kalia, S.S.; Adelman, K.; Bale, S.J.; Chung, W.K.; Eng, C.; Evans, J.P.; Herman, G.E.; Hufnagel, S.B.; Klein, T.E.; Korf, B.R.; et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American of Medical Genetics and Genomics. Genet. Med. 2017, 19, 249–255. [Google Scholar] [CrossRef]
- Miller, D.T.; Lee, K.; Chung, W.K.; Gordon, A.S.; Herman, G.E.; Klein, T.E.; Stewart, D.R.; Amendola, L.M.; Adelman, K.; Bale, S.J.; et al. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 1381–1390. [Google Scholar] [CrossRef]
- Jang, M.A.; Lee, S.H.; Kim, N.; Ki, C.S. Frequency and spectrum of actionable pathogenic secondary findings in 196 Korean exomes. Genet. Med. 2015, 17, 1007–1011. [Google Scholar] [CrossRef]
- Yu, M.H.C.; Mak, C.C.Y.; Fung, J.L.F.; Lee, M.; Tsang, M.H.Y.; Chau, J.F.T.; Chung, P.H.-Y.; Yang, W.; Chan, G.C.F.; Lee, S.L.; et al. Actionable secondary findings in 1116 Hong Kong Chinese based on exome sequencing data. J. Hum. Genet. 2021, 66, 637–641. [Google Scholar] [CrossRef]
- Jalkh, N.; Mehawej, C.; Chouery, E. Actionable Exomic Secondary Findings in 280 Lebanese Participants. Front. Genet. 2020, 11, 208. [Google Scholar] [CrossRef]
- Chetruengchai, W.; Shotelersuk, V. Actionable secondary findings in the 73 ACMG-recommended genes in 1559 Thai exomes. J. Hum. Genet. 2021, 67, 137–142. [Google Scholar] [CrossRef]
- Elfatih, A.; Mifsud, B.; Syed, N.; Badii, R.; Mbarek, H.; Abbaszadeh, F.; Qatar Genome Program Research Consortium; Estivill, X. Actionable genomic variants in 6045 participants from the Qatar Genome Program. Hum. Mutat. 2021. [Google Scholar] [CrossRef]
- Arslan Ateş, E.; Türkyilmaz, A.; Yıldırım, Ö.; Alavanda, C.; Polat, H.; Demir, Ş.; Çebi, A.H.; Geçkinli, B.B.; Güney, A.İ.; Ata, P.; et al. Secondary findings in 622 Turkish clinical exome sequencing data. J. Hum. Genet. 2021, 66, 1113–1119. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, Y.; Liu, S.; Song, X.; Yang, X.Z.; Fan, Y.; Chen, W.; Akdemir, Z.C.; Yan, Z.; Zuo, Y.; et al. The coexistence of copy number variations (CNVs) and single nucleotide polymorphisms (SNPs) at a locus can result in distorted calculations of the significance in associating SNPs to disease. Hum. Genet. 2018, 137, 553–567. [Google Scholar] [CrossRef]
- Chen, N.; Zhao, S.; Jolly, A.; Wang, L.; Pan, H.; Yuan, J.; Chen, S.; Koch, A.; Ma, C.; Tian, W.; et al. Perturbations of genes essential for Müllerian duct and Wölffian duct development in Mayer-Rokitansky-Küster-Hauser syndrome. Am. J. Hum. Genet. 2021, 108, 337–345. [Google Scholar] [CrossRef]
- Fan, X.; Zhao, S.; Yu, C.; Wu, D.; Yan, Z.; Fan, L.; Song, Y.; Wang, Y.; Li, C.; Ming, Y.; et al. Exome sequencing reveals genetic architecture in patients with isolated or syndromic short stature. J. Genet. Genom. 2021, 48, 396–402. [Google Scholar] [CrossRef]
- Sun, L.; Huang, Y.; Zhao, S.; Zhao, J.; Yan, Z.; Guo, Y.; Lin, M.; Zhong, W.; Yin, Y.; Chen, Z.; et al. Deciphering the mutational signature of congenital limb malformations. Mol. Ther. Nucleic Acids 2021, 24, 961–970. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Li, Q.; Wang, K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am. J. Hum. Genet. 2017, 100, 267–280. [Google Scholar] [CrossRef]
- Loibl, S.; Poortmans, P.; Morrow, M.; Denkert, C.; Curigliano, G. Breast cancer. Lancet 2021, 397, 1750–1769. [Google Scholar] [CrossRef]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef]
- Lawrence, L.; Sincan, M.; Markello, T.; Adams, D.R.; Gill, F.; Godfrey, R.; Golas, G.; Groden, C.; Landis, D.; Nehrebecky, M.; et al. The implications of familial incidental findings from exome sequencing: The NIH Undiagnosed Diseases Program experience. Genet. Med. 2014, 16, 741–750. [Google Scholar] [CrossRef]
- Kwak, S.H.; Chae, J.; Choi, S.; Kim, M.J.; Choi, M.; Chae, J.H.; Cho, E.H.; Hwang, T.J.; Jang, S.S.; Kim, J.I.; et al. Findings of a 1303 Korean whole-exome sequencing study. Exp. Mol. Med. 2017, 49, e356. [Google Scholar] [CrossRef] [Green Version]
- Tang, C.S.; Dattani, S.; So, M.T.; Cherny, S.S.; Tam, P.K.H.; Sham, P.C.; Garcia-Barcelo, M.M. Actionable secondary findings from whole-genome sequencing of 954 East Asians. Hum. Genet. 2018, 137, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.W.; Hwu, W.L.; Chien, Y.H.; Hsu, C.; Hung, M.Z.; Lin, I.L.; Lai, F.; Lee, N.-C. Frequency and spectrum of actionable pathogenic secondary findings in Taiwanese exomes. Mol. Genet. Genom. Med. 2020, 8, e1455. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi-Kabata, Y.; Yasuda, J.; Tanabe, O.; Suzuki, Y.; Kawame, H.; Fuse, N.; Nagasaki, M.; Kawai, Y.; Kojima, K.; Katsuoka, F.; et al. Evaluation of reported pathogenic variants and their frequencies in a Japanese population based on a whole-genome reference panel of 2049 individuals. J. Hum. Genet. 2018, 63, 213–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Gene | Variant Name | Nucleotide Change | Protein Change | Carriers | Clinical Characteristics |
|---|---|---|---|---|---|
| BRCA1 | chr17_41244976_G_A | c.2572C > T | p.Gln858Ter | CSS16261000102 | Mother dx benign breast tumor; maternal grandmother dx benign breast tumor |
| BRCA2 | chr13_32914982_C_T | c.6490C > T | p.Gln2164Ter | AIS18008000102 | Great-grandmother dx breast cancer |
| BRCA2 | chr13_32929364_CA_C | c.7379del | p.Asn2460ThrfsTer7 | AIS18004900102 | Mother dx hyperplasia of mammary gland |
| MYH7 | chr14_23896019_G_A | c.2011C > T | p.Arg671Cys | OSS17011000102 | Grandfather dx hypertrophic cardiomyopathy |
| TTN | chr2_179505980_T_A | c.40621A > T | p.Lys13541Ter | CSS16210400102 | Grandfather dx myocardial infarction; great-grandfather dx cardiomyopathy |
| KCNQ1 | chr11_2594217_G_T | c.921 + 1G > T | p.? | AIS18008000102 | Carriers dx atrial septal defect |
| SCN5A | chr3_38662333_C_T | c.611 + 1G > A | p.? | CSS16156400102 | Carriers dx mitral valve abnormality |
| PKP2 | chr12_32949111_G_T | c.2421C > A | p.Tyr807Ter | OSS17011300102 | Father dx hypertrophic cardiomyopathy combined with tricuspid valve abnormality |
| TTN | chr2_179454531_G_A | c.61921C > T | p.Arg20641Ter | AIS19013300102 | Carriers dx mitral valve abnormality |
| Study | Sample Size | Ethnicity | Genes Included | Frequency with SFs (%) |
|---|---|---|---|---|
| Kwak et al., 2017 [20] | 1303 | Korean | ACMG 56 (v1) | 2.46 |
| Lawrence et al., 2014 [19] | 543 | Mixed | ACMG 56 (v1) | 8.8 |
| Tang et al., 2018 [21] | 954 | Chinese and Vietnamese | ACMG 59 (v2) | 2.50 |
| Yamaguchi-Kabata et al., 2018 [23] | 2049 | Japanese | autosomal genes (v2) in ACMG 59 | 21 |
| Nadine et al., 2020 [7] | 280 | Lebanese | ACMG 59 (v2) | 6 |
| Ersa et al., 2021 [10] | 622 | Turkish | ACMG 59 (v2) | 2.1 |
| Amal et al., 2021 [9] | 6045 | Qatari | ACMG 59 (v2) | 2.3 |
| Wanna et al., 2021 [8] | 1559 | Thai | ACMG 73 (v3) | 11.9 |
| This study | 2987 | Chinese | ACMG 73 (v3) | 5.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, Y.; Liu, B.; Shi, J.; Zhao, S.; Xu, K.; Sun, L.; Chen, N.; Tian, W.; Zhang, J.; Wu, N. Landscape of Secondary Findings in Chinese Population: A Practice of ACMG SF v3.0 List. J. Pers. Med. 2022, 12, 1503. https://doi.org/10.3390/jpm12091503
Huang Y, Liu B, Shi J, Zhao S, Xu K, Sun L, Chen N, Tian W, Zhang J, Wu N. Landscape of Secondary Findings in Chinese Population: A Practice of ACMG SF v3.0 List. Journal of Personalized Medicine. 2022; 12(9):1503. https://doi.org/10.3390/jpm12091503
Chicago/Turabian StyleHuang, Yingzhao, Bowen Liu, Jile Shi, Sen Zhao, Kexin Xu, Liying Sun, Na Chen, Wen Tian, Jianguo Zhang, and Nan Wu. 2022. "Landscape of Secondary Findings in Chinese Population: A Practice of ACMG SF v3.0 List" Journal of Personalized Medicine 12, no. 9: 1503. https://doi.org/10.3390/jpm12091503