
Prenatal Diagnosis of Uniparental Disomy in Cases of Rare Autosomal Trisomies Detected Using Noninvasive Prenatal Test: A Case of Prader–Willi Syndrome
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Subjects
2.2. NIPT
2.3. Methylation Analysis
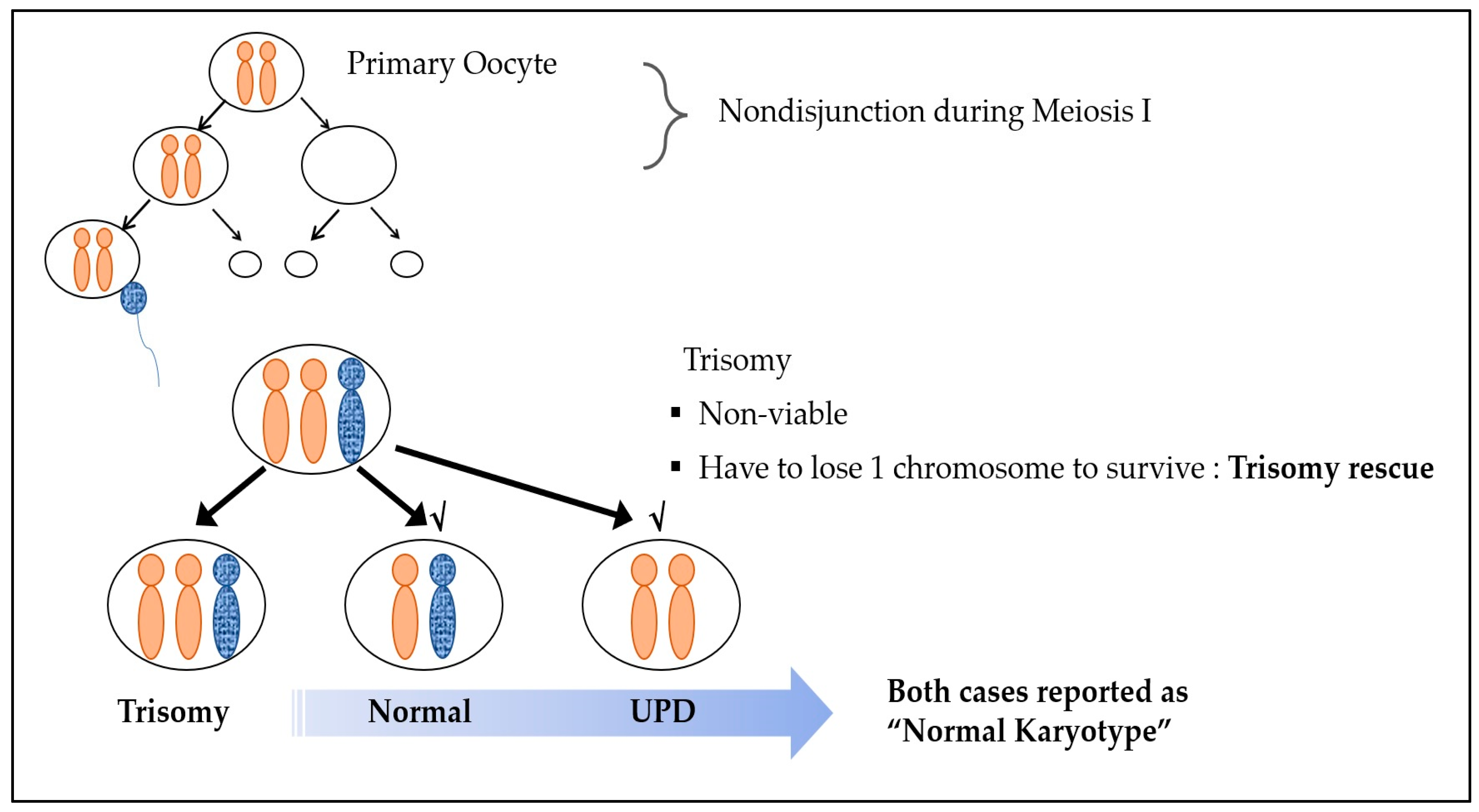
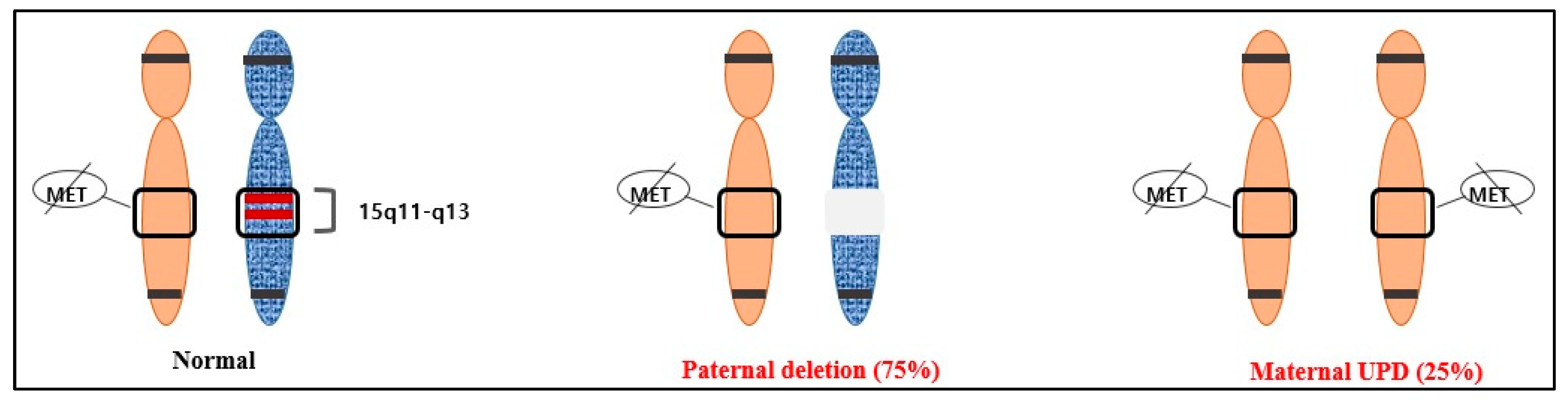
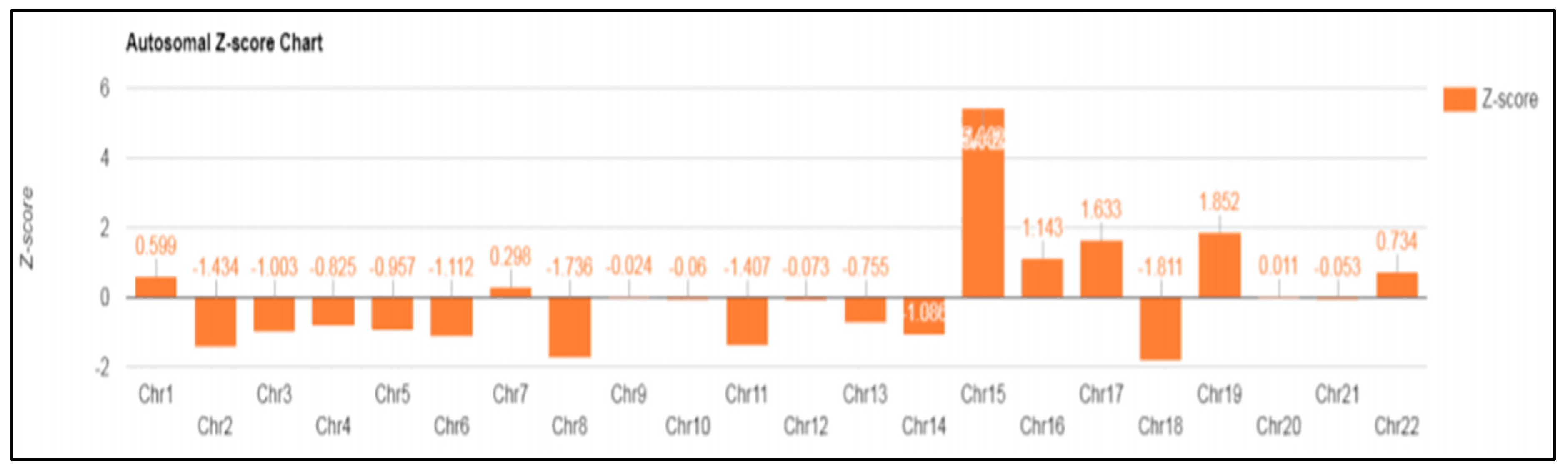
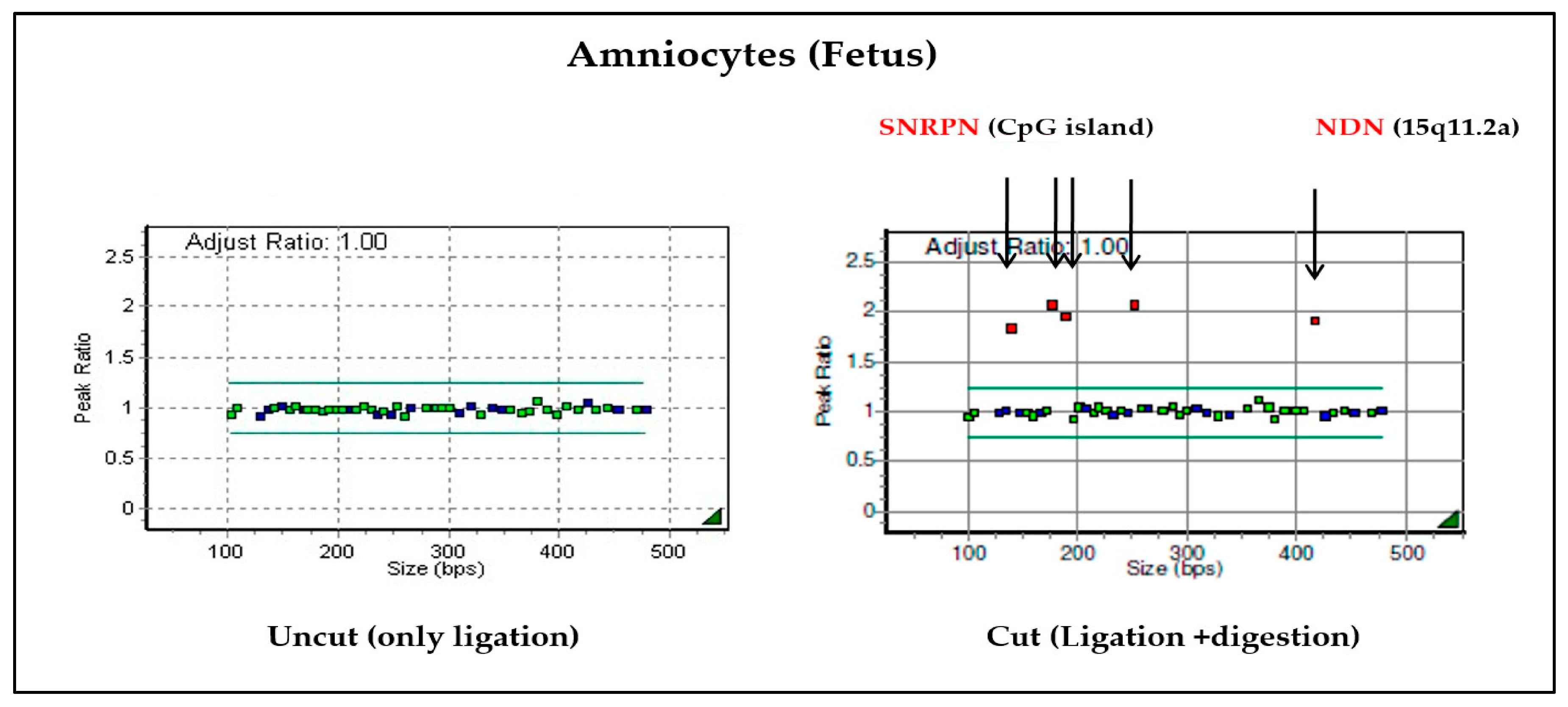
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gil, M.M.; Accurti, V.; Santacruz, B.; Plana, M.N.; Nicolaides, K.H. Analysis of cell-free DNA in maternal blood in screening for aneuploidies: Updated meta-analysis. Ultrasound Obstet. Gynecol. 2017, 50, 302–314. [Google Scholar] [CrossRef]
- Liang, D.; Cram, D.S.; Tan, H.; Linpeng, S.; Liu, Y.; Sun, H.; Zhang, Y.; Tian, F.; Zhu, H.; Xu, M.; et al. Clinical utility of noninvasive prenatal screening for expanded chromosome disease syndromes. Genet. Med. 2019, 21, 1998–2006. [Google Scholar] [CrossRef]
- Benn, P.; Grati, F.R. Genome-wide non-invasive prenatal screening for all cytogenetically visible imbalances. Ultrasound Obstet. Gynecol. 2018, 51, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, S.; Sagaser, K.; Nelson, Z.; Frey, J.; Wardrop, J.; Boomer, T.; McCullough, R.; Schwartz, S. Deletion rescue resulting in segmental homozygosity: A mechanism underlying discordant NIPT results. Am. J. Med. Genet. A 2020, 182, 2666–2670. [Google Scholar] [CrossRef] [PubMed]
- Soster, E.; Boomer, T.; Hicks, S.; Caldwell, S.; Dyr, B.; Chibuk, J.; Almasri, E. Three years of clinical experience with a genome-wide cfDNA screening test for aneuploidies and copy-number variants. Genet. Med. 2021, 23, 1349–1355. [Google Scholar] [CrossRef]
- Pescia, G.; Guex, N.; Iseli, C.; Brennan, L.; Osteras, M.; Xenarios, I.; Farinelli, L.; Conrad, B. Cell-free DNA testing of an extended range of chromosomal anomalies: Clinical experience with 6388 consecutive cases. Genet. Med. 2017, 19, 169–175. [Google Scholar] [CrossRef] [PubMed]
- ACOG Statement on FDA Warning on Genetic Non-Invasive Prenatal Screening Tests 2022. Available online: https://www.acog.org/news/news-releases/2022/04/statement-on-fda-warning-genetic-non-invasive-prenatal-screening-tests (accessed on 20 April 2022).
- Grati, F.R.; Ferreira, J.; Benn, P.; Izzi, C.; Verdi, F.; Vercellotti, E.; Dalpiaz, C.; D’Ajello, P.; Filippi, E.; Volpe, N.; et al. Outcomes in pregnancies with a confined placental mosaicism and implications for prenatal screening using cell-free DNA. Genet. Med. 2020, 22, 309–316. [Google Scholar] [CrossRef]
- Mardy, A.H.; Norton, M.E. Diagnostic testing after positive results on cell free DNA screening: CVS or Amnio? Prenat. Diagn. 2021, 41, 1249–1254. [Google Scholar] [CrossRef]
- Liehr, T. False-positives and false-negatives in non-invasive prenatal testing (NIPT): What can we learn from a meta-analyses on >750,000 tests? Mol. Cytogenet. 2022, 15, 36. [Google Scholar] [CrossRef]
- Del Gaudio, D.; Shinawi, M.; Astbury, C.; Tayeh, M.K.; Deak, K.L.; Raca, G. ACMG Laboratory Quality Assurance Committee. Diagnostic testing for uniparental disomy: A points to consider statement from the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2020, 22, 1133–1141. [Google Scholar] [CrossRef] [Green Version]
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-Willi syndrome. Genet. Med. 2012, 14, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, S.C.; Clayton-Smith, J.; Birch, R.; Buiting, K. Practice guidelines for the molecular analysis of Prader-Willi and Angelman syndromes. BMC Med. Genet. 2010, 11, 70. [Google Scholar] [CrossRef]
- Crolla, J.A.; Harvey, J.F.; Sitch, F.L.; Dennis, N.R. Supernumerary marker 15 chromosomes: A clinical, molecular and FISH approach to diagnosis and prognosis. Hum. Genet. 1995, 95, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Gillessen-Kaesbach, G.; Robinson, W.; Lohmann, D.; Kaya-Westerloh, S.; Passarge, E.; Horsthemke, B. Genotype-phenotype correlation in a series of 167 deletion and non-deletion patients with Prader-Willi syndrome. Hum. Genet. 1995, 96, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.K.; Kim, S.H.; Park, J.E.; Jang, H.Y.; Go, M.K.; Yang, S.H.; Ryu, S.W.; Bae, S.M.; Cha, D.H.; Shim, S.H. Inconsistency between non-invasive prenatal testing(NIPT) and conventional prenatal diagnosis due to confined placental and fetal mosaicism: Two case reports. Front. Med. 2022, 9, 1063480. [Google Scholar] [CrossRef]
- Kaneko-Ishino, T.; Kohda, T.; Ishino, F. The regulation and biological significance of genomic imprinting in mammals. J. Biochem. 2003, 133, 699–711. [Google Scholar] [CrossRef]
- Glenn, C.C.; Saitoh, S.; Jong, M.T.; Filbrandt, M.M.; Surti, U.; Driscoll, D.J.; Nicholls, R.D. Gene structure, DNA methylation, and imprinted expression of the human SNRPN gene. Am. J. Hum. Genet. 1996, 58, 335–346. [Google Scholar] [PubMed]
- Kubota, T.; Das, S.; Christian, S.L.; Baylin, S.B.; Herman, J.G.; Ledbetter, D.H. Methylation-specific PCR simplifies imprinting analysis. Nat. Genet. 1997, 16, 16–17. [Google Scholar] [CrossRef]
- Schouten, J.P.; McElgunn, C.J.; Waaijer, R.; Zwijnenburg, D.; Diepvens, F.; Pals, G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002, 30, e57. [Google Scholar] [CrossRef]
- Procter, M.; Chou, L.S.; Tang, W.; Jama, M.; Mao, R. Molecular diagnosis of Prader-Willi and Angelman syndromes by methylation-specific melting analysis and methylation-specific multiplex ligation-dependent probe amplification. Clin. Chem. 2006, 52, 1276–1283. [Google Scholar] [CrossRef] [Green Version]
- Bittel, D.C.; Kibiryeva, N.; Butler, M.G. Methylation-specific multiplex ligation-dependent probe amplification analysis of subjects with chromosome 15 abnormalities. Genet. Test 2007, 11, 467–475. [Google Scholar] [CrossRef] [PubMed]
- @US_FDA, FDA Warns of Risks Associated with Non-Invasive Prenatal Screening Tests|FDA. 2022. Available online: https://www.fda.gov/news-events/press-announcements/fda-warns-risks-associated-non-invasive-prenatal-screening-tests (accessed on 19 April 2022).
- Dungan, J.S.; Klugman, S.; Darilek, S.; Malinowsk, J.; Akkari, Y.M.N.; Monaghan, K.G.; Erwin, A.; Best, R.G. Noninvasive prenatal screening (NIPS) for fetal chromosome abnormalities in a general-risk population: An evidence-based clinical guideline of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2022, 13, 100336. [Google Scholar] [CrossRef] [PubMed]
- Benn, P. Uniparental disomy: Origin, frequency, and clinical significance. Prenat. Diagn. 2021, 41, 564–572. [Google Scholar] [CrossRef]
- Rose, N.C.; Benn, P.; Milunsky, A. Current controversies in prenatal diagnosis 1: Should NIPT routinely include microdeletions/microduplications? Prenat. Diagn. 2016, 36, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.I.; Wapner, R.J.; Berkowitz, R.L. Noninvasive prenatal screening or advanced diagnostic testing: Caveat emptor. Am J. Obstet. Gynecol. 2016, 215, 298–305. [Google Scholar] [CrossRef]
- Ferreira, J.C.; Grati, F.R.; Bajaj, K.; Malvestiti, F.; Grimi, M.B.; Trotta, A.; Liuti, R.; Milani, S.; Branca, L.; Hartman, J.; et al. Frequency of fetal karyotype abnormalities in women undergoing invasive testing in the absence of ultrasound and other high-risk indications. Prenat. Diagn. 2016, 36, 1146–1155. [Google Scholar] [CrossRef]
- Pertile, M.D.; Halks-Miller, M.; Flowers, N.; Barbacioru, C.; Kinnings, S.L.; Vavrek, D.; Seltzer, W.K.; Bianchi, D.W. Rare autosomal trisomies, revealed by maternal plasma DNA sequencing, suggest increased risk of feto-placental disease. Sci. Transl. Med. 2017, 9, eaan1240. [Google Scholar] [CrossRef] [PubMed]
- Van Opstal, D.; van Maarle, M.C.; Lichtenbelt, K.; Weiss, M.M.; Schuring-Blom, H.; Bhola, S.L.; Hoffer, M.J.V.; Huijsdens-van Amsterdam, K.; Macville, M.V.; Kooper, A.J.A.; et al. Origin and clinical relevance of chromosomal aberrations other than the common trisomies detected by genome-wide NIPS: Results of the TRIDENT study. Genet. Med. 2018, 20, 480–485. [Google Scholar] [CrossRef]
- Fiorentino, F.; Bono, S.; Pizzuti, F.; Duca, S.; Polverari, A.; Faieta, M.; Baldi, M.; Diano, L.; Spinella, F. The clinical utility of genome-wide non invasive prenatal screening. Prenat. Diagn. 2017, 37, 593–601. [Google Scholar] [CrossRef]
- Scott, F.; Bonifacio, M.; Sandow, R.; Ellis, K.; Smet, M.E.; McLennan, A. Rare autosomal trisomies: Important and not so rare. Prenat. Diagn. 2018, 38, 765–771. [Google Scholar] [CrossRef] [Green Version]
- Benn, P.; Malvestiti, F.; Grimi, B.; Maggi, F.; Simoni, G.; Grati, F.R. Rare autosomal trisomies: Comparison of detection through cell-free DNA analysis and direct chromosome preparation of chorionic villus samples. Ultrasound Obstet. Gynecol. 2019, 54, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Mossfield, T.; Soster, E.; Menezes, M.; Agenbag, G.; Dubois, M.L.; Gekas, J.; Hardy, T.; Jurkowska, M.; Kleinfinger, P.; Loggenberg, K.; et al. Global Expanded NIPT Consortium. Multisite assessment of the impact of cell-free DNA-based screening for rare autosomal aneuploidies on pregnancy management and outcomes. Front. Genet. 2022, 13, 975987. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G.; Sturich, J.; Myers, S.E.; Gold, J.A.; Kimonis, V.; Driscoll, D.J. Is gestation in Prader-Willi syndrome affected by the genetic subtype? J. Assist. Reprod. Genet. 2009, 26, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Shubina, J.; Barkov, I.Y.; Stupko, O.K.; Kuznetsova, M.V.; Goltsov, A.Y.; Kochetkova, T.O.; Trofimov, D.Y.; Sukhikh, G.T. Prenatal diagnosis of Prader-Willi syndrome due to uniparental disomy with NIPS: Case report and literature review. Mol. Genet. Genomic. Med. 2020, 8, e1448. [Google Scholar] [CrossRef]
- Smith, A.; Hung, D. The dilemma of diagnostic testing for Prader-Willi syndrome. Transl. Pediatr. 2017, 6, 46–56. [Google Scholar] [CrossRef]
- Papenhausen, P.; Schwartz, S.; Risheg, H.; Keitges, E.; Gadi, I.; Burnside, R.D.; Jaswaney, V.; Pappas, J.; Pasion, R.; Friedman, K.; et al. UPD detection using homozygosity profiling with a SNP genotyping microarray. Am. J. Med. Genet. A 2011, 155A, 757–768. [Google Scholar] [CrossRef]
- Kearney, H.M.; Kearney, J.B.; Conlin, L.K. Diagnostic implications of excessive homozygosity detected by SNP-based microarrays: Consanguinity, uniparental disomy, and recessive single-gene mutations. Clin. Lab. Med. 2011, 31, 595–613, ix. [Google Scholar] [CrossRef]
- Wang, W.; Law, H.Y.; Chong, S.S. Detection and discrimination between deletional and non-deletional Prader-Willi and Angelman syndromes by methylation-specific PCR and quantitative melting curve analysis. J. Mol. Diagn. 2009, 11, 446–449. [Google Scholar] [CrossRef]
- Acreman, M.L.; Bussolaro, S.; Raymond, Y.C.; Fantasia, I.; Rolnik, D.L.; Da Silva Costa, F. The predictive value of prenatal cell-free DNA testing for rare autosomal trisomies: A systematic review and meta-analysis. Am. J. Obstet. Gynecol. 2022; in press. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
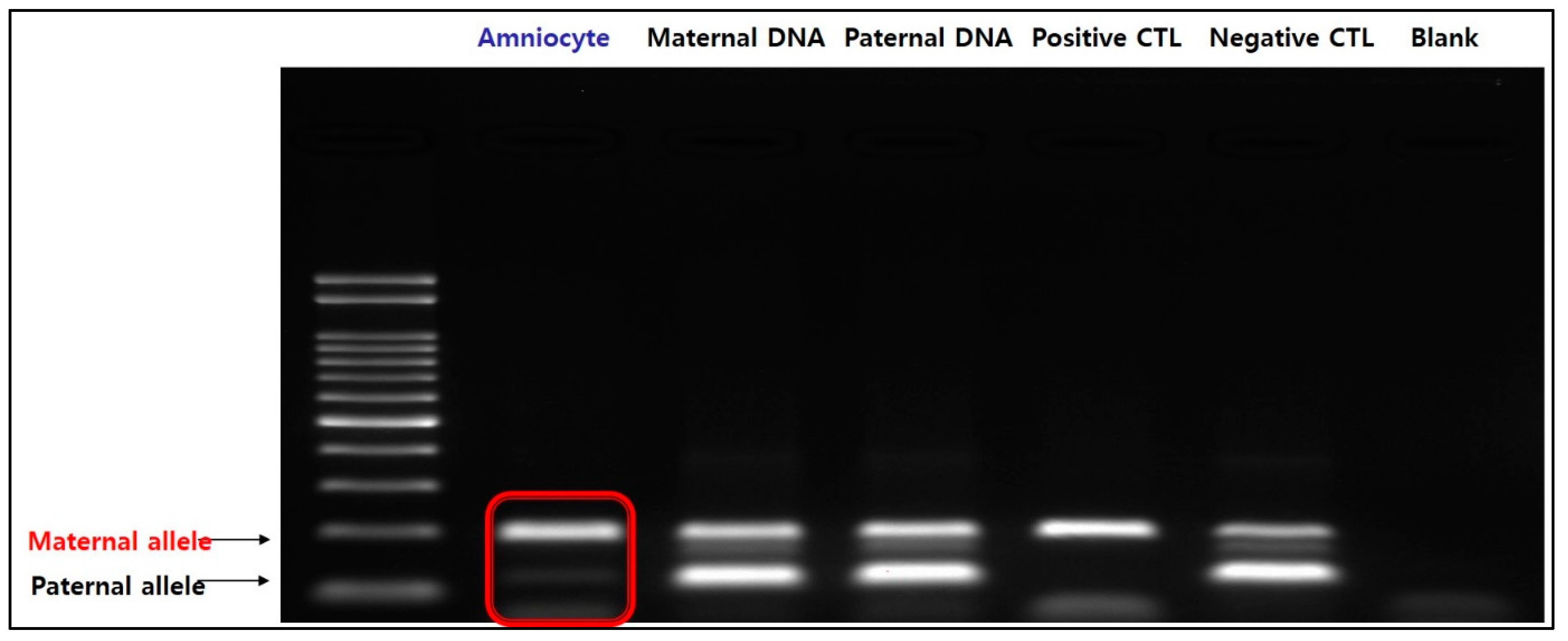{kind=link}
| NIPT Result | Karyotyping Confirmed Using Amniocentesis | Method | UPD Test Result | |
|---|---|---|---|---|
| Case 1 | Trisomy 7 | Normal | MS-MLPA analysis. Probe set: ME032-A1 UPD7-UPD114/MS-PCR | No genomic imbalances |
| Case 2 | Trisomy 7 | Normal | MS-MLPA analysis. Probe set: ME032-A1 UPD7-UPD114/MS-PCR | No genomic imbalances |
| Case 3 | Trisomy 8 | Normal | STR markers: D8S264, D8S1106, D8S1104, D8S591, D8S1127, D8S1179 | UPD not detected |
| Case 4 | Trisomy 8 | Normal | STR markers: D8S264, D8S1106, D8S1104, D8S591, D8S1127 | UPD not detected |
| Case 5 | Trisomy 15 | Normal | MS-MLPA analysis. Probe set: P245 microdeletion/ME028 Prader-Willi/Angelman MS-PCR (Region: exon1 5-CpG island of the SNRPN gene) | No genomic imbalances |
| Case 6 | Trisomy 15 | Normal | MS-MLPA analysis. Probe set: P245 microdeletion/ME028 Prader-Willi/Angelman MS-PCR (Region: exon1 5-CpG island of the SNRPN gene) | Prader–Willi syndrome (maternal UPD) |
| Genetic Mechanism | Incidence | Recurrence Risk |
|---|---|---|
| De novo deletion of 15q11-q13 in the paternal chromosome | 75–80% | <1% |
| Maternal UPD | 20–25% | <1% |
| Imprinting defects (IDs) without deletion in the IC | ≈1% | <1% |
| Imprinting center (IC) deletion | ≈10–15% of patients with an ID | Up to 50% (if father also has an IC deletion) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, D.K.; Park, J.E.; Kang, K.M.; Shim, S.H.; Shim, S.H.; Han, Y.J.; Cho, H.Y.; Cha, D.H. Prenatal Diagnosis of Uniparental Disomy in Cases of Rare Autosomal Trisomies Detected Using Noninvasive Prenatal Test: A Case of Prader–Willi Syndrome. Diagnostics 2023, 13, 580. https://doi.org/10.3390/diagnostics13040580
Hong DK, Park JE, Kang KM, Shim SH, Shim SH, Han YJ, Cho HY, Cha DH. Prenatal Diagnosis of Uniparental Disomy in Cases of Rare Autosomal Trisomies Detected Using Noninvasive Prenatal Test: A Case of Prader–Willi Syndrome. Diagnostics. 2023; 13(4):580. https://doi.org/10.3390/diagnostics13040580
Chicago/Turabian StyleHong, Da Kyung, Ji Eun Park, Kyung Min Kang, Sung Han Shim, So Hyun Shim, You Jung Han, Hee Young Cho, and Dong Hyun Cha. 2023. "Prenatal Diagnosis of Uniparental Disomy in Cases of Rare Autosomal Trisomies Detected Using Noninvasive Prenatal Test: A Case of Prader–Willi Syndrome" Diagnostics 13, no. 4: 580. https://doi.org/10.3390/diagnostics13040580