
Implementation of Exome Sequencing in Prenatal Diagnostics: Chances and Challenges

 and
and
Abstract
:1. Introduction
2. Methods
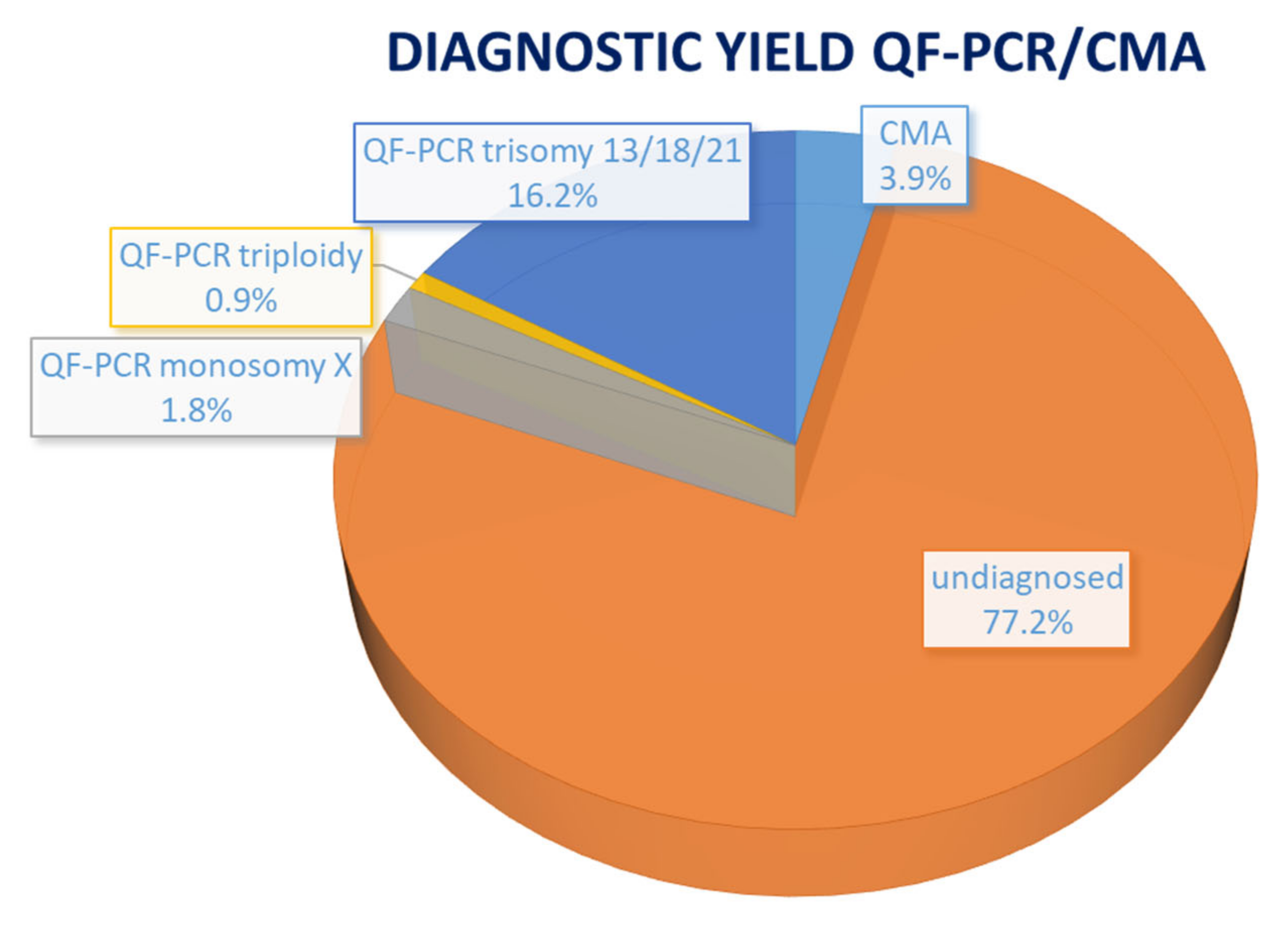
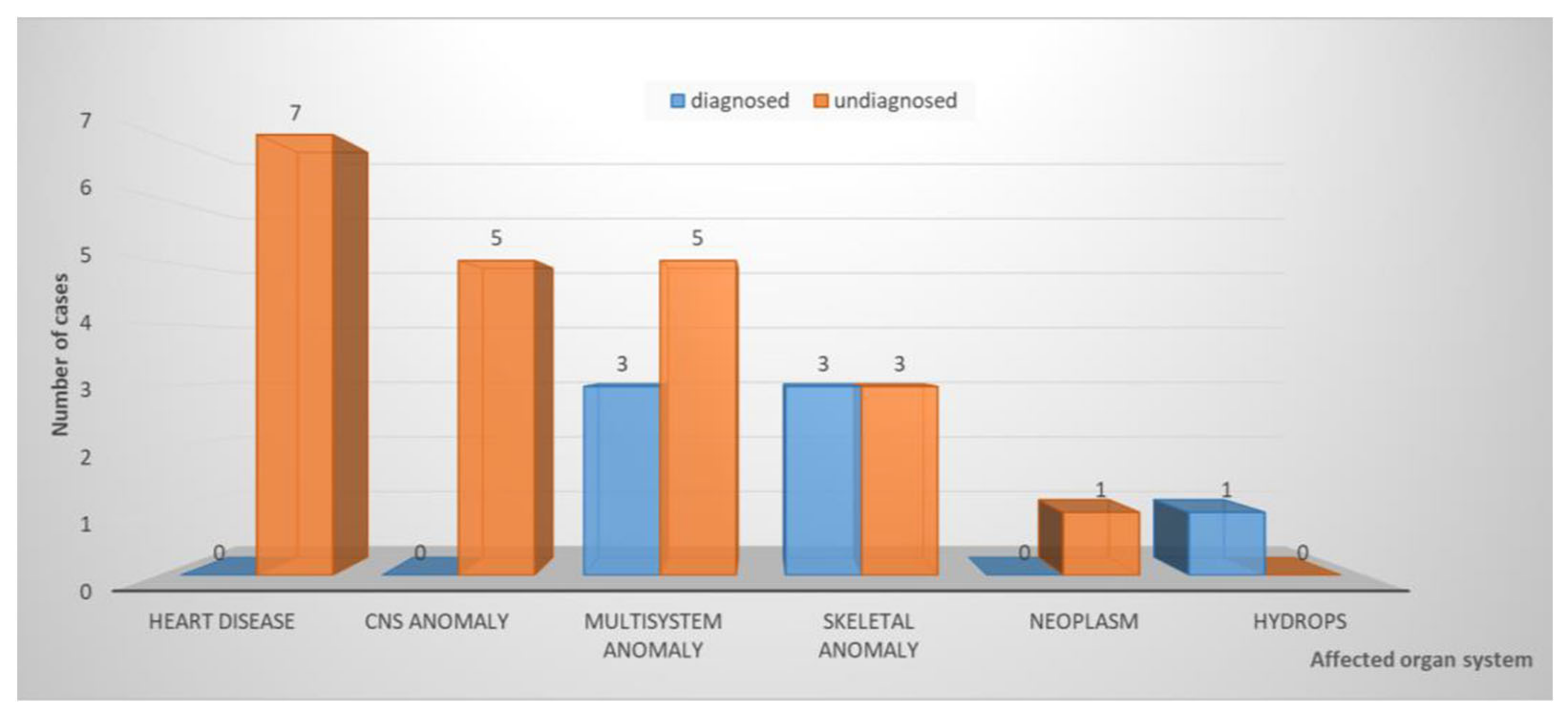
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boyd, P.A.; Tonks, A.M.; Rankin, J.; Rounding, C.; Wellesley, D.; Draper, E.S. Monitoring the prenatal detection of structural fetal congenital anomalies in England and Wales: Register-based study. J. Med. Screen. 2011, 18, 2–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calzolari, E.; Barisic, I.; Loane, M.; Morris, J.; Wellesley, D.; Dolk, H.; Addor, M.; Arriola, L.; Bianchi, F.; Neville, A.J.; et al. Epidemiology of multiple congenital anomalies in Europe: A EUROCAT population-based registry study. Birth Defects Res. Part A Clin. Mol. Teratol. 2014, 100, 270–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wapner, R.J.; Martin, C.L.; Levy, B.; Ballif, B.C.; Eng, C.M.; Zachary, J.M.; Savage, M.; Platt, L.D.; Saltzman, D.; Grobman, W.A.; et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N. Engl. J. Med. 2012, 367, 2175–2184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellis, R.; Oprych, K.; Scotchman, E.; Hill, M.; Chitty, L.S. Diagnostic yield of exome sequencing for prenatal diagnosis of fetal structural anomalies: A systematic review and meta-analysis. Prenat. Diagn. 2022, 42, 662–685. [Google Scholar] [CrossRef]
- Pauta, M.; Martinez-Portilla, R.J.; Borrell, A. Diagnostic yield of exome sequencing in fetuses with multisystem malformations: Systematic review and meta-analysis. Ultrasound Obstet. Gynecol. 2022, 59, 715–722. [Google Scholar] [CrossRef]
- Mone, F.; Mellis, R.; Gabriel, H.; Baptiste, C.; Giordano, J.; Wapner, R.; Chitty, L.S. Should we offer prenatal exome sequencing for intrauterine growth restriction or short long bones? A systematic review and meta-analysis. Am. J. Obstet. Gynecol. 2022. online ahead of print. [Google Scholar] [CrossRef]
- Mellis, R.; Eberhardt, R.Y.; Hamilton, S.J.; PAGE Consortium; McMullan, D.J.; Kilby, M.D.; Maher, E.R.; Hurles, M.E.; Giordano, J.L.; Aggarwal, V.; et al. Fetal exome sequencing for isolated increased nuchal translucency: Should we be doing it? BJOG 2022, 129, 52–61. [Google Scholar] [CrossRef]
- Pauta, M.; Martinez-Portilla, E.J.; Borrell, A. Prenatal Exome Sequencing in Recurrent Fetal Structural Anomalies: Systematic Review and Meta-Analysis. J. Clin. Med. 2021, 10, 4739. [Google Scholar] [CrossRef]
- Van den Veyver, I.B.; Chandler, N.; Wilkins-Haug, L.E.; Wapner, R.J.; Chitty, L.S. International Society for Prenatal Diagnosis Updated Position Statement on the use of genome-wide sequencing for prenatal diagnosis. Prenat. Diagn. 2022, 42, 796–803. [Google Scholar] [CrossRef]
- Vanakker, O.; Vilain, C.; Janssens, K.; Van der Aa, N.; Smits, G.; Bandelier, C.; Blaumeiser, B.; Bulk, S.; Caberg, J.H.; De Leener, A.; et al. Implementation of genomic arrays in prenatal diagnosis: The Belgian approach to meet the challenges. Eur. J. Med. Genet. 2014, 57, 151–156. [Google Scholar] [CrossRef]
- Vandeweyer, G.; Van Laer, L.; Loeys, B.; Van den Bulcke, T.; Kooy, R.F. VariantDB: A flexible annotation and filtering portal for next generation sequencing data. Genome Med. 2014, 6, 74. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vears, D.; Amor, D.J. A framework for reporting secondary and incidental findings in prenatal sequencing: When and for whom? Prenat. Diagn. 2022, 42, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.T.; Lee, K.; Chung, W.K.; Gordon, A.S.; Herman, G.E.; Klein, T.E.; Stewart, D.R.; Amendola, L.M.; Adelman, K.; Bale, S.J.; et al. ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2021, 23, 1381–1390. [Google Scholar] [CrossRef]
- Winsor, E.J.; Silver, M.P.; Theve, R.; Wright, M.; Ward, B.E. Maternal cell contamination in uncultured amniotic fluid. Prenat. Diagn. 1996, 16, 49–54. [Google Scholar] [CrossRef]
- Gray, K.J.; Wilkins-Haug, L.E.; Herrig, N.J.; Vora, N.L. Fetal phenotypes emerge as genetic technologies become robust. Prenat. Diagn. 2019, 39, 811–817. [Google Scholar] [CrossRef]
- Giordano, J.L.; Wapner, R.J. The fetal sequencing consortium: The value of multidisciplinary dialog and collaboration. Prenat. Diagn. 2022, 42, 807–810. [Google Scholar] [CrossRef]
- Lamouroux, A.; Dauge, C.; Wells, C.; Mousty, E.; Pinson, L.; Cavé, H.; Capri, Y.; Faure, J.M.; Grosjean, F.; Sauvestre, F.; et al. Extending the prenatal Noonan’s phenotype by review of ultrasound and autopsy data. Prenat. Diagn. 2022, 42, 574–582. [Google Scholar] [CrossRef]
- Westenius, E.; Sahlin, E.; Conner, P.; Lindstrand, A.; Iwarsson, E. Diagnostic yield using whole-genome sequencing and in-silico panel of 281 genes associated with non-immune hydrops fetalis in clinical setting. Ultrasound Obstet. Gynecol. 2022, 60, 487–493. [Google Scholar] [CrossRef]
- Das, S.; Sharma, C.; Gothwal, M.; Tada, N. Whole exome sequencing, clinical exome or targeted gene panels: What to choose for suspected lethal skeletal dysplasia (short rib thoracic dysplasia type IV). BMJ Case Rep. 2022, 15, e251118. [Google Scholar] [CrossRef]
- Cao, J.; Chen, A.; Tian, L.; Yan, L.; Li, H.; Zhou, B. Application of whole exome sequencing in fetal cases with skeletal abnormalities. Heliyon 2022, 8, e09819. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, M.; Wang, H. Prenatal trio-based whole exome sequencing in fetuses with abnormalities of the skeletal system. Mol. Genet. Genom. 2022, 297, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- So, P.L.; Luk, H.M.; Cheung, K.W.; Hui, W.; Chung, M.Y.; Mak, A.S.; Lok, W.Y.; Yu, K.P.T.; Cheng, S.S.; Hau, E.W.; et al. Prenatal phenotype of Kabuki syndrome: A case series and literature review. Prenat. Diagn. 2021, 41, 1089–1100. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, A.D. Fanconi anemia and its diagnosis. Mutat. Res. 2009, 668, 4–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, M.; Rac, M.W.F.; McKinney, J. Radial Ray Malformation. Am. J. Obstet. Gynecol. 2019, 221, B16–B18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennelly, M.M.; Moran, P. A clinical algorithm of prenatal diagnosis of Radial Ray Defects with two and three dimensional ultrasound. Prenat. Diagn. 2007, 27, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Ruaud, L.; Roux, N.; Boutaud, L.; Bessières, B.; Ageorges, F.; Achaiaa, A.; Bole, C.; Nitschke, P.; Masson, C.; Vekemans, M.; et al. Biallelic THOC6 pathogenic variants: Prenatal phenotype and review of the literature. Birth Defects Res. 2022, 114, 499–504. [Google Scholar] [CrossRef]
- Tjon, J.K.; Tan-Sindhunata, G.M.; Bugiani, M.; Witbreuk, M.M.; van der Sluijs, J.A.; Weiss, M.M.; van de Pol, L.A.; van Weissenbruch, M.M.; van der Knoop, B.J.; de Vries, J.I. Fetal akinesia deformation sequence, arthrogryposis multiplex congenita, and bilateral clubfeet: Is motor assessment of additional value for in utero diagnosis? A 10-year cohort study. Prenat. Diagn. 2019, 39, 219–231. [Google Scholar] [CrossRef] [Green Version]
- Barros, F.; Rolo, L.; Nardozza, L. Prenatal Diagnosis of Lethal Multiple Pterygium Syndrome Using Two-and Three-Dimensional Ultrasonography. J. Clin. Imaging Sci. 2012, 2, 65. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case No. | Phenotype | Phenotypic Group | Gene | Variant | Inheritance | Classification | Associated Syndrome | Outcome |
|---|---|---|---|---|---|---|---|---|
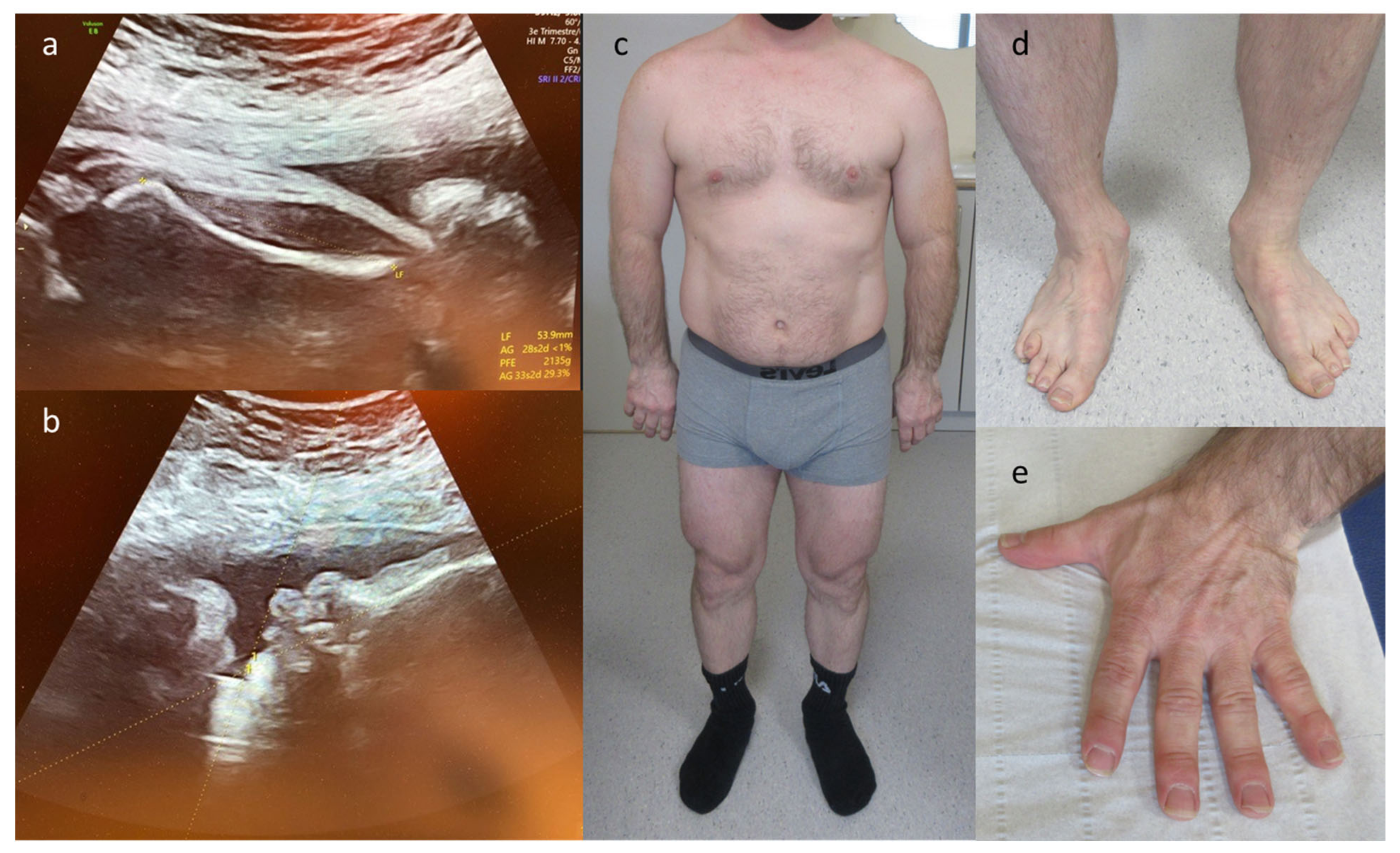
| 1 | Abnormal ears, bilateral talipes equinovarus | skeletal | KMT2D | c.450G > A p.(Trp150*) | AD–de novo | path | Kabuki syndrome 1 (OMIM# 147920) | TOP |
| 8 | IUGR, oligodactyly left hand, hypoplastic ray right hand | skeletal | FANCG | c.115C > T p.(Arg39*) | AR–hom | path | Fanconi anemia, complementation group G (OMIM# 614082) | TOP |
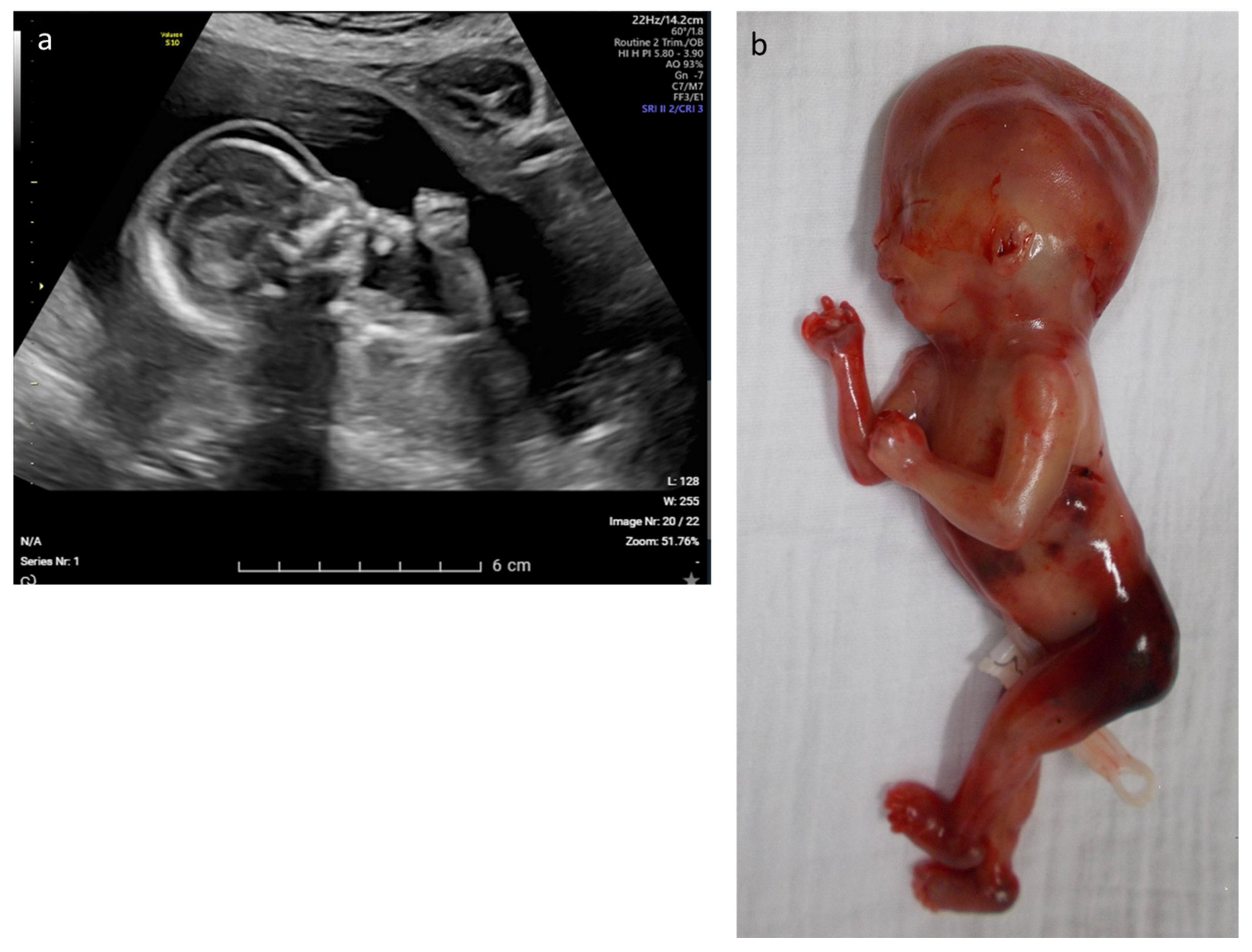
| 10 | Rhizomelic shortening and bowing of the long bones, microretrognathia and clenched hands | skeletal | COL2A1 | c.2710C > T p.(Arg904Cys) | AD–pat | path | Stickler syndrome type I (OMIM# 108300) | LB |
| 12 | Edema, rocker bottom foot, retrognathia, abnormal thorax and ribs, increased NT | multisystem | CHRNA1 | c.548A > G p.(Asp183Gly) | AR–hom | likely path | Multiple pterygium syndrome, lethal type (OMIM# 253290) | TOP |
| 23 | Hydrops, acites, hydrothorax | hydrops | RIT1 | c.297T > A p.(Phe99Leu) | AD–de novo | path | Noonan syndroom 8 (OMIM# 615355) | TOP |
| STXBP1 | c.875G > A p.(Arg292His) | AD–de novo | path (IF) | Developmental and epileptic encephalopathy type 4 (OMIM# 612164) | ||||
| 26 | Olivopontocerebellar hypoplasia, tetralogy of Fallot, hypospadias | multisystem | THOC6 | c.298T > A p.(Trp100Arg) | AR–het (mat) | path | Beaulieu-Boycott-Innes syndrome (OMIM# 613680) | TOP |
| THOC6 | c.700G > C p.(Val234Leu) | AR–het (mat) | path | |||||
| THOC6 | c.824G > A p.(Gly275Asp) | AR–het(mat) | path | |||||
| THOC6 | c.569G > A p.(Gly190Glu) | AR–het (pat) | path | |||||
| 28 | Fetal akinesia, hypotonia, rocker-bottom feet, hydrops, hydrothorax, ascites | multisystem | MUSK | c.2201G > T p.(Gly734Val) | AR–hom | likely path | Fetal akinesia deformation sequence 1 (OMIM# 208150) | TOP |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janicki, E.; De Rademaeker, M.; Meunier, C.; Boeckx, N.; Blaumeiser, B.; Janssens, K. Implementation of Exome Sequencing in Prenatal Diagnostics: Chances and Challenges. Diagnostics 2023, 13, 860. https://doi.org/10.3390/diagnostics13050860
Janicki E, De Rademaeker M, Meunier C, Boeckx N, Blaumeiser B, Janssens K. Implementation of Exome Sequencing in Prenatal Diagnostics: Chances and Challenges. Diagnostics. 2023; 13(5):860. https://doi.org/10.3390/diagnostics13050860
Chicago/Turabian StyleJanicki, Ewa, Marjan De Rademaeker, Colombine Meunier, Nele Boeckx, Bettina Blaumeiser, and Katrien Janssens. 2023. "Implementation of Exome Sequencing in Prenatal Diagnostics: Chances and Challenges" Diagnostics 13, no. 5: 860. https://doi.org/10.3390/diagnostics13050860