
Role of microRNAs in Disorders of Gut–Brain Interactions: Clinical Insights and Therapeutic Alternatives


Abstract
:1. Introduction
2. Functional Role of miRNAs in Gut Physiological Mechanisms
3. Impact of miRNAs in DGBIs
3.1. miRNA Dysregulation in GI Cells Contribute to the Pathogenesis of DGBIs
3.1.1. ICC-Specific miRNA Dysregulation
3.1.2. SMC-Specific miRNA Dysregulation
3.1.3. Enteric Neuron-Specific miRNA Dysregulation
3.1.4. Gut Immune Cell-Specific miRNA Dysregulation
3.2. miRNA Dysregulation in Pathophysiological Mechanisms of DGBIs
3.2.1. miRNA Dysregulation and Immune Dysfunction
3.2.2. miRNA Dysregulation and Visceral Hypersensitivity
3.2.3. miRNA Dysregulation and Impaired GI Barrier Function
3.2.4. miRNA Dysregulation and Serotonergic Signaling
3.3. miRNA Dysregulation and DGBIs
3.3.1. miRNA Dysregulation in IBS
3.3.2. miRNA Dysregulation in FD
3.3.3. miRNA Dysregulation in Gastroparesis
3.3.4. miRNA Dysregulation in STC
4. miRNA-GPCR Interactions in DGBIs
5. Clinical Insight and Therapeutic Alternatives for DGBIs
5.1. Gut Microbiota and DGBIs
5.2. Gut Microbiota-Derived Molecules and DGBIs
5.3. Gut Microbiota Regulated Modulation of the Gut–Brain Axis in DGBIs
5.4. Gut Microbiome Modulation through Probiotic Intervention and Personized Nutrition
6. miRNAs as a Potential Therapeutic Option
7. Current Challenges and Solutions for Mirna Therapeutics
8. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sperber, A.D.; Bangdiwala, S.I.; Drossman, D.A.; Ghoshal, U.C.; Simren, M.; Tack, J.; Whitehead, W.E.; Dumitrascu, D.L.; Fang, X.; Fukudo, S.; et al. Worldwide prevalence and burden of functional gastrointestinal disorders, results of rome foundation global study. Gastroenterology 2021, 160, 99–114.e3. [Google Scholar] [CrossRef]
- Black, C.J.; Drossman, D.A.; Talley, N.J.; Ruddy, J.; Ford, A.C. Functional gastrointestinal disorders: Advances in understanding and management. Lancet 2020, 396, 1664–1674. [Google Scholar] [CrossRef]
- Tack, J.; Drossman, D.A. What’s new in Rome IV? Neurogastroenterol. Motil. 2017, 29. [Google Scholar] [CrossRef]
- Keefer, L.; Ko, C.W.; Ford, A.C. AGA clinical practice update on management of chronic gastrointestinal pain in disorders of gut-brain interaction: Expert review. Clin. Gastroenterol. Hepatol. 2021. [Google Scholar] [CrossRef]
- Parkman, H.P.; Yates, K.; Hasler, W.L.; Nguyen, L.; Pasricha, P.J.; Snape, W.J.; Farrugia, G.; Koch, K.L.; Abell, T.L.; McCallum, R.W.; et al. Clinical features of idiopathic gastroparesis vary with sex, body mass, symptom onset, delay in gastric emptying, and gastroparesis severity. Gastroenterology 2011, 140, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Zikos, T.A.; Kamal, A.N.; Neshatian, L.; Triadafilopoulos, G.; Clarke, J.O.; Nandwani, M.; Nguyen, L.A. High prevalence of slow transit constipation in patients with gastroparesis. J. Neurogastroenterol. Motil. 2019, 25, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Black, C.J.; Ford, A.C. Global burden of irritable bowel syndrome: Trends, predictions and risk factors. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 473–486. [Google Scholar] [CrossRef]
- Ghoshal, U.C. Marshall and warren lecture 2019: A paradigm shift in pathophysiological basis of irritable bowel syndrome and its implication on treatment. J. Gastroenterol. Hepatol. 2020, 35, 712–721. [Google Scholar] [CrossRef] [Green Version]
- Ford, A.C.; Mahadeva, S.; Carbone, M.F.; Lacy, B.E.; Talley, N.J. Functional dyspepsia. Lancet 2020, 396, 1689–1702. [Google Scholar] [CrossRef]
- Grover, M.; Farrugia, G.; Stanghellini, V. Gastroparesis: A turning point in understanding and treatment. Gut 2019, 68, 2238–2250. [Google Scholar] [CrossRef] [PubMed]
- Enck, P.; Aziz, Q.; Barbara, G.; Farmer, A.D.; Fukudo, S.; Mayer, E.A.; Niesler, B.; Quigley, E.M.; Rajilic-Stojanovic, M.; Schemann, M.; et al. Irritable bowel syndrome. Nat. Rev. Dis. Prim. 2016, 2, 16014. [Google Scholar] [CrossRef] [Green Version]
- Enck, P.; Azpiroz, F.; Boeckxstaens, G.; Elsenbruch, S.; Feinle-Bisset, C.; Holtmann, G.; Lackner, J.M.; Ronkainen, J.; Schemann, M.; Stengel, A.; et al. Functional dyspepsia. Nat. Rev. Dis. Prim. 2017, 3, 17081. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, M.; Chedid, V.; Ford, A.C.; Haruma, K.; Horowitz, M.; Jones, K.L.; Low, P.A.; Park, S.Y.; Parkman, H.P.; Stanghellini, V. Gastroparesis. Nat. Rev. Dis. Prim. 2018, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, U.C. Pros and cons while looking through an Asian window on the Rome IV criteria for irritable bowel syndrome: Pros. J. Neurogastroenterol. Motil. 2017, 23, 334–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford, A.C.; Sperber, A.D.; Corsetti, M.; Camilleri, M. Irritable bowel syndrome. Lancet 2020, 396, 1675–1688. [Google Scholar] [CrossRef]
- Singh, R.; Zogg, H.; Wei, L.; Bartlett, A.; Ghoshal, U.C.; Rajender, S.; Ro, S. Gut microbial dysbiosis in the pathogenesis of gastrointestinal dysmotility and metabolic disorders. J. Neurogastroenterol. Motil. 2021, 27, 19–34. [Google Scholar] [CrossRef]
- Yoo, B.B.; Mazmanian, S.K. The enteric network: Interactions between the immune and nervous systems of the gut. Immunity 2017, 46, 910–926. [Google Scholar] [CrossRef] [Green Version]
- Sanders, K.M.; Koh, S.D.; Ro, S.; Ward, S.M. Regulation of gastrointestinal motility—Insights from smooth muscle biology. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 633–645. [Google Scholar] [CrossRef] [Green Version]
- Grover, M.; Farrugia, G.; Lurken, M.S.; Bernard, C.E.; Faussone-Pellegrini, M.S.; Smyrk, T.C.; Parkman, H.P.; Abell, T.L.; Snape, W.J.; Hasler, W.L.; et al. Cellular changes in diabetic and idiopathic gastroparesis. Gastroenterology 2011, 140, 1575–1585.e8. [Google Scholar] [CrossRef] [Green Version]
- Farrugia, G. Interstitial cells of Cajal in health and disease. Neurogastroenterol. Motil. 2008, 20 (Suppl. 1), 54–63. [Google Scholar] [CrossRef]
- Grover, M.; Gibbons, S.J.; Nair, A.A.; Bernard, C.E.; Zubair, A.S.; Eisenman, S.T.; Wilson, L.A.; Miriel, L.; Pasricha, P.J.; Parkman, H.P.; et al. Transcriptomic signatures reveal immune dysregulation in human diabetic and idiopathic gastroparesis. BMC Med. Genom. 2018, 11, 62. [Google Scholar] [CrossRef]
- Cipriani, G.; Gibbons, S.J.; Kashyap, P.C.; Farrugia, G. Intrinsic gastrointestinal macrophages: Their phenotype and role in gastrointestinal motility. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 120–130.e1. [Google Scholar] [CrossRef] [Green Version]
- Mazzone, A.; Strege, P.R.; Gibbons, S.J.; Alcaino, C.; Joshi, V.; Haak, A.J.; Tschumperlin, D.J.; Bernard, C.E.; Cima, R.R.; Larson, D.W.; et al. microRNA overexpression in slow transit constipation leads to reduced NaV1.5 current and altered smooth muscle contractility. Gut 2020, 69, 868–876. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Costinean, S.; Croce, C.M.; Brasier, A.R.; Merwat, S.; Larson, S.A.; Basra, S.; Verne, G.N. MicroRNA 29 targets nuclear factor-kappaB-repressing factor and Claudin 1 to increase intestinal permeability. Gastroenterology 2015, 148, 158–169.e8. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Yang, L.; Larson, S.; Basra, S.; Merwat, S.; Tan, A.; Croce, C.; Verne, G.N. Decreased miR-199 augments visceral pain in patients with IBS through translational upregulation of TRPV1. Gut 2016, 65, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Martinez, C.; Rodino-Janeiro, B.K.; Lobo, B.; Stanifer, M.L.; Klaus, B.; Granzow, M.; Gonzalez-Castro, A.M.; Salvo-Romero, E.; Alonso-Cotoner, C.; Pigrau, M.; et al. miR-16 and miR-125b are involved in barrier function dysregulation through the modulation of claudin-2 and cingulin expression in the jejunum in IBS with diarrhoea. Gut 2017, 66, 1537–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.; Ha, S.E.; Wei, L.; Jin, B.; Zogg, H.; Poudrier, S.M.; Jorgensen, B.G.; Park, C.; Ronkon, C.F.; Bartlett, A.; et al. miR-10b-5p rescues diabetes and gastrointestinal dysmotility. Gastroenterology 2021, 160, 1662–1678.e18. [Google Scholar] [CrossRef]
- Mahurkar-Joshi, S.; Rankin, C.R.; Videlock, E.J.; Soroosh, A.; Verma, A.; Khandadash, A.; Iliopoulos, D.; Pothoulakis, C.; Mayer, E.A.; Chang, L. The colonic mucosal MicroRNAs, MicroRNA-219a-5p, and MicroRNA-338-3p are downregulated in irritable bowel syndrome and are associated with barrier function and MAPK signaling. Gastroenterology 2021, 160, 2409–2422.e19. [Google Scholar] [CrossRef] [PubMed]
- Wouters, M.M. Novel insight in diarrhoea-predominant IBS: miRNAs modulate barrier function. Gut 2017, 66, 1537–1538. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Wei, L.; Ghoshal, U.C. Micro-organic basis of functional gastrointestinal (GI) disorders: Role of microRNAs in GI pacemaking cells. Indian J. Gastroenterol. 2021, 40, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Margolis, K.G.; Gershon, M.D.; Bogunovic, M. Cellular organization of neuroimmune interactions in the gastrointestinal tract. Trends Immunol. 2016, 37, 487–501. [Google Scholar] [CrossRef] [Green Version]
- Verheijden, S.; Boeckxstaens, G.E. Neuroimmune interaction and the regulation of intestinal immune homeostasis. Am. J. Physiol. Liver Physiol. 2018, 314, G75–G80. [Google Scholar] [CrossRef] [PubMed]
- Boeckxstaens, G.E.; Wouters, M.M. Neuroimmune factors in functional gastrointestinal disorders: A focus on irritable bowel syndrome. Neurogastroenterol. Motil. 2017, 29. [Google Scholar] [CrossRef] [Green Version]
- Wouters, M.M.; Boeckxstaens, G.E. Neuroimmune mechanisms in functional bowel disorders. Neth. J. Med. 2011, 69, 55–61. [Google Scholar] [PubMed]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug. Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- To, K.K.W.; Fong, W.; Tong, C.W.S.; Wu, M.; Yan, W.; Cho, W.C.S. Advances in the discovery of microRNA-based anticancer therapeutics: Latest tools and developments. Expert Opin. Drug. Discov. 2020, 15, 63–83. [Google Scholar] [CrossRef] [PubMed]
- Winkle, M.; El-Daly, S.M.; Fabbri, M.; Calin, G.A. Noncoding RNA therapeutics—Challenges and potential solutions. Nat. Rev. Drug. Discov. 2021, 20, 629–651. [Google Scholar] [CrossRef]
- Michaels, Y.S.; Barnkob, M.B.; Barbosa, H.; Baeumler, T.A.; Thompson, M.K.; Andre, V.; Colin-York, H.; Fritzsche, M.; Gileadi, U.; Sheppard, H.M.; et al. Precise tuning of gene expression levels in mammalian cells. Nat. Commun 2019, 10, 818. [Google Scholar] [CrossRef] [Green Version]
- Sevignani, C.; Calin, G.A.; Siracusa, L.D.; Croce, C.M. Mammalian microRNAs: A small world for fine-tuning gene expression. Mamm. Genome 2006, 17, 189–202. [Google Scholar] [CrossRef] [Green Version]
- Catalanotto, C.; Cogoni, C.; Zardo, G. MicroRNA in control of gene expression: An overview of nuclear functions. Int. J. Mol. Sci. 2016, 17, 1712. [Google Scholar] [CrossRef] [Green Version]
- McGeary, S.E.; Lin, K.S.; Shi, C.Y.; Pham, T.M.; Bisaria, N.; Kelley, G.M.; Bartel, D.P. The biochemical basis of microRNA targeting efficacy. Science 2019, 366. [Google Scholar] [CrossRef]
- Ro, S.; Park, C.; Young, D.; Sanders, K.M.; Yan, W. Tissue-dependent paired expression of miRNAs. Nucleic Acids Res. 2007, 35, 5944–5953. [Google Scholar] [CrossRef]
- Lee, Y.; Ahn, C.; Han, J.; Choi, H.; Kim, J.; Yim, J.; Lee, J.; Provost, P.; Radmark, O.; Kim, S.; et al. The nuclear RNase III Drosha initiates microRNA processing. Nature 2003, 425, 415–419. [Google Scholar] [CrossRef]
- Yi, R.; Qin, Y.; Macara, I.G.; Cullen, B.R. Exportin-5 mediates the nuclear export of pre-microRNAs and short hairpin RNAs. Genes Dev. 2003, 17, 3011–3016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dragomir, M.P.; Knutsen, E.; Calin, G.A. SnapShot: Unconventional miRNA functions. Cell 2018, 174, 1038.e1. [Google Scholar] [CrossRef] [PubMed]
- Forman, J.J.; Legesse-Miller, A.; Coller, H.A. A search for conserved sequences in coding regions reveals that the let-7 microRNA targets Dicer within its coding sequence. Proc. Natl. Acad. Sci. USA 2008, 105, 14879–14884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Sung, Y.M.; Park, J.; Kim, S.; Kim, J.; Park, J.; Ha, H.; Bae, J.Y.; Kim, S.; Baek, D. General rules for functional microRNA targeting. Nat. Genet. 2016, 48, 1517–1526. [Google Scholar] [CrossRef]
- Jens, M.; Rajewsky, N. Competition between target sites of regulators shapes post-transcriptional gene regulation. Nat. Rev. Genet. 2015, 16, 113–126. [Google Scholar] [CrossRef]
- Yu, C.; Zang, L.; Feng, B.; Zhang, L.; Xue, P.; Sun, J.; Dong, F.; Ma, J.; Zheng, M. Coexpression network analysis identified specific miRNAs and genes in association with slowtransit constipation. Mol. Med. Rep. 2020, 22, 4696–4706. [Google Scholar] [CrossRef]
- Vicario, M.; Martinez, C.; Santos, J. Role of microRNA in IBS with increased gut permeability. Gut 2010, 59, 710–712. [Google Scholar] [CrossRef]
- Mahurkar-Joshi, S.; Chang, L. Epigenetic mechanisms in irritable bowel syndrome. Front. Psychiatry 2020, 11, 805. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.N.; Hata, A. Regulation of MicroRNA biogenesis: A miRiad of mechanisms. Cell Commun. Signal. 2009, 7, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilczynska, A.; Bushell, M. The complexity of miRNA-mediated repression. Cell Death Differ. 2015, 22, 22–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, H.; Liu, Y.J.; Chen, Z.C.; Fan, G.Q. miR-222 regulates cell growth, apoptosis, and autophagy of interstitial cells of Cajal isolated from slow transit constipation rats by targeting c-kit. Indian J. Gastroenterol. 2021, 40, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.J.; Li, J.; Yang, X.J.; Zhang, W.; Su, T.; Xiong, T.W.; Song, J.; Liu, P. miR-551b-5p increases intracellular Ca(2+) concentration but does not alter c-Kit expression in rat interstitial cells of Cajal. Int. J. Clin. Exp. Pathol. 2017, 10, 7578–7585. [Google Scholar]
- Park, C.; Lee, M.Y.; Slivano, O.J.; Park, P.J.; Ha, S.; Berent, R.M.; Fuchs, R.; Collins, N.C.; Yu, T.J.; Syn, H.; et al. Loss of serum response factor induces microRNA-mediated apoptosis in intestinal smooth muscle cells. Cell Death Dis. 2015, 6, e2011. [Google Scholar] [CrossRef] [Green Version]
- Gangula, P.R.; Challagundla, K.B.; Ravella, K.; Mukhopadhyay, S.; Chinnathambi, V.; Mittal, M.K.; Sekhar, K.R.; Sampath, C. Sepiapterin alleviates impaired gastric nNOS function in spontaneous diabetic female rodents through NRF2 mRNA turnover and miRNA biogenesis pathway. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G980–G990. [Google Scholar] [CrossRef]
- Nezami, B.G.; Mwangi, S.M.; Lee, J.E.; Jeppsson, S.; Anitha, M.; Yarandi, S.S.; Farris, A.B., 3rd; Srinivasan, S. MicroRNA 375 mediates palmitate-induced enteric neuronal damage and high-fat diet-induced delayed intestinal transit in mice. Gastroenterology 2014, 146, 473–483.e3. [Google Scholar] [CrossRef] [Green Version]
- Ren, H.X.; Zhang, F.C.; Luo, H.S.; Zhang, G.; Liang, L.X. Role of mast cell-miR-490-5p in irritable bowel syndrome. World J. Gastroenterol. 2017, 23, 93–102. [Google Scholar] [CrossRef]
- Ji, L.J.; Li, F.; Zhao, P.; Weng, L.P.; Wei, J.; Yan, J.; Liu, L.N. Silencing interleukin 1alpha underlies a novel inhibitory role of miR-181c-5p in alleviating low-grade inflammation of rats with irritable bowel syndrome. J. Cell Biochem. 2019, 120, 15268–15279. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zhang, Q.; Li, S.; Li, L.; Ding, Z.; Qian, Q.; Fan, L.; Jiang, C. The relationship between colonic macrophages and MicroRNA-128 in the pathogenesis of slow transit constipation. Dig. Dis. Sci. 2015, 60, 2304–2315. [Google Scholar] [CrossRef]
- Wohlfarth, C.; Schmitteckert, S.; Hartle, J.D.; Houghton, L.A.; Dweep, H.; Fortea, M.; Assadi, G.; Braun, A.; Mederer, T.; Pohner, S.; et al. miR-16 and miR-103 impact 5-HT4 receptor signalling and correlate with symptom profile in irritable bowel syndrome. Sci. Rep. 2017, 7, 14680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapeller, J.; Houghton, L.A.; Monnikes, H.; Walstab, J.; Moller, D.; Bonisch, H.; Burwinkel, B.; Autschbach, F.; Funke, B.; Lasitschka, F.; et al. First evidence for an association of a functional variant in the microRNA-510 target site of the serotonin receptor-type 3E gene with diarrhea predominant irritable bowel syndrome. Hum. Mol. Genet. 2008, 17, 2967–2977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, X.J.; Mao, W.M.; Wang, Q.; Yang, G.G.; Wu, W.J.; Shao, S.X. MicroRNA-24 inhibits serotonin reuptake transporter expression and aggravates irritable bowel syndrome. Biochem. Biophys. Res. Commun. 2016, 469, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Fei, L.; Wang, Y. microRNA-495 reduces visceral sensitivity in mice with diarrhea-predominant irritable bowel syndrome through suppression of the PI3K/AKT signaling pathway via PKIB. IUBMB Life 2020, 72, 1468–1480. [Google Scholar] [CrossRef] [PubMed]
- Hou, Q.; Huang, Y.; Zhang, C.; Zhu, S.; Li, P.; Chen, X.; Hou, Z.; Liu, F. MicroRNA-200a targets cannabinoid receptor 1 and serotonin transporter to increase visceral hyperalgesia in diarrhea-predominant irritable bowel syndrome rats. J. Neurogastroenterol. Motil. 2018, 24, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Xiao, X.; Shi, Y.; Wu, Y.; Huang, Y.; Li, D.; Xiong, F.; He, G.; Chai, Y.; Tang, H. Inhibition of miRNA-29a regulates intestinal barrier function in diarrhea-predominant irritable bowel syndrome by upregulating ZO-1 and CLDN1. Exp. Ther. Med. 2020, 20, 155. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Souba, W.W.; Croce, C.M.; Verne, G.N. MicroRNA-29a regulates intestinal membrane permeability in patients with irritable bowel syndrome. Gut 2010, 59, 775–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, D.; Guo, S.; Al-Sadi, R.; Ma, T.Y. MicroRNA regulation of intestinal epithelial tight junction permeability. Gastroenterology 2011, 141, 1323–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Q.; Huang, Y.; Zhu, S.; Li, P.; Chen, X.; Hou, Z.; Liu, F. MiR-144 increases intestinal permeability in IBS-D rats by targeting OCLN and ZO1. Cell. Physiol. Biochem. 2017, 44, 2256–2268. [Google Scholar] [CrossRef]
- Nakata, K.; Sugi, Y.; Narabayashi, H.; Kobayakawa, T.; Nakanishi, Y.; Tsuda, M.; Hosono, A.; Kaminogawa, S.; Hanazawa, S.; Takahashi, K. Commensal microbiota-induced microRNA modulates intestinal epithelial permeability through the small GTPase ARF4. J. Biol. Chem. 2017, 292, 15426–15433. [Google Scholar] [CrossRef] [Green Version]
- Xing, Y.; Xue, S.; Wu, J.; Zhou, J.; Xing, F.; Li, T.; Nie, X. Serum exosomes derived from irritable bowel syndrome patient increase cell permeability via regulating miR-148b-5p/RGS2 signaling in human colonic epithelium cells. Gastroenterol. Res. Pr. 2021, 2021, 6655900. [Google Scholar] [CrossRef]
- Sanders, K.M.; Ward, S.M.; Koh, S.D. Interstitial cells: Regulators of smooth muscle function. Physiol. Rev. 2014, 94, 859–907. [Google Scholar] [CrossRef]
- Deng, J.J.; Lai, M.Y.; Tan, X.; Yuan, Q. Acupuncture protects the interstitial cells of Cajal by regulating miR-222 in a rat model of post-operative ileus. Acupunct. Med. 2019, 37, 125–132. [Google Scholar] [CrossRef]
- Park, C.; Hennig, G.W.; Sanders, K.M.; Cho, J.H.; Hatton, W.J.; Redelman, D.; Park, J.K.; Ward, S.M.; Miano, J.M.; Yan, W.; et al. Serum response factor-dependent MicroRNAs regulate gastrointestinal smooth muscle cell phenotypes. Gastroenterology 2011, 141, 164–175. [Google Scholar] [CrossRef] [Green Version]
- Ro, S. Multi-phenotypic role of serum response factor in the gastrointestinal system. J. Neurogastroenterol. Motil. 2016, 22, 193–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beyder, A.; Farrugia, G. Targeting ion channels for the treatment of gastrointestinal motility disorders. Ther. Adv. Gastroenterol. 2012, 5, 5–21. [Google Scholar] [CrossRef] [Green Version]
- Holm, A.N.; Rich, A.; Miller, S.M.; Strege, P.; Ou, Y.; Gibbons, S.; Sarr, M.G.; Szurszewski, J.H.; Rae, J.L.; Farrugia, G. Sodium current in human jejunal circular smooth muscle cells. Gastroenterology 2002, 122, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H. MicroRNA regulation of cardiac conduction and arrhythmias. Transl. Res. 2013, 161, 381–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, M.; Gershon, M.D. Enteric nervous system development: What could possibly go wrong? Nat. Rev. Neurosci. 2018, 19, 552–565. [Google Scholar] [CrossRef]
- Spencer, N.J.; Hu, H. Enteric nervous system: Sensory transduction, neural circuits and gastrointestinal motility. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Karnati, H.K.; Panigrahi, M.K.; Gutti, R.K.; Greig, N.H.; Tamargo, I.A. miRNAs: Key players in neurodegenerative disorders and epilepsy. J. Alzheimers Dis. 2015, 48, 563–580. [Google Scholar] [CrossRef] [Green Version]
- Niesler, B.; Kuerten, S.; Demir, I.E.; Schafer, K.H. Disorders of the enteric nervous system—A holistic view. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Burns, G.; Pryor, J.; Holtmann, G.; Walker, M.M.; Talley, N.J.; Keely, S. Immune activation in functional gastrointestinal disorders. Gastroenterol. Hepatol. (N. Y.) 2019, 15, 539–548. [Google Scholar]
- Keely, S.; Walker, M.M.; Marks, E.; Talley, N.J. Immune dysregulation in the functional gastrointestinal disorders. Eur. J. Clin. Invest. 2015, 45, 1350–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbara, G.; Cremon, C.; Carini, G.; Bellacosa, L.; Zecchi, L.; De Giorgio, R.; Corinaldesi, R.; Stanghellini, V. The immune system in irritable bowel syndrome. J. Neurogastroenterol. Motil. 2011, 17, 349–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wouters, M.M.; Vicario, M.; Santos, J. The role of mast cells in functional GI disorders. Gut 2016, 65, 155–168. [Google Scholar] [CrossRef] [Green Version]
- O’Connell, R.M.; Rao, D.S.; Chaudhuri, A.A.; Baltimore, D. Physiological and pathological roles for microRNAs in the immune system. Nat. Rev. Immunol. 2010, 10, 111–122. [Google Scholar] [CrossRef]
- Hirschberger, S.; Hinske, L.C.; Kreth, S. MiRNAs: Dynamic regulators of immune cell functions in inflammation and cancer. Cancer Lett. 2018, 431, 11–21. [Google Scholar] [CrossRef]
- Boldin, M.P.; Taganov, K.D.; Rao, D.S.; Yang, L.; Zhao, J.L.; Kalwani, M.; Garcia-Flores, Y.; Luong, M.; Devrekanli, A.; Xu, J.; et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J. Exp. Med. 2011, 208, 1189–1201. [Google Scholar] [CrossRef]
- Androulidaki, A.; Iliopoulos, D.; Arranz, A.; Doxaki, C.; Schworer, S.; Zacharioudaki, V.; Margioris, A.N.; Tsichlis, P.N.; Tsatsanis, C. The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity 2009, 31, 220–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.M.; Splinter, P.L.; O’Hara, S.P.; LaRusso, N.F. A cellular micro-RNA, let-7i, regulates Toll-like receptor 4 expression and contributes to cholangiocyte immune responses against Cryptosporidium parvum infection. J. Biol. Chem. 2007, 282, 28929–28938. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, A.; Vigorito, E.; Clare, S.; Warren, M.V.; Couttet, P.; Soond, D.R.; van Dongen, S.; Grocock, R.J.; Das, P.P.; Miska, E.A.; et al. Requirement of bic/microRNA-155 for normal immune function. Science 2007, 316, 608–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbara, G.; Feinle-Bisset, C.; Ghoshal, U.C.; Quigley, E.M.; Santos, J.; Vanner, S.; Vergnolle, N.; Zoetendal, E.G. The intestinal microenvironment and functional gastrointestinal disorders. Gastroenterology 2016, 150, 1305–1318.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perna, E.; Aguilera-Lizarraga, J.; Florens, M.V.; Jain, P.; Theofanous, S.A.; Hanning, N.; De Man, J.G.; Berg, M.; De Winter, B.; Alpizar, Y.A.; et al. Effect of resolvins on sensitisation of TRPV1 and visceral hypersensitivity in IBS. Gut 2021, 70, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Piche, T. Tight junctions and IBS—The link between epithelial permeability, low-grade inflammation, and symptom generation? Neurogastroenterol. Motil. 2014, 26, 296–302. [Google Scholar] [CrossRef]
- Turcotte, J.F.; Kao, D.; Mah, S.J.; Claggett, B.; Saltzman, J.R.; Fedorak, R.N.; Liu, J.J. Breaks in the wall: Increased gaps in the intestinal epithelium of irritable bowel syndrome patients identified by confocal laser endomicroscopy (with videos). Gastrointest. Endosc. 2013, 77, 624–630. [Google Scholar] [CrossRef]
- Fritscher-Ravens, A.; Schuppan, D.; Ellrichmann, M.; Schoch, S.; Rocken, C.; Brasch, J.; Bethge, J.; Bottner, M.; Klose, J.; Milla, P.J. Confocal endomicroscopy shows food-associated changes in the intestinal mucosa of patients with irritable bowel syndrome. Gastroenterology 2014, 147, 1012–1020.e4. [Google Scholar] [CrossRef]
- Bertrand, J.; Ghouzali, I.; Guerin, C.; Bole-Feysot, C.; Gouteux, M.; Dechelotte, P.; Ducrotte, P.; Coeffier, M. Glutamine restores tight junction protein claudin-1 expression in colonic mucosa of patients with diarrhea-predominant irritable bowel syndrome. JPEN J. Parenter. Enter. Nutr. 2016, 40, 1170–1176. [Google Scholar] [CrossRef]
- Cichon, C.; Sabharwal, H.; Ruter, C.; Schmidt, M.A. MicroRNAs regulate tight junction proteins and modulate epithelial/endothelial barrier functions. Tissue Barriers 2014, 2, e944446. [Google Scholar] [CrossRef] [Green Version]
- Mawe, G.M.; Hoffman, J.M. Serotonin signalling in the gut—Functions, dysfunctions and therapeutic targets. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 473–486. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Singh, R.; Ha, S.E.; Martin, A.M.; Jones, L.A.; Jin, B.; Jorgensen, B.G.; Zogg, H.; Chervo, T.; Gottfried-Blackmore, A.; et al. Serotonin deficiency is associated with delayed gastric emptying. Gastroenterology 2021, 160, 2451–2466.e19. [Google Scholar] [CrossRef]
- Gershon, M.D. Serotonin is a sword and a shield of the bowel: Serotonin plays offense and defense. Trans. Am. Clin. Climatol. Assoc. 2012, 123, 268–280, discussion 280. [Google Scholar]
- Gershon, M.D. Review article: Serotonin receptors and transporters—Roles in normal and abnormal gastrointestinal motility. Aliment. Pharmacol. Ther. 2004, 20 (Suppl. 7), 3–14. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.; Ha, S.E.; Wei, L.; Singh, R.; Zogg, H.; Clemmensen, B.; Heredia, D.J.; Gould, T.W.; Sanders, K.M.; Ro, S. Colonic moatility is improved by the activation of 5-HT2B receptors on interstitial cells of Cajal in diabetic mice. Gastroenterology 2021, 161, 608–622.e7. [Google Scholar] [CrossRef] [PubMed]
- Nefla, M.; Sudre, L.; Denat, G.; Priam, S.; Andre-Leroux, G.; Berenbaum, F.; Jacques, C. The pro-inflammatory cytokine 14-3-3epsilon is a ligand of CD13 in cartilage. J. Cell Sci. 2015, 128, 3250–3262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fourie, N.H.; Peace, R.M.; Abey, S.K.; Sherwin, L.B.; Rahim-Williams, B.; Smyser, P.A.; Wiley, J.W.; Henderson, W.A. Elevated circulating miR-150 and miR-342-3p in patients with irritable bowel syndrome. Exp. Mol. Pathol. 2014, 96, 422–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Verne, G.N. miRNA-based therapies for the irritable bowel syndrome. Expert Opin. Biol. Ther. 2011, 11, 991–995. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, F.; Takashima, S.; Nadatani, Y.; Otani, K.; Hosomi, S.; Kamata, N.; Taira, K.; Nagami, Y.; Tanigawa, T.; Fukumoto, S.; et al. Exosomal hsa-miR-933 in gastric juice as a potential biomarker for functional dyspepsia. Dig. Dis. Sci. 2020, 65, 3493–3501. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Zhou, X.; Xiang, X.; Ou, Y.; He, J. Effect of miRNA-19a on gastrointestinal motility in rats with functional dyspepsia. Exp. Ther. Med. 2018, 15, 4875–4879. [Google Scholar] [CrossRef]
- Arisawa, T.; Tahara, T.; Fukuyama, T.; Hayashi, R.; Matsunaga, K.; Hayashi, N.; Nakamura, M.; Toshikuni, N.; Shiroeda, H.; Shibata, T. Genetic polymorphism of pri-microRNA 325, targeting SLC6A4 3′-UTR, is closely associated with the risk of functional dyspepsia in Japan. J. Gastroenterol. 2012, 47, 1091–1098. [Google Scholar] [CrossRef]
- Saito, Y.; Suzuki, H.; Tsugawa, H.; Suzuki, S.; Matsuzaki, J.; Hirata, K.; Hibi, T. Dysfunctional gastric emptying with down-regulation of muscle-specific microRNAs in Helicobacter pylori-infected mice. Gastroenterology 2011, 140, 189–198. [Google Scholar] [CrossRef]
- Narayanan, S.P.; O’Brien, D.; Sharma, M.; Miller, K.; Adams, P.; Passos, J.F.; Eirin, A.; Ordog, T.; Bharucha, A.E. Duodenal mucosal mitochondrial gene expression is associated with delayed gastric emptying in diabetic gastroenteropathy. JCI Insight 2021, 6. [Google Scholar] [CrossRef]
- Zhao, S.; Chen, Q.; Kang, X.; Kong, B.; Wang, Z. Aberrantly expressed genes and miRNAs in slow transit constipation based on RNA-Seq analysis. Biomed. Res. Int. 2018, 2018, 2617432. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Mittal, B.; Ghoshal, U.C. Functional dyspepsia is associated with GNbeta3 C825T and CCK-AR T/C polymorphism. Eur. J. Gastroenterol. Hepatol. 2016, 28, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Adam, B.; Liebregts, T.; Holtmann, G. Mechanisms of disease: Genetics of functional gastrointestinal disorders—Searching the genes that matter. Nat. Clin. Pr. Gastroenterol. Hepatol. 2007, 4, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Rozengurt, E. Mitogenic signaling pathways induced by G protein-coupled receptors. J. Cell Physiol. 2007, 213, 589–602. [Google Scholar] [CrossRef]
- Holzer, P. Opioid receptors in the gastrointestinal tract. Regul. Pept. 2009, 155, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Law, I.K.M.; Padua, D.M.; Iliopoulos, D.; Pothoulakis, C. Role of G protein-coupled receptors-microRNA interactions in gastrointestinal pathophysiology. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 313, G361–G372. [Google Scholar] [CrossRef] [Green Version]
- Tache, Y.; Bonaz, B. Corticotropin-releasing factor receptors and stress-related alterations of gut motor function. J. Clin. Investig. 2007, 117, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Dothel, G.; Chang, L.; Shih, W.; Barbaro, M.R.; Cremon, C.; Stanghellini, V.; De Ponti, F.; Mayer, E.A.; Barbara, G.; Sternini, C. micro-opioid receptor, beta-endorphin, and cannabinoid receptor-2 are increased in the colonic mucosa of irritable bowel syndrome patients. Neurogastroenterol. Motil. 2019, 31, e13688. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, T.; Mano, A.; Shibasaki, T. miR-449a contributes to glucocorticoid-induced CRF-R1 downregulation in the pituitary during stress. Mol. Endocrinol. 2013, 27, 1593–1602. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, T.; Mano, A.; Shibasaki, T. Increased expression of miR-325-3p by urocortin 2 and its involvement in stress-induced suppression of LH secretion in rat pituitary. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E781–E787. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, M. Alvimopan, a selective peripherally acting mu-opioid antagonist. Neurogastroenterol. Motil. 2005, 17, 157–165. [Google Scholar] [CrossRef]
- Sobczak, M.; Salaga, M.; Storr, M.A.; Fichna, J. Physiology, signaling, and pharmacology of opioid receptors and their ligands in the gastrointestinal tract: Current concepts and future perspectives. J. Gastroenterol. 2014, 49, 24–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Yang, C.; Kirkmire, C.M.; Wang, Z.J. Regulation of opioid tolerance by let-7 family microRNA targeting the mu opioid receptor. J. Neurosci. 2010, 30, 10251–10258. [Google Scholar] [CrossRef] [Green Version]
- Lu, Z.; Xu, J.; Xu, M.; Pasternak, G.W.; Pan, Y.X. Morphine regulates expression of mu-opioid receptor MOR-1A, an intron-retention carboxyl terminal splice variant of the mu-opioid receptor (OPRM1) gene via miR-103/miR-107. Mol. Pharmacol. 2014, 85, 368–380. [Google Scholar] [CrossRef] [Green Version]
- Ni, J.; Gao, Y.; Gong, S.; Guo, S.; Hisamitsu, T.; Jiang, X. Regulation of mu-opioid type 1 receptors by microRNA134 in dorsal root ganglion neurons following peripheral inflammation. Eur. J. Pain. 2013, 17, 313–323. [Google Scholar] [CrossRef]
- Liu, S.; da Cunha, A.P.; Rezende, R.M.; Cialic, R.; Wei, Z.; Bry, L.; Comstock, L.E.; Gandhi, R.; Weiner, H.L. The host shapes the gut microbiota via fecal MicroRNA. Cell Host Microbe 2016, 19, 32–43. [Google Scholar] [CrossRef] [Green Version]
- Moloney, G.M.; Viola, M.F.; Hoban, A.E.; Dinan, T.G.; Cryan, J.F. Faecal microRNAs: Indicators of imbalance at the host-microbe interface? Benef. Microbes 2018, 9, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Weiner, H.L. Control of the gut microbiome by fecal microRNA. Microb. Cell 2016, 3, 176–177. [Google Scholar] [CrossRef]
- Aguilar, C.; Mano, M.; Eulalio, A. MicroRNAs at the Host-Bacteria Interface: Host Defense or Bacterial Offense. Trends Microbiol. 2019, 27, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zeng, Y.; Zeng, D.; Wang, H.; Zhou, M.; Sun, N.; Xin, J.; Khalique, A.; Rajput, D.S.; Pan, K.; et al. Probiotics and MicroRNA: Their roles in the host-microbe interactions. Front. Microbiol. 2020, 11, 604462. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D. Gut microbiota—At the intersection of everything? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 321–322. [Google Scholar] [CrossRef] [PubMed]
- Backhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [Green Version]
- Lynch, S.V.; Pedersen, O. The human intestinal microbiome in health and disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D. Gut microbiota: Changes in gut microbes and host metabolism: Squaring the circle? Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 563–564. [Google Scholar] [CrossRef]
- Schroeder, B.O.; Backhed, F. Signals from the gut microbiota to distant organs in physiology and disease. Nat. Med. 2016, 22, 1079–1089. [Google Scholar] [CrossRef]
- Malinen, E.; Rinttila, T.; Kajander, K.; Matto, J.; Kassinen, A.; Krogius, L.; Saarela, M.; Korpela, R.; Palva, A. Analysis of the fecal microbiota of irritable bowel syndrome patients and healthy controls with real-time PCR. Am. J. Gastroenterol. 2005, 100, 373–382. [Google Scholar] [CrossRef]
- Shukla, R.; Ghoshal, U.; Dhole, T.N.; Ghoshal, U.C. Fecal microbiota in patients with irritable bowel syndrome compared with healthy controls using real-time polymerase chain reaction: An evidence of dysbiosis. Dig. Dis. Sci. 2015, 60, 2953–2962. [Google Scholar] [CrossRef]
- Tap, J.; Derrien, M.; Tornblom, H.; Brazeilles, R.; Cools-Portier, S.; Dore, J.; Storsrud, S.; Le Neve, B.; Ohman, L.; Simren, M. Identification of an intestinal microbiota signature associated with severity of irritable bowel syndrome. Gastroenterology 2017, 152, 111–123.e8. [Google Scholar] [CrossRef] [Green Version]
- Pittayanon, R.; Lau, J.T.; Yuan, Y.; Leontiadis, G.I.; Tse, F.; Surette, M.; Moayyedi, P. Gut microbiota in patients with irritable bowel syndrome—A systematic review. Gastroenterology 2019, 157, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Ghoshal, U.C.; Srivastava, D.; Misra, A. A randomized double-blind placebo-controlled trial showing rifaximin to improve constipation by reducing methane production and accelerating colon transit: A pilot study. Indian J. Gastroenterol. 2018, 37, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Rodino-Janeiro, B.K.; Vicario, M.; Alonso-Cotoner, C.; Pascua-Garcia, R.; Santos, J. A review of microbiota and irritable bowel syndrome: Future in therapies. Adv. Ther. 2018, 35, 289–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, L.; Shanahan, E.R.; Raj, A.; Koloski, N.A.; Fletcher, L.; Morrison, M.; Walker, M.M.; Talley, N.J.; Holtmann, G. Dyspepsia and the microbiome: Time to focus on the small intestine. Gut 2017, 66, 1168–1169. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, M.; Nakae, H.; Matsuoka, T.; Takahashi, S.; Hisada, T.; Tomita, J.; Koga, Y. Alteration in the gastric microbiota and its restoration by probiotics in patients with functional dyspepsia. BMJ Open Gastroenterol. 2017, 4, e000144. [Google Scholar] [CrossRef]
- Sekirov, I.; Russell, S.L.; Antunes, L.C.; Finlay, B.B. Gut microbiota in health and disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergman, E.N. Energy contributions of volatile fatty acids from the gastrointestinal tract in various species. Physiol. Rev. 1990, 70, 567–590. [Google Scholar] [CrossRef] [Green Version]
- Samuel, B.S.; Shaito, A.; Motoike, T.; Rey, F.E.; Backhed, F.; Manchester, J.K.; Hammer, R.E.; Williams, S.C.; Crowley, J.; Yanagisawa, M.; et al. Effects of the gut microbiota on host adiposity are modulated by the short-chain fatty-acid binding G protein-coupled receptor, Gpr41. Proc. Natl. Acad. Sci. USA 2008, 105, 16767–16772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Q.; Jia, Q.; Song, L.; Duan, L. Alterations in fecal short-chain fatty acids in patients with irritable bowel syndrome: A systematic review and meta-analysis. Medicine 2019, 98, e14513. [Google Scholar] [CrossRef] [PubMed]
- Reigstad, C.S.; Salmonson, C.E.; Rainey, J.F., 3rd; Szurszewski, J.H.; Linden, D.R.; Sonnenburg, J.L.; Farrugia, G.; Kashyap, P.C. Gut microbes promote colonic serotonin production through an effect of short-chain fatty acids on enterochromaffin cells. FASEB J. 2015, 29, 1395–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fung, T.C.; Vuong, H.E.; Luna, C.D.G.; Pronovost, G.N.; Aleksandrova, A.A.; Riley, N.G.; Vavilina, A.; McGinn, J.; Rendon, T.; Forrest, L.R.; et al. Intestinal serotonin and fluoxetine exposure modulate bacterial colonization in the gut. Nat. Microbiol. 2019, 4, 2064–2073. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.B.; Van Benschoten, A.H.; Cimermancic, P.; Donia, M.S.; Zimmermann, M.; Taketani, M.; Ishihara, A.; Kashyap, P.C.; Fraser, J.S.; Fischbach, M.A. Discovery and characterization of gut microbiota decarboxylases that can produce the neurotransmitter tryptamine. Cell Host Microbe 2014, 16, 495–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattarai, Y.; Williams, B.B.; Battaglioli, E.J.; Whitaker, W.R.; Till, L.; Grover, M.; Linden, D.R.; Akiba, Y.; Kandimalla, K.K.; Zachos, N.C.; et al. Gut microbiota-produced tryptamine activates an epithelial G-protein-coupled receptor to increase colonic secretion. Cell Host Microbe 2018, 23, 775–785.e775. [Google Scholar] [CrossRef] [Green Version]
- Quigley, E.M.M. Microbiota-brain-gut axis and neurodegenerative diseases. Curr. Neurol. Neurosci. Rep. 2017, 17, 94. [Google Scholar] [CrossRef]
- Rhee, S.H.; Pothoulakis, C.; Mayer, E.A. Principles and clinical implications of the brain-gut-enteric microbiota axis. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 306–314. [Google Scholar] [CrossRef] [Green Version]
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The gut-brain axis: Interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 2015, 28, 203–209. [Google Scholar]
- Salzman, N.H.; Underwood, M.A.; Bevins, C.L. Paneth cells, defensins, and the commensal microbiota: A hypothesis on intimate interplay at the intestinal mucosa. Semin. Immunol. 2007, 19, 70–83. [Google Scholar] [CrossRef]
- Needham, B.D.; Kaddurah-Daouk, R.; Mazmanian, S.K. Gut microbial molecules in behavioural and neurodegenerative conditions. Nat. Rev. Neurosci. 2020, 21, 717–731. [Google Scholar] [CrossRef]
- Van Oudenhove, L.; Crowell, M.D.; Drossman, D.A.; Halpert, A.D.; Keefer, L.; Lackner, J.M.; Murphy, T.B.; Naliboff, B.D.; Levy, R.L. Biopsychosocial aspects of functional gastrointestinal disorders. Gastroenterology 2016, 150, 1355–1367.e2. [Google Scholar] [CrossRef] [PubMed]
- Zamani, M.; Alizadeh-Tabari, S.; Zamani, V. Systematic review with meta-analysis: The prevalence of anxiety and depression in patients with irritable bowel syndrome. Aliment. Pharmacol. Ther. 2019, 50, 132–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aragon, G.; Graham, D.B.; Borum, M.; Doman, D.B. Probiotic therapy for irritable bowel syndrome. Gastroenterol. Hepatol. (N. Y.) 2010, 6, 39–44. [Google Scholar]
- Koga, Y.; Ohtsu, T.; Kimura, K.; Asami, Y.; Probiotic, L. Gasseri strain (LG21) for the upper gastrointestinal tract acting through improvement of indigenous microbiota. BMJ Open Gastroenterol. 2019, 6, e000314. [Google Scholar] [CrossRef] [Green Version]
- Bubnov, R.; Babenko, L.; Lazarenko, L.; Kryvtsova, M.; Shcherbakov, O.; Zholobak, N.; Golubnitschaja, O.; Spivak, M. Can tailored nanoceria act as a prebiotic? Report on improved lipid profile and gut microbiota in obese mice. EPMA J. 2019, 10, 317–335. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gibson, G.R.; Sailer, M.; Theis, S.; Rastall, R.A. Prebiotics inhibit proteolysis by gut bacteria in a host diet-dependent manner: A three-stage continuous in vitro gut model experiment. Appl. Environ. Microbiol. 2020, 86. [Google Scholar] [CrossRef] [PubMed]
- Bubnov, R.V.; Babenko, L.P.; Lazarenko, L.M.; Mokrozub, V.V.; Spivak, M.Y. Specific properties of probiotic strains: Relevance and benefits for the host. EPMA J. 2018, 9, 205–223. [Google Scholar] [CrossRef]
- Zmora, N.; Suez, J.; Elinav, E. You are what you eat: Diet, health and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 35–56. [Google Scholar] [CrossRef]
- Delzenne, N.M.; Bindels, L.B. Food for thought about manipulating gut bacteria. Nature 2020, 577, 32–34. [Google Scholar] [CrossRef] [Green Version]
- Sonnenburg, J.L.; Backhed, F. Diet-microbiota interactions as moderators of human metabolism. Nature 2016, 535, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Mehtab, W.; Agarwal, A.; Singh, N.; Malhotra, A.; Makharia, G.K. All that a physician should know about FODMAPs. Indian J. Gastroenterol. 2019, 38, 378–390. [Google Scholar] [CrossRef]
- Mars, R.A.T.; Frith, M.; Kashyap, P.C. Functional gastrointestinal disorders and the microbiome—What is the best strategy for moving microbiome-based therapies for functional gastrointestinal disorders into the clinic? Gastroenterology 2021, 160, 538–555. [Google Scholar] [CrossRef]
- Staudacher, H.M.; Whelan, K. The low FODMAP diet: Recent advances in understanding its mechanisms and efficacy in IBS. Gut 2017, 66, 1517–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altobelli, E.; Del Negro, V.; Angeletti, P.M.; Latella, G. Low-FODMAP diet improves irritable bowel syndrome symptoms: A meta-analysis. Nutrients 2017, 9, 940. [Google Scholar] [CrossRef]
- Tomova, A.; Bukovsky, I.; Rembert, E.; Yonas, W.; Alwarith, J.; Barnard, N.D.; Kahleova, H. The effects of vegetarian and vegan diets on gut microbiota. Front. Nutr. 2019, 6, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonneau, E.; Neveu, B.; Kostantin, E.; Tsongalis, G.J.; De Guire, V. How close are miRNAs from clinical practice? A perspective on the diagnostic and therapeutic market. EJIFCC 2019, 30, 114–127. [Google Scholar]
- Brennecke, J.; Stark, A.; Russell, R.B.; Cohen, S.M. Principles of microRNA-target recognition. PLoS Biol. 2005, 3, e85. [Google Scholar] [CrossRef] [PubMed]
- Krek, A.; Grun, D.; Poy, M.N.; Wolf, R.; Rosenberg, L.; Epstein, E.J.; MacMenamin, P.; da Piedade, I.; Gunsalus, K.C.; Stoffel, M.; et al. Combinatorial microRNA target predictions. Nat. Genet. 2005, 37, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, G.; Tian, C.; Jiang, W.; Jin, L.; Zhang, C.; Chen, X. Exosome-mediated small RNA delivery for gene therapy. Wiley Interdiscip. Rev. RNA 2016, 7, 758–771. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Y.; Liu, H.; Tang, W.H. Exosomes: Biogenesis, biologic function and clinical potential. Cell Biosci. 2019, 9, 19. [Google Scholar] [CrossRef]
- Baumann, V.; Winkler, J. miRNA-based therapies: Strategies and delivery platforms for oligonucleotide and non-oligonucleotide agents. Futur. Med. Chem. 2014, 6, 1967–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crooke, S.T.; Seth, P.P.; Vickers, T.A.; Liang, X.H. The interaction of phosphorothioate-containing RNA targeted drugs with proteins is a critical determinant of the therapeutic effects of these agents. J. Am. Chem. Soc. 2020, 142, 14754–14771. [Google Scholar] [CrossRef] [PubMed]
- Dzierlega, K.; Yokota, T. Optimization of antisense-mediated exon skipping for Duchenne muscular dystrophy. Gene Ther. 2020, 27, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Dowdy, S.F. Overcoming cellular barriers for RNA therapeutics. Nat. Biotechnol. 2017, 35, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Chugh, P.; Dittmer, D.P. Potential pitfalls in microRNA profiling. Wiley Interdiscip. Rev. RNA 2012, 3, 601–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, M. MicroRNA profiling: Separating signal from noise. Nat. Methods 2010, 7, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.; Rajpurohit, A.; Burke, E.E.; Williams, C.; Collado-Torres, L.; Kimos, M.; Brandon, N.J.; Cross, A.J.; Jaffe, A.E.; Weinberger, D.R.; et al. Comprehensive assessment of multiple biases in small RNA sequencing reveals significant differences in the performance of widely used methods. BMC Genom. 2019, 20, 513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hucker, S.M.; Fehlmann, T.; Werno, C.; Weidele, K.; Luke, F.; Schlenska-Lange, A.; Klein, C.A.; Keller, A.; Kirsch, S. Single-cell microRNA sequencing method comparison and application to cell lines and circulating lung tumor cells. Nat. Commun. 2021, 12, 4316. [Google Scholar] [CrossRef]
- Kalla, R.; Ventham, N.T.; Kennedy, N.A.; Quintana, J.F.; Nimmo, E.R.; Buck, A.H.; Satsangi, J. MicroRNAs: New players in IBD. Gut 2015, 64, 504–517. [Google Scholar] [CrossRef] [Green Version]
- Riffo-Campos, A.L.; Riquelme, I.; Brebi-Mieville, P. Tools for sequence-based miRNA target prediction: What to choose? Int. J. Mol. Sci. 2016, 17, 1987. [Google Scholar] [CrossRef] [Green Version]
- Kashyap, P.C.; Marcobal, A.; Ursell, L.K.; Larauche, M.; Duboc, H.; Earle, K.A.; Sonnenburg, E.D.; Ferreyra, J.A.; Higginbottom, S.K.; Million, M.; et al. Complex interactions among diet, gastrointestinal transit, and gut microbiota in humanized mice. Gastroenterology 2013, 144, 967–977. [Google Scholar] [CrossRef] [Green Version]
- Tarallo, S.; Ferrero, G.; De Filippis, F.; Francavilla, A.; Pasolli, E.; Panero, V.; Cordero, F.; Segata, N.; Grioni, S.; Pensa, R.G.; et al. Stool microRNA profiles reflect different dietary and gut microbiome patterns in healthy individuals. Gut 2021. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Ren, Y.; Sayed, M.; Hu, X.; Lei, C.; Kumar, A.; Hutchins, E.; Mu, J.; Deng, Z.; Luo, C.; et al. Plant-derived exosomal MicroRNAs shape the gut microbiota. Cell Host Microbe 2018, 24, 637–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, H. Gut microbiota: Host faecal miRNA regulates gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 122–123. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Singh, R.; Ro, S.; Ghoshal, U.C. Gut microbiota dysbiosis in functional gastrointestinal disorders: Underpinning the symptoms and pathophysiology. JGH Open 2021, 5, 976–987. [Google Scholar] [CrossRef]
- Mars, R.A.T.; Yang, Y.; Ward, T.; Houtti, M.; Priya, S.; Lekatz, H.R.; Tang, X.; Sun, Z.; Kalari, K.R.; Korem, T.; et al. Longitudinal multi-omics reveals subset-specific mechanisms underlying irritable bowel syndrome. Cell 2020, 182, 1460–1473.e17. [Google Scholar] [CrossRef]


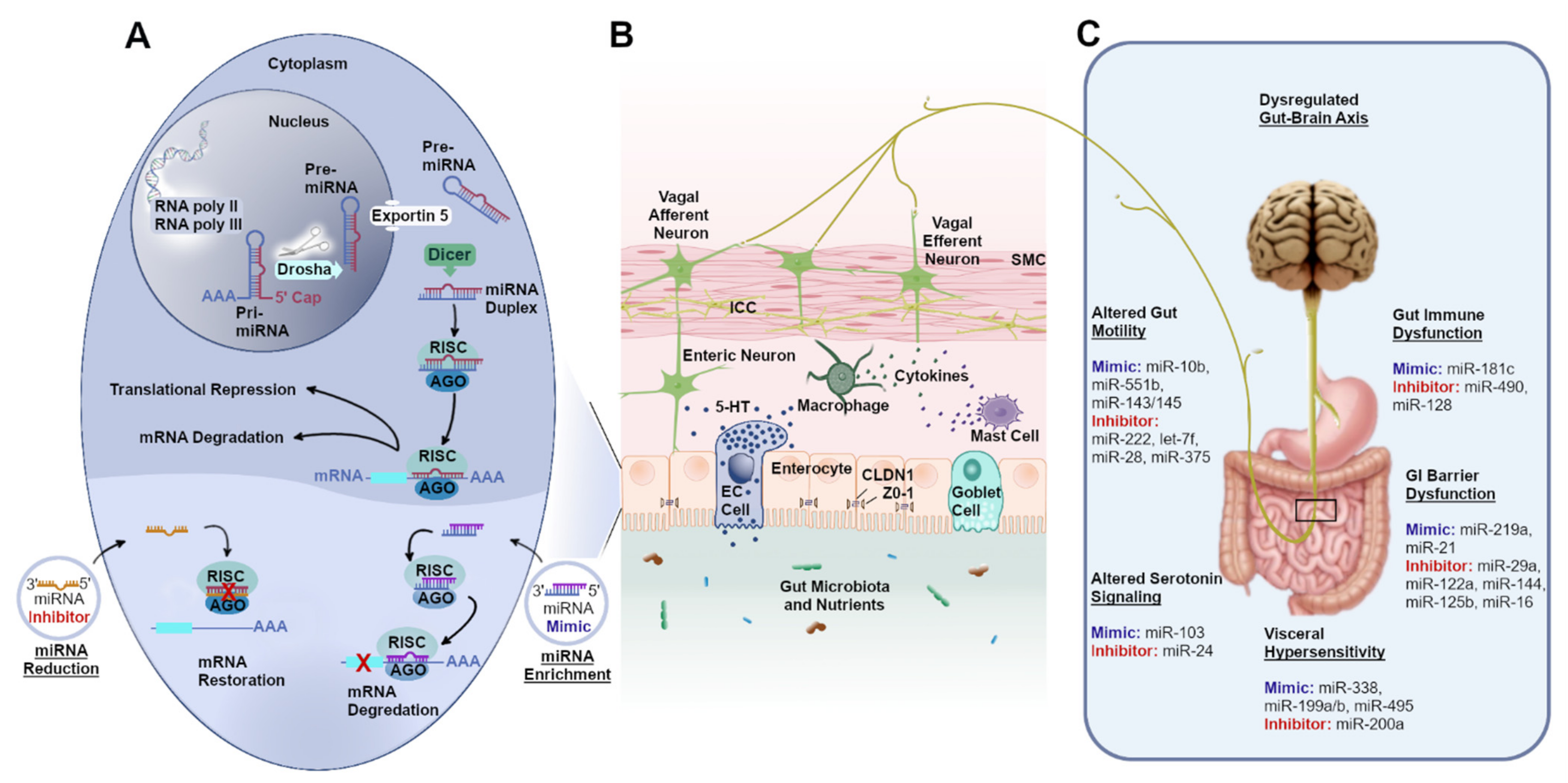{kind=link}
| Disease | miRNA (Expression) | Target | Key Findings | References | ||
|---|---|---|---|---|---|---|
| Gene | Cell | Pathophysiological Mechanisms | ||||
| Gastroparesis, STC | miR-10b ↓ | KLF11, KIT | ICCs | GI dysmotility | Deficiency of miR-10b in ICCs led to gastroparesis and STC, while injection of miR-10b rescued the dysmotility. | [28] |
| STC | miR-222 ↑ | KIT, SCF | ICCs | GI dysmotility | Overexpression of miR-222 in ICCs diminished ICC proliferation and accelerated autophagy, whereas inhibition of miR-222 prevented apoptosis of ICCs. | [55] |
| Delayed gut transit | miR-551b ↓ | KIT | ICCs | GI dysmotility | miR-551b mimic increased intracellular Ca2+ concentration in ICCs. | [56] |
| STC | let-7f ↑ | NaV1.5 | SMCs | GI dysmotility | Upregulation of let-7f resulted in decreased NaV1.5 expression, current density, and reduced motility of GI smooth muscle. | [23] |
| Delayed gut transit | miR-143/145 ↓ | SRF | SMCs | GI dysmotility | Deficiency of Dicer in SMCs resulted in degeneration of SMCs in GI smooth muscle. SRF-induced miR-143 and miR-145 expression promoted GI SMC differentiation and suppression of proliferation. | [57] |
| Delayed gut transit | miR-28 ↑ | NRF2 | Enteric neurons | GI dysmotility | Elevated miR-28 levels in enteric neurons delayed gastric motility by modulating nNOSα dimerization. | [58] |
| Delayed gut transit | miR-375 ↑ | Pdk1 | Enteric neurons | GI dysmotility | Overexpression of miR-375 in enteric neurons resulted in neuronal cell apoptosis while injection of a miR-375 inhibitor prevented the neuronal cell apoptosis and improved gut motility. | [59] |
| IBS-D | miR-490 ↑ | Tryptase, PAR-2 | Mast cells | Gut immune dysfunction | Overexpression of miR-490 in mast cells resulted in increased proliferation of mast cells while inhibition of miR-490 expression promoted apoptosis and inhibited proliferation of mast cells. | [60] |
| IBS model | miR-181c ↓ | IL-1A | Colonic biopsy | Gut immune dysfunction | Inhibition of miR-181c resulted in increased IL-1A levels while overexpressed miR-18c silenced IL-1A and inhibited low-grade inflammation in IBS rats. | [61] |
| STC | miR-128 ↑ | MAPK-14 | Colonic macrophages | Gut immune dysfunction | miR-128 expression negatively correlated with macrophage number, suggesting a miR-128 inhibitor might be a potential therapeutic candidate for a subset of patients with STC having gut immune dysfunction. | [62] |
| IBS-D | miR-16, miR-103/107 ↓ | 5HTR4 | Jejunal biopsy | Altered serotonin signaling | miR-16 and miR-103/107 were downregulated in jejunum biopsies from IBS-D patients and were negatively correlated with IBS symptoms. | [63] |
| IBS-D | miR-510 ↑ | 5HTR3E | IECs | Altered serotonin signaling | miR-510 expression was upregulated in enterocytes and myenteric plexuses of colon sections from patients with IBS-D and resulted in altered serotonin signaling via 5HTR3. | [64] |
| IBS | miR-24 ↑ | SERT | IECs | Altered serotonin signaling/visceral hypersensitivity | miR-24 expression was upregulated in colonic biopsies from IBS patients. Treatment with a miR-24 inhibitor increased nociceptive threshold levels and reduced MPO activity in the proximal colon of IBS mice, and upregulated expression levels of SERT in IECs. | [65] |
| IBS-D | miR-199a/b ↓ | TRPV1 | Colonic biopsy | Visceral hypersensitivity | Decreased colonic miR-199a/b correlated with visceral pain in patients with IBS-D. Administration of miR-199 (lenti-miR-199 precursor) reversed visceral nociception in a rat model of visceral hypersensitivity. | [26] |
| IBS-D | miR-495 ↓ | PI3K, AKT, PKB | Rectal biopsy | Visceral hypersensitivity | miR-495 upregulation reduced visceral sensitivity in IBS-D mice via inhibition of the PI3K/AKT signaling pathway by targeting PKIB. | [66] |
| IBS-D | miR-200a ↑ | CNR1, SERT | Colonic biopsy | Visceral hypersensitivity | Upregulation of miR-200a induced visceral hyperalgesia by targeting CNR1 and SERT. A miR-200a mimic markedly inhibited the expression of CNR1/SERT in IBS-D rats. | [67] |
| IBS | miR-338 ↓ | MAPK, threonine kinase | IECs | Visceral hypersensitivity | Inhibition of miR-338 increased MAPK or protein serine/threonine kinase pathway genes leading to increased visceral sensation. | [29] |
| IBS-D | miR-29a ↑ | ZO-1, CLDN1 | Colonic biopsy | Intestinal barrier dysfunction | Upregulation of miR-29a downregulates ZO-1 and CLDN1 expression resulting in leaky gut. Treatment with miRNA-29a inhibitor downregulated D-LA and DAO activity, and increased the expression of ZO-1 and CLDN1 in the intestinal mucosal epithelium. | [68] |
| IBS-D | miR-29a ↑ | GLUL | Colonic and duodenal biopsy | Intestinal barrier dysfunction | Upregulation of miR-29a led to reduced GLUL levels resulting in impaired intestinal membrane permeability in patients with IBS-D. | [69] |
| IBS | miR-219a ↓ | TJP1/ZO-1, E-CDH1, CEACAM5, CTNND1 | IECs | Intestinal barrier dysfunction | Inhibition of miR-219a-5p in intestinal epithelial cells led to hyperpermeability as TEER was reduced and dextran flux was increased. | [29] |
| IBS model | miR-122a ↑ | TNF-α | IECs | Intestinal barrier dysfunction | miR-122a upregulation led to intestinal barrier dysfunction via TNF-α-mediated degradation of occludin. | [70] |
| IBS model | miR-144 ↑ | OCLN, ZO-1 | Colonic biopsy | Intestinal barrier dysfunction | miR-144 upregulation led to intestinal hyperpermeability, while inhibition of miR-144 improved intestinal barrier function in IBS-D rat colonic epithelial cells. | [71] |
| IBS model | miR-21 ↓ | PTEN, PDCD4, ARF4 | IECs | Intestinal barrier dysfunction | Inhibition of miR-21-5p in IECs led to intestinal epithelial hyperpermeability. | [72] |
| IBS-D | miR-125b, ↑ miR-16 | CGN, CLDN2 | IECs | Intestinal barrier dysfunction | Upregulation of miR-125b and miR-16 downregulated CGN and CLDN2 and resulted in intestinal barrier dysfunction. | [27] |
| IBS | miR-148b ↑ | RGS2 | HT-29 cells cultured with IBS-derived serum exosomes | Intestinal barrier dysfunction | miR-148b overexpression increased cell permeability and downregulated RGS2 expression. | [73] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, R.; Zogg, H.; Ro, S. Role of microRNAs in Disorders of Gut–Brain Interactions: Clinical Insights and Therapeutic Alternatives. J. Pers. Med. 2021, 11, 1021. https://doi.org/10.3390/jpm11101021
Singh R, Zogg H, Ro S. Role of microRNAs in Disorders of Gut–Brain Interactions: Clinical Insights and Therapeutic Alternatives. Journal of Personalized Medicine. 2021; 11(10):1021. https://doi.org/10.3390/jpm11101021
Chicago/Turabian StyleSingh, Rajan, Hannah Zogg, and Seungil Ro. 2021. "Role of microRNAs in Disorders of Gut–Brain Interactions: Clinical Insights and Therapeutic Alternatives" Journal of Personalized Medicine 11, no. 10: 1021. https://doi.org/10.3390/jpm11101021