
Recent Insights into the Role of DNA Methylation and Histone Modifications in Systemic Sclerosis: A Scoping Review
 , and
, and
Abstract
:1. Introduction
2. Methods
2.1. Search Strategy
2.2. Eligibility and Selection of Studies
2.3. Data Charting
3. Results
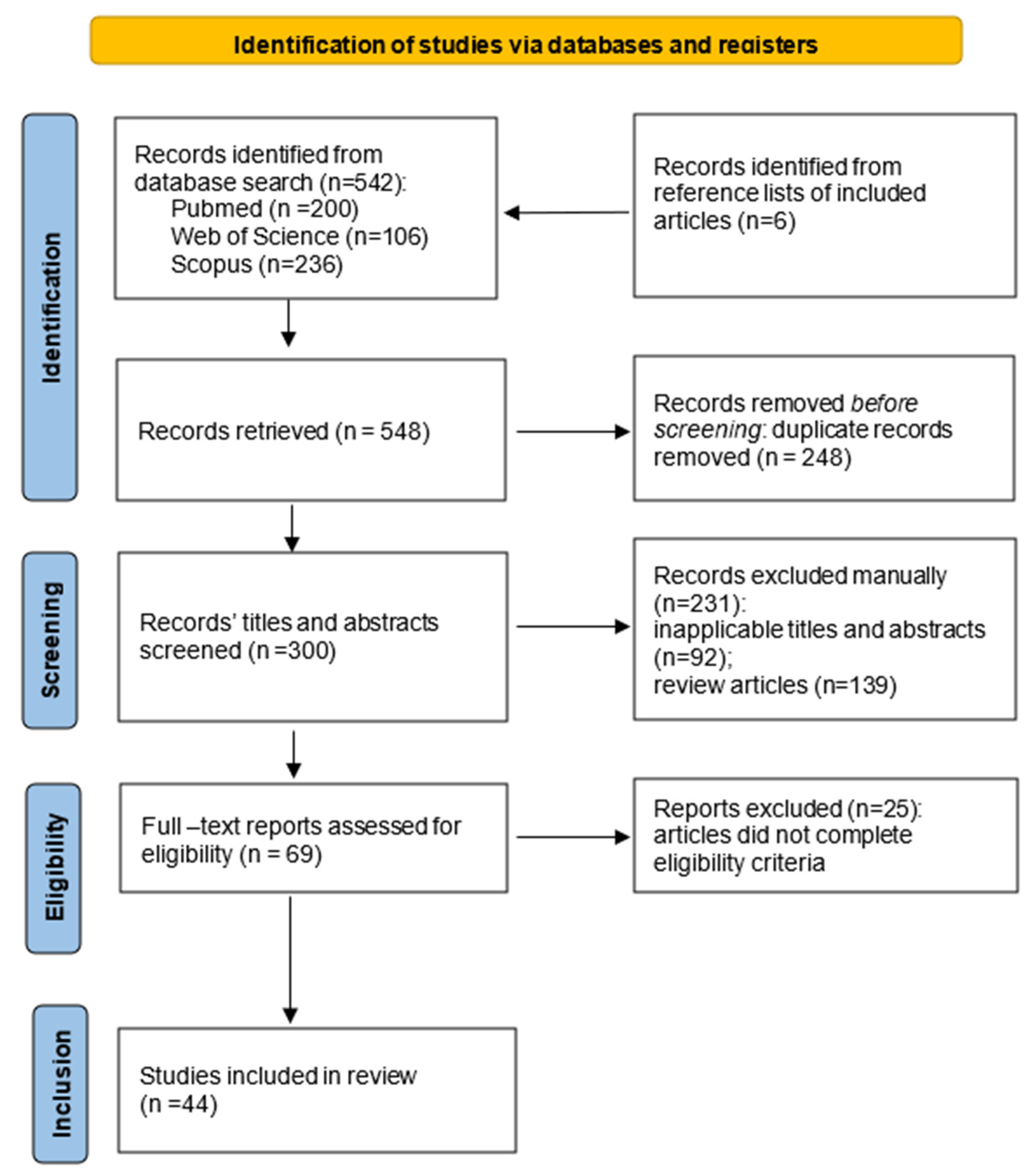
3.1. Selection of Sources
3.2. Characteristics of Sources and Results from Individual Sources
3.3. Synthesis of Results
3.4. New Insights into DNA Methylation and Role in Pathogenesis
3.4.1. In Immune Cells
3.4.2. In Endothelial Cells
3.5. New Insights into Histone PTMs, Associated Enzymes and Chromatin Landscape and Role in Pathogenesis
3.5.1. In FBs
3.5.2. Immune Cells
4. Discussion
Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Denton, C.P.; Khanna, D. Systemic Sclerosis. Lancet 2017, 390, 1685–1699. [Google Scholar] [CrossRef] [PubMed]
- Allanore, Y.; Simms, R.; Distler, O.; Trojanowska, M.; Pope, J.; Denton, C.P.; Varga, J. Systemic Sclerosis. Nat. Rev. Dis. Primers 2015, 1, 15002. [Google Scholar] [CrossRef] [PubMed]
- Tsou, P.S.; Varga, J.; O’Reilly, S. Advances in epigenetics in systemic sclerosis: Molecular mechanisms and therapeutic potential. Nat. Rev. Rheumatol. 2021, 17, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Broen, J.C.A.; Coenen, M.J.H.; Radstake, T.R.D.J. Genetics of Systemic Sclerosis: An Update. Curr. Rheumatol. Rep. 2012, 14, 11–21. [Google Scholar] [CrossRef]
- Agarwal, S.K.; Tan, F.K.; Arnett, F.C. Genetics and genomic studies in scleroderma (systemic sclerosis). Rheum. Dis. Clin. North Am. 2008, 34, 17–40. [Google Scholar] [CrossRef] [PubMed]
- Angiolilli, C.; Marut, W.; van der Kroef, M.; Chouri, E.; Reedquist, K.A.; Radstake, T.R. New insights into the genetics and epigenetics of systemic sclerosis. Nat. Rev. Rheumatol. 2018, 14, 657–673. [Google Scholar] [CrossRef]
- Arnett, F.C.; Cho, M.; Chatterjee, S.; Aguilar, M.B.; Reveille, J.D.; Mayes, M.D. Familial occurrence frequencies and relative risks for systemic sclerosis (scleroderma) in three United States cohorts. Arthritis Rheum. 2001, 44, 1359–1362. [Google Scholar] [CrossRef]
- Feghali-Bostwick, C.; Medsger, T.A., Jr.; Wright, T.M. Analysis of systemic sclerosis in twins reveals low concordance for disease and high concordance for the presence of antinuclear antibodies. Arthritis Rheum. 2003, 48, 1956–1963. [Google Scholar] [CrossRef]
- Altorok, N.; Almeshal, N.; Wang, Y.; Kahaleh, B. Epigenetics, the Holy Grail in the Pathogenesis of Systemic Sclerosis. Rheumatology 2015, 54, 1759–1770. [Google Scholar] [CrossRef]
- Ramos, P.S. Epigenetics of scleroderma: Integrating genetic, ethnic, age, and environmental effects. J. Scleroderma Relat. Disord. 2019, 4, 238–250. [Google Scholar] [CrossRef]
- Toraño, E.G.; García, M.G.; Fernández-Morera, J.L.; Niño-García, P.; Fernández, A.F. The Impact of External Factors on the Epigenome: In Utero and over Lifetime. BioMed Res. Int. 2016, 2016, 2568635. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Millán-Zambrano, G.; Burton, A.; Bannister, A.J.; Schneider, R. Histone post-translational modifications-cause and consequence of genome function. Nat. Rev. Genet. 2022, 23, 563–580. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Kraus, W.L. From Discovery to Function: The Expanding Roles of Long NonCoding RNAs in Physiology and Disease. Endocr. Rev. 2015, 36, 25–64. [Google Scholar] [CrossRef] [PubMed]
- Tricco, A.C.; Lillie, E.; Zarin, W.; O’Brien, K.K.; Colquhoun, H.; Levac, D.; Moher, D.; Peters, M.D.; Horsley, T.; Weeks, L.; et al. PRISMA Extension for Scoping Reviews (PRISMA-ScR): Checklist and Explanation. Ann. Intern. Med. 2018, 169, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Peters, M.D.J.; Godfrey, C.M.; Khalil, H.; McInerney, P.; Parker, D.; Soares, C.B. Guidance for Conducting Systematic Scoping Reviews. Int. J. Evid. Based Healthc. 2015, 13, 141–146. [Google Scholar] [CrossRef]
- Liu, Q.; Zaba, L.; Satpathy, A.T.; Longmire, M.; Zhang, W.; Li, K.; Granja, J.; Guo, C.; Lin, J.; Li, R.; et al. Chromatin Accessibility Landscapes of Skin Cells in Systemic Sclerosis Nominate Dendritic Cells in Disease Pathogenesis. Nat. Commun. 2020, 11, 5843. [Google Scholar] [CrossRef]
- Shin, J.Y.; Beckett, J.D.; Bagirzadeh, R.; Creamer, T.J.; Shah, A.A.; McMahan, Z.; Paik, J.J.; Sampedro, M.M.; MacFarlane, E.G.; Beer, M.A.; et al. Epigenetic Activation and Memory at a TGFB2 Enhancer in Systemic Sclerosis. Sci. Transl. Med. 2019, 11, eaaw0790. [Google Scholar] [CrossRef]
- Noda, S.; Asano, Y.; Nishimura, S.; Taniguchi, T.; Fujiu, K.; Manabe, I.; Nakamura, K.; Yamashita, T.; Saigusa, R.; Akamata, K.; et al. Simultaneous Downregulation of KLF5 and Fli1 Is a Key Feature Underlying Systemic Sclerosis. Nat. Commun. 2014, 5, 5797. [Google Scholar] [CrossRef]
- Tsou, P.; Campbell, P.; Amin, M.; Coit, P.; Miller, S.; Fox, D.; Khanna, D.; Sawalha, A. Inhibition of EZH2 Prevents Fibrosis and Restores Normal Angiogenesis in Scleroderma. Proc. Natl. Acad. Sci. USA 2019, 116, 3695–3702. [Google Scholar] [CrossRef]
- Krämer, M.; Dees, C.; Huang, J.; Schlottmann, I.; Palumbo-Zerr, K.; Zerr, P.; Gelse, K.; Beyer, C.; Distler, A.; Marquez, V.; et al. Inhibition of H3K27 Histone Trimethylation Activates Fibroblasts and Induces Fibrosis. Ann. Rheum. Dis. 2013, 72, 614–620. [Google Scholar] [CrossRef]
- Van Der Kroef, M.; Castellucci, M.; Mokry, M.; Cossu, M.; Garonzi, M.; Bossini-Castillo, L.M.; Chouri, E.; Wichers, C.G.; Beretta, L.; Trombetta, E.; et al. Histone Modifications Underlie Monocyte Dysregulation in Patients with Systemic Sclerosis, Underlining the Treatment Potential of Epigenetic Targeting. Ann. Rheum. Dis. 2019, 78, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Bhattacharyya, S.; Lafyatis, R.; Farina, G.; Yu, J.; Thimmapaya, B.; Wei, J.; Varga, J. P300 Is Elevated in Systemic Sclerosis and Its Expression Is Positively Regulated by TGF-β: Epigenetic Feed-Forward Amplification of Fibrosis. J. Investig. Dermatol. 2013, 133, 1302–1310. [Google Scholar] [CrossRef] [PubMed]
- Ciechomska, M.; O’Reilly, S.; Przyborski, S.; Oakley, F.; Bogunia-Kubik, K.; Van Laar, J.M. Histone Demethylation and Toll-like Receptor 8-Dependent Cross-Talk in Monocytes Promotes Transdifferentiation of Fibroblasts in Systemic Sclerosis Via Fra-2. Arthritis Rheumatol. 2016, 68, 1493–1504. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Ghosh, A.K.; Chu, H.; Fang, F.; Hinchcliff, M.E.; Wang, J.; Marangoni, R.G.; Varga, J. The Histone Deacetylase Sirtuin 1 Is Reduced in Systemic Sclerosis and Abrogates Fibrotic Responses by Targeting Transforming Growth Factor β Signaling. Arthritis Rheumatol. 2015, 67, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Zerr, P.; Palumbo-Zerr, K.; Huang, J.; Tomcik, M.; Sumova, B.; Distler, O.; Schett, G.; Distler, J.H.W. Sirt1 Regulates Canonical TGF-β Signalling to Control Fibroblast Activation and Tissue Fibrosis. Ann. Rheum. Dis. 2016, 75, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C.; Brandt, A.; Merlevede, B.; Hallenberger, L.; Dees, C.; Wohlfahrt, T.; Pötter, S.; Zhang, Y.; Chen, C.W.; Mallano, T.; et al. The Histone Demethylase Jumonji Domain-Containing Protein 3 (JMJD3) Regulates Fibroblast Activation in Systemic Sclerosis. Ann. Rheum. Dis. 2018, 77, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Tsou, P.-S.; Palisoc, P.J.; Ali, M.; Khanna, D.; Sawalha, A.H. Genome-Wide Reduction in Chromatin Accessibility and Unique Transcription Factor Footprints in Endothelial Cells and Fibroblasts in Scleroderma Skin. Arthritis Rheumatol. 2021, 73, 1501–1513. [Google Scholar] [CrossRef]
- Tsou, P.-S.; Wren, J.D.; Amin, M.A.; Schiopu, E.; Fox, D.A.; Khanna, D.; Sawalha, A.H. Histone Deacetylase 5 Is Overexpressed in Scleroderma Endothelial Cells and Impairs Angiogenesis via Repression of Proangiogenic Factors. Arthritis Rheumatol. 2016, 68, 2975–2985. [Google Scholar] [CrossRef]
- Zehender, A.; Li, Y.N.; Lin, N.Y.; Stefanica, A.; Nüchel, J.; Chen, C.W.; Hsu, H.H.; Zhu, H.; Ding, X.; Huang, J.; et al. TGFβ Promotes Fibrosis by MYST1-Dependent Epigenetic Regulation of Autophagy. Nat. Commun. 2021, 12, 4404. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, Y.; Luo, Y.; Yin, Y.; Wang, Q.; Li, Y.; Kanekura, T.; Wang, J.; Liang, G.; Zhao, M.; et al. Aberrant Histone Modification in Peripheral Blood B Cells from Patients with Systemic Sclerosis. Clin. Immunol. 2013, 149, 46–54. [Google Scholar] [CrossRef]
- Wang, Q.; Xiao, Y.; Shi, Y.; Luo, Y.; Li, Y.; Zhao, M.; Lu, Q.; Xiao, R. Overexpression of JMJD3 May Contribute to Demethylation of H3K27me3 in CD4+ T Cells from Patients with Systemic Sclerosis. Clin. Immunol. 2015, 161, 396–399. [Google Scholar] [CrossRef]
- Papazoglou, A.; Huang, M.; Bulik, M.; Lafyatis, A.; Tabib, T.; Morse, C.; Sembrat, J.; Rojas, M.; Valenzi, E.; Lafyatis, R. Epigenetic Regulation of Profibrotic Macrophages in Systemic Sclerosis–Associated Interstitial Lung Disease. Arthritis Rheumatol. 2022, 74, 2003–2014. [Google Scholar] [CrossRef]
- Palumbo-Zerr, K.; Zerr, P.; Distler, A.; Fliehr, J.; Mancuso, R.; Huang, J.; Mielenz, D.; Tomcik, M.; Fürnrohr, B.G.; Scholtysek, C.; et al. Orphan Nuclear Receptor NR4A1 Regulates Transforming Growth Factor-β Signaling and Fibrosis. Nat. Med. 2015, 21, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Altorok, N.; Tsou, P.-S.; Coit, P.; Khanna, D.; Sawalha, A.H. Genome-Wide DNA Methylation Analysis in Dermal Fibroblasts from Patients with Diffuse and Limited Systemic Sclerosis Reveals Common and Subset-Specific DNA Methylation Aberrancies. Ann. Rheum. Dis. 2015, 74, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Baker Frost, D.; da Silveira, W.; Hazard, E.S.; Atanelishvili, I.; Wilson, R.C.; Flume, J.; Day, K.L.; Oates, J.C.; Bogatkevich, G.S.; Feghali-Bostwick, C.; et al. Differential DNA Methylation Landscape in Skin Fibroblasts from African Americans with Systemic Sclerosis. Genes 2021, 12, 129. [Google Scholar] [CrossRef] [PubMed]
- Hattori, M.; Yokoyama, Y.; Hattori, T.; Motegi, S.; Amano, H.; Hatada, I.; Ishikawa, O. Global DNA Hypomethylation and Hypoxia-Induced Expression of the Ten Eleven Translocation (TET) Family, TET1, in Scleroderma Fibroblasts. Exp. Dermatol. 2015, 24, 841–846. [Google Scholar] [CrossRef]
- Dees, C.; Pötter, S.; Zhang, Y.; Bergmann, C.; Zhou, X.; Luber, M.; Wohlfahrt, T.; Karouzakis, E.; Ramming, A.; Gelse, K.; et al. TGF-β-Induced Epigenetic Deregulation of SOCS3 Facilitates STAT3 Signaling to Promote Fibrosis. J. Clin. Investig. 2020, 130, 2347–2363. [Google Scholar] [CrossRef]
- Dees, C.; Schlottmann, I.; Funke, R.; Distler, A.; Palumbo-Zerr, K.; Zerr, P.; Lin, N.Y.; Beyer, C.; Distler, O.; Schett, G.; et al. The Wnt Antagonists DKK1 and SFRP1 Are Downregulated by Promoter Hypermethylation in Systemic Sclerosis. Ann. Rheum. Dis. 2014, 73, 1232–1239. [Google Scholar] [CrossRef]
- Zhang, Y.; Pötter, S.; Chen, C.-W.; Liang, R.; Gelse, K.; Ludolph, I.; Horch, R.E.; Distler, O.; Schett, G.; Distler, J.H.W.; et al. Poly(ADP-Ribose) Polymerase-1 Regulates Fibroblast Activation in Systemic Sclerosis. Ann. Rheum. Dis. 2018, 77, 744–751. [Google Scholar] [CrossRef]
- He, Y.; Tsou, P.-S.; Khanna, D.; Sawalha, A.H. Methyl-CpG-Binding Protein 2 Mediates Antifibrotic Effects in Scleroderma Fibroblasts. Ann. Rheum. Dis. 2018, 77, 1209–1219. [Google Scholar] [CrossRef] [PubMed]
- Henderson, J.; Brown, M.; Horsburgh, S.; Duffy, L.; Wilkinson, S.; Worrell, J.; Stratton, R.; O’Reilly, S. Methyl Cap Binding Protein 2: A Key Epigenetic Protein in Systemic Sclerosis. Rheumatology 2019, 58, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Klein, K.O.; Colmegna, I.; Lora, M.; Greenwood, C.M.T.; Hudson, M. Whole-Genome Bisulfite Sequencing in Systemic Sclerosis Provides Novel Targets to Understand Disease Pathogenesis. BMC Med. Genom. 2019, 12, 144. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Pu, W.; Wang, L.; Jiang, S.; Zhou, X.; Tu, W.; Yu, L.; Zhang, J.; Guo, S.; Liu, Q.; et al. Genome-Wide DNA Methylation Analysis in Systemic Sclerosis Reveals Hypomethylation of IFN-Associated Genes in CD4+ and CD8+ T Cells. J. Investig. Dermatol. 2018, 138, 1069–1077. [Google Scholar] [CrossRef]
- Li, T.; Ortiz-Fernández, L.; Andrés-León, E.; Ciudad, L.; Javierre, B.; López-Isac, E.; Guillén-Del-Castillo, A.; Simeón-Aznar, C.; Ballestar, E.; Martin, J. Epigenomics and Transcriptomics of Systemic Sclerosis CD4+T Cells Reveal Long-Range Dysregulation of Key Inflammatory Pathways Mediated by Disease-Associated Susceptibility Loci. Genome Med. 2020, 12, 81. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shu, Y.; Xiao, Y.; Wang, Q.; Kanekura, T.; Li, Y.; Wang, J.; Zhao, M.; Lu, Q.; Xiao, R. Hypomethylation and Overexpression of ITGAL (CD11a) in CD4+ T Cells in Systemic Sclerosis. Clin. Epigenetics 2014, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Wang, Q.; Sun, X.H.; Liu, R.Z.; Shu, Y.; Kanekura, T.; Huang, J.H.; Li, Y.P.; Wang, J.C.; Zhao, M.; et al. DNA Hypermethylation of the Forkhead Box Protein 3 (FOXP3) Promoter in CD4+ T Cells of Patients with Systemic Sclerosis. Br. J. Dermatol. 2014, 171, 39–47. [Google Scholar] [CrossRef]
- Zeng, Z.; Wang, Y.; Xiao, Y.; Zheng, J.; Liu, R.; He, X.; Yu, J.; Tang, B.; Qiu, X.; Tang, R.; et al. Overexpression of OASL Upregulates TET1 to Induce Aberrant Activation of CD4+ T Cells in Systemic Sclerosis via IRF1 Signaling. Arthritis Res. Ther. 2022, 24, 50. [Google Scholar] [CrossRef]
- Allen, P.C.; Smith, S.; Wilson, R.C.; Wirth, J.R.; Wilson, N.H.; Baker Frost, D.; Flume, J.; Gilkeson, G.S.; Cunningham, M.A.; Langefeld, C.D.; et al. Distinct Genome-Wide DNA Methylation and Gene Expression Signatures in Classical Monocytes from African American Patients with Systemic Sclerosis. Clin. Epigenetics 2023, 15, 25. [Google Scholar] [CrossRef]
- Affandi, A.J.; Carvalheiro, T.; Ottria, A.; Broen, J.C.; Bossini-Castillo, L.; Tieland, R.G.; Van Bon, L.; Chouri, E.; Rossato, M.; Mertens, J.S.; et al. Low RUNX3 Expression Alters Dendritic Cell Function in Patients with Systemic Sclerosis and Contributes to Enhanced Fibrosis. Ann. Rheum. Dis. 2019, 78, 1249–1259. [Google Scholar] [CrossRef]
- Zhu, H.; Zhu, C.; Mi, W.; Chen, T.; Zhao, H.; Zuo, X.; Luo, H.; Li, Q.-Z. Integration of Genome-Wide DNA Methylation and Transcription Uncovered Aberrant Methylation-Regulated Genes and Pathways in the Peripheral Blood Mononuclear Cells of Systemic Sclerosis. Int. J. Rheumatol. 2018, 2018, 7342472. [Google Scholar] [CrossRef]
- Ugor, E.; Simon, D.; Almanzar, G.; Pap, R.; Najbauer, J.; Németh, P.; Balogh, P.; Prelog, M.; Czirják, L.; Berki, T. Increased Proportions of Functionally Impaired Regulatory T Cell Subsets in Systemic Sclerosis. Clin. Immunol. 2017, 184, 54–62. [Google Scholar] [CrossRef]
- Rezaei, R.; Mahmoudi, M.; Gharibdoost, F.; Kavosi, H.; Dashti, N.; Imeni, V.; Jamshidi, A.; Aslani, S.; Mostafaei, S.; Vodjgani, M. IRF7 Gene Expression Profile and Methylation of Its Promoter Region in Patients with Systemic Sclerosis. Int. J. Rheum. Dis. 2017, 20, 1551–1561. [Google Scholar] [CrossRef]
- Almanzar, G.; Klein, M.; Schmalzing, M.; Hilligardt, D.; El Hajj, N.; Kneitz, H.; Wild, V.; Rosenwald, A.; Benoit, S.; Hamm, H.; et al. Disease Manifestation and Inflammatory Activity as Modulators of Th17/Treg Balance and RORC/FoxP3 Methylation in Systemic Sclerosis. Int. Arch. Allergy Immunol. 2016, 171, 141–154. [Google Scholar] [CrossRef]
- Nada, S.; Kahaleh, B.; Altorok, N. Genome-Wide DNA Methylation Pattern in Systemic Sclerosis Microvascular Endothelial Cells: Identification of Epigenetically Affected Key Genes and Pathways. J. Scleroderma Relat. Disord. 2022, 7, 71–81. [Google Scholar] [CrossRef]
- Wang, Y.; Kahaleh, B. Epigenetic Repression of Bone Morphogenetic Protein Receptor II Expression in Scleroderma. J. Cell. Mol. Med. 2013, 17, 1291–1299. [Google Scholar] [CrossRef]
- Matucci-Cerinic, M.; Kahaleh, B.; Wigley, F.M. Review: Evidence That Systemic Sclerosis Is a Vascular Disease. Arthritis Rheum. 2013, 65, 1953–1962. [Google Scholar] [CrossRef]
- Lei, W.; Luo, Y.; Lei, W.; Luo, Y.; Yan, K.; Zhao, S.; Li, Y.; Qiu, X.; Zhou, Y.; Long, H.; et al. Abnormal DNA Methylation in CD4+ T Cells from Patients with Systemic Lupus Erythematosus, Systemic Sclerosis, and Dermatomyositis. Scand. J. Rheumatol. 2009, 38, 369–374. [Google Scholar] [CrossRef]
- Lian, X.; Xiao, R.; Hu, X.; Kanekura, T.; Jiang, H.; Li, Y.; Wang, Y.; Yang, Y.; Zhao, M.; Lu, Q. DNA demethylation of CD40l in CD4+ T cells from women with systemic sclerosis: A possible explanation for female susceptibility. Arthritis Rheum. 2012, 64, 2338–2345. [Google Scholar] [CrossRef]
- Jiang, H.; Xiao, R.; Lian, X.; Kanekura, T.; Luo, Y.; Yin, Y.; Zhang, G.; Yang, Y.; Wang, Y.; Zhao, M.; et al. Demethylation of TNFSF7 Contributes to CD70 Overexpression in CD4+ T Cells from Patients with Systemic Sclerosis. Clin. Immunol. 2012, 143, 39–44. [Google Scholar] [CrossRef]
- Yu, J.; Zeng, Y.; Zhao, K.; Lu, T.; Klein, K.; Colmegna, I.; Lora, M.; Bhatnagar, S.; Leask, A.; Greenwood, C.; et al. Novel Insights into Systemic Sclerosis Using a Sensitive Computational Method to Analyze Whole-Genome Bisulfite Sequencing Data. Clin. Epigenetics 2023, 15, 96. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C.; Distler, J.H. Epigenetic factors as drivers of fibrosis in systemic sclerosis. Epigenomics 2017, 9, 463–477. [Google Scholar] [CrossRef]
- Wang, Y.; Fan, P.S.; Kahaleh, B. Association between enhanced type I collagen expression and epigenetic repression of the FLI1 gene in scleroderma fibroblasts. Arthritis Rheum. 2006, 54, 2271–2279. [Google Scholar] [CrossRef] [PubMed]
- Malaab, M.; Renaud, L.; Takamura, N.; Zimmerman, K.D.; da Silveira, W.A.; Ramos, P.S.; Haddad, S.; Peters-Golden, M.; Penke, L.R.; Wolf, B.; et al. Antifibrotic Factor KLF4 Is Repressed by the miR-10/ TFAP2A/TBX5 Axis in Dermal Fibroblasts: Insights from Twins Discordant for Systemic Sclerosis. Ann. Rheum. Dis. 2022, 81, 268–277. [Google Scholar] [CrossRef]
- Ramos, P.; Zimmerman, K.; Haddad, S.; Langefeld, C.; Medsger, T.; Feghali-Bostwick, C. Integrative Analysis of DNA Methylation in Discordant Twins Unveils Distinct Architectures of Systemic Sclerosis Subsets. Clin. Epigenetics 2019, 11, 58. [Google Scholar] [CrossRef] [PubMed]
- Dal-Bekar, N.; Siomek-Gorecka, A.; Gackowski, D.; Koken-Avsar, A.; Yarkan-Tugsal, H.; Birlik, M.; Islekel, H. Global Hypomethylation Pattern in Systemic Sclerosis: An Application for Absolute Quantification of Epigenetic DNA Modification Products by 2D-UPLC-MS/MS. Clin. Immunol. 2022, 239, 108997. [Google Scholar] [CrossRef]


{kind=link}
| Cell Type/Model | Method | Results | Conclusion | Authors, Year |
|---|---|---|---|---|
| Clinically affected and unaffected SSc skin (8 resident cell types) | ATAC-seq | Significantly more differential peaks in DCs than the other resident cells. DCs display the most upregulated receptor/ligand interactions with other cell types. SSc-associated SNPs are predominantly enriched in DCs. | DCs possess the greatest disease-associated changes in chromatin accessibility. | Liu et al., 2020 [17] |
| Human dermal FBs | ATAC-seq | Higher accessibility at one of the enhancers of TGFB2 with a correlation between the extent of chromatin accessibility and TGFB2 mRNA expression. The enhancer exhibits epigenetic marks—H3K27ac and occupancy by EP300 (enhancer activity). Inhibition of NF-kB or BRD4 achieved sustained inhibition of TGFB2 enhancer activity and mitigated pro-fibrotic gene expression. | Activation of a newly found enhancer of TGFB2 maintains a profibrotic state and is regulated epigenetically. | Shin et al., 2019 [18] |
| Human dermal FBs; murine models | ChIP assay | Hypoacetylation of H3 and H4 on the KLF5 promoter. KLF5 and Fli1 synergistically repress CTGF transcription. Simultaneous downregulation of both KLF5 and Fli1 is a hallmark of SSc | Epigenetic downregulation of the antifibrotic factor KLF5. | Noda et al., 2014 [19] |
| Human dermal FBs and ECs; murine models | ChIP-seq, addition of DZNep, GSK126 | Increased levels of EZH2 and H3K27me3. DZNep dose-dependently decreased EZH2, H3K27me3 and profibrotic genes. Overexpression of EZH2 stimulates cell migration, gel contraction and profibrotic genes. EZH2 inhibits angiogenesis by repressing the Notch signaling pathway. Enrichment of EZH2 binding and H3K27me3 marks at the promoter region of DLL4 in EC. | EZH2 is a key epigenetic factor that promotes fibrosis and inhibits angiogenesis in SSc. | Tsou et al., 2019 [20] |
| Human dermal FBs, murine models | Addition of DZNep, RT-PCR, Western blot, immunohistochemistry | Increased levels of H3K27me3. Inhibition of H3K27me3 with DZNep stimulates the release of collagen. DZNep exacerbates experimental fibrosis. Inhibition of H3K27me3 exerts its profibrotic effects by induction of FRA-2. | H3K27me3 acts as a negative regulator of tissue fibrosis by repressing the expression of FRA-2. | Krämer et al., 2013 [21] |
| Monocytes | ChIP-seq of H3K4me3 and H3K27ac | 1046 and 534 genomic loci have aberrant H3K4me3 and H3K27ac marks. Gene expression significantly correlates and is proportional to the levels of these chromatin marks near gene transcription start sites. Upregulated genes are enriched in monocyte activation, IFN response and cytokine signaling pathways. Strong enrichment of binding sites for STAT and IRF TFs in the hypermethylated and hyperacetylated regions. | Alterations of the chromatin landscape impacting the transcriptome and gene expression of monocytes, correlating with their IFN signature. | Van der Kroef et al., 2019 [22] |
| Human dermal FBs | Immunostaining, semi-quantitative PCR, Western blot, immunofluorescence, ChIP | TGF-β stimulates the transcription of the HAT p300, thus, leading to its overexpression. This is independent of Smads and involves Egr-1. TGF-β leads to p300-dependent histone H4 hyperacetylation at the COL1A2 locus. | Histone acetylation mediated by p300 is an important epigenetic mechanism in fibrogenesis. | Ghosh et al., 2013 [23] |
| Human dermal FBs, human monocytes; murine models | ChIP assay | Fra-2 overexpression in skin biopsy samples from SSc patients and bleomycin-treated mice. TIMP-1 overexpression is induced by TLR-8 and mediated via Fra-2. Treatment with DZNep and the addition of TLR-8 agonist significantly increases Fra-2 and TIMP-1 expression in monocytes and induces transdifferentiation of FBs to myofibroblasts. | Epigenetic changes induced by DZNep have a role in TIMP-1 production mediated by Fra-2 in monocytes. | Ciechomska et al., 2016 [24] |
| Human dermal FBs, murine models | ChIP assay | Underexpression of SIRT1 in skin biopsy samples and in FBs. Activation of SIRT1 significantly attenuates the TGFβ-induced stimulation of FB contractility and migration. SIRT1 blocked Smad-dependent responses partly by the downregulation of the HAT p300 in explanted dermal FBs. | SIRT1 is underexpressed and it exerts potent antifibrotic effects by blocking Smad-dependent transcription. | Wei et al., 2015 [25] |
| Human dermal FBs, animal models | RT-PCR, western blot and immunohistochemistry | SIRT1 is downregulated in fibrotic skin mediated by enhanced TGF-β activation. SIRT1 activation stimulates TGF-β-induced FB activation and the release of collagen. Effective inactivation of SIRT1 in FBs exerts potent antifibrotic effects in murine models of experimental fibrosis. | SIRT1 is a positive regulator of TGF-β/Smad signaling. Downregulation of SIRT1 by TGF-β acts as an endogenous negative feedback mechanism to decrease TGF-β signaling in FBs. | Zerr et al., 2016 [26] |
| Human dermal FBs, murine models | ChIP assay | TGF-β-dependent overexpression of JMJD3 in SSc skin and in experimental fibrosis. JMJD3 promotes FB activation via FRA2. Inactivation of JMJD3 reverses the activated phenotype of FBs and promotes the accumulation of H3K27me3 at the FRA2 promoter, thus, reducing its expression. Pharmacological inhibition of JMJD3 ameliorated experimental fibrosis. | JMJD3 is profibrotic and modulates FB activation by regulating the levels of H3K27me3 at the promoter of FRA2. | Bergmann et al., 2017 [27] |
| Human dermal FBs and ECs | ATAC-seq | Chromatin accessibility is broadly decreased. Identification of differentially accessible chromatin loci enriched in pathways involved in the nervous system, cell membrane projections, cilia mobility, nitric oxide, and others. Increased chromatin binding of SNAI2, ETV2 and ELF1 in ECs, RUNX1 and RUNX2 in FBs. Upregulation of SNAI2 and ETV2 affects angiogenesis in ECs, the downregulation of ENTPD1 affects the profibrotic properties of FBs. | Global reduction in chromatin accessibility in ECs and FBs in dcSSc. Pathways related to neurons might play a role in the dysregulated angiogenesis and fibrosis. | Tsou et al., 2021 [28] |
| Dermal ECs | ATAC-seq | HDAC5 is overexpressed. HDAC5 knockdown increased tube formation in SSc ECs. ATAC-seq after HDAC5 knockdown identifies HDAC-5 regulated genes involved in angiogenesis and fibrosis (FSTL1, PVRL2 and CYR61). HDAC5 mediates its anti-angiogenic effects partly by modulating chromatin accessibility. | Increased expression of HDAC5 represses several pro-angiogenic factors contributing to impaired angiogenesis in SSc. | Tsou et al., 2016 [29] |
| Human dermal FBs, murine models | Inhibition by TSA, inhibition of HMT | TGF-β promotes the activation of autophagy mediated by canonical TGF-β/SMAD3 signaling and repression of the H4K16 HAT MYST1. The latter regulates ATG7 and BECLIN1. Activation of autophagy stimulates FBt activation and induces fibrosis. Overexpression of MYST1 re-establishes the epigenetic control of autophagy. | The epigenetic control of autophagy is altered by a TGF-β dependent downregulation of MYST1. | Zehender et al., 2021 [30] |
| Human B-cells | Global histone H3/H4 acetylation and H3K4/H3K9 methylation assay | Global histone H4 hyperacetylation with global histone H3K9 hypomethylation. HDAC2, HDAC7 and SUV39H2 are significantly downregulated in contrast to JHDM2A which is upregulated. Global histone H4 acetylation was positively correlated with SDAI. | Global histone H4 hyperacetylation associated with disease severity and significant changes in the expression of genes that regulate histone acetylation. | Wang et al., 2013 [31] |
| Human CD4+ T-cells | Colorimetric H3K27 quantification assay | Global H3K27me3 levels are significantly lower with an inverse correlation with the levels of JMJD3. No differences between the expression levels of UTX, EZH1 and EZH2. | Global reduction in a gene-repressive mark. | Wang et al., 2015 [32] |
| Macrophages in lung samples | ScATAC-seq | Increased number of subpopulation of macrophages with the upregulated expression of SPP1 and MMP9. Increased accessibility of SPP1 and MMP9 in SPP1-macrophages. FABP4 gene shows more accessible chromatin in FABPR-macrophages. Transcription binding sites enriched in open chromatin identify multiple TFs: ATF5, TFEB, BCL11A, ETV5, JUN and others. | Identification of transcription factors in the profibrotic macrophages. | Papazoglou et al., 2022 [33] |
| Human dermal FBs, murine models | ChIP assay | Increased levels of NR4A1 in fibrotic skin. Short-term stimulation with TGF-β upregulates NR4A1 mediated by Smad signaling and the TF SP1. NR4A1 recruits the SP1-SIN3A-CoREST-LSD1-HDAC1 complex to reduce the expression of TGF-β target genes. Exposure of FBs to TGF-β for prolonged periods results in rapidly declining levels of NR4A1 mRNA and pan-NR4A1 protein. Rapid acetylation of histones H3 and H4 at the NR4A1 promoter upon TGF-β stimulation. Incubation with selective HDAC I and II inhibitors demonstrated that the desensitization of NR4A1 transcription is dependent on HDAC4, HDAC5, HDAC7 and HDAC10. | The persistently active TGF-β signaling uses HDAC-mediated epigenetic repression and AKT-induced phosphorylation to inhibit the NR4A1 negative feedback loop. | Palumbo-Zerr et al., 2015 [34] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kostova, T.; Karalilova, R.; Batalov, Z.; Kazakova, M.; Sarafian, V.; Batalov, A. Recent Insights into the Role of DNA Methylation and Histone Modifications in Systemic Sclerosis: A Scoping Review. Diagnostics 2024, 14, 652. https://doi.org/10.3390/diagnostics14060652
Kostova T, Karalilova R, Batalov Z, Kazakova M, Sarafian V, Batalov A. Recent Insights into the Role of DNA Methylation and Histone Modifications in Systemic Sclerosis: A Scoping Review. Diagnostics. 2024; 14(6):652. https://doi.org/10.3390/diagnostics14060652
Chicago/Turabian StyleKostova, Tsvetelina, Rositsa Karalilova, Zguro Batalov, Maria Kazakova, Victoria Sarafian, and Anastas Batalov. 2024. "Recent Insights into the Role of DNA Methylation and Histone Modifications in Systemic Sclerosis: A Scoping Review" Diagnostics 14, no. 6: 652. https://doi.org/10.3390/diagnostics14060652