
A Nanoplex PCR Assay for the Simultaneous Detection of Vancomycin- and Linezolid-Resistant Genes in Enterococcus

Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Reference Strains and Clinical Isolates
2.1.1. Reference Strains
2.1.2. Clinical Isolates
2.1.3. Synthetic dsDNA
2.2. Primers
2.3. Preparation of DNA Templates from Clinical Isolates
2.4. Development of Nanoplex PCR
3. Results
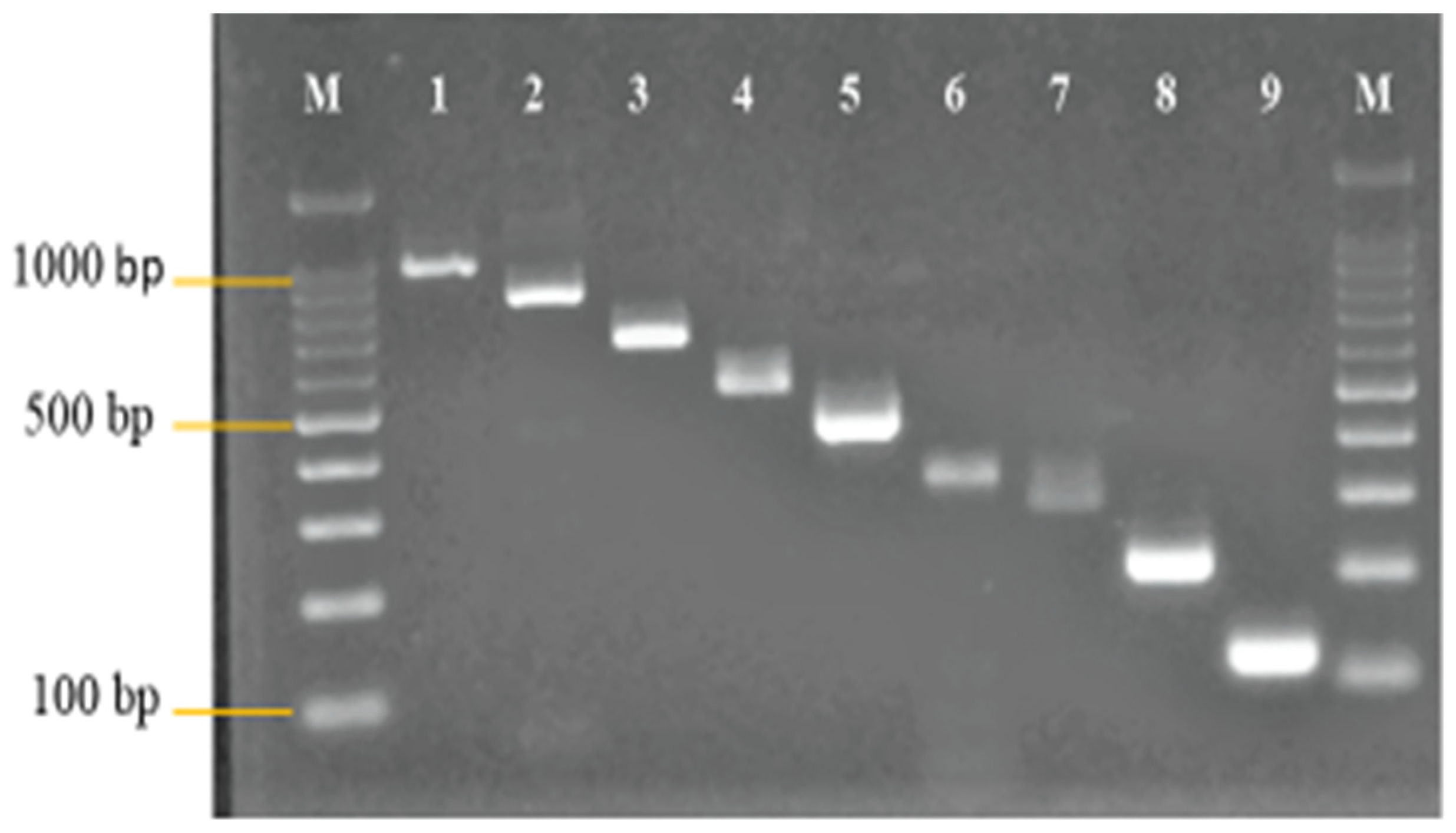
3.1. Primer Design and Analysis
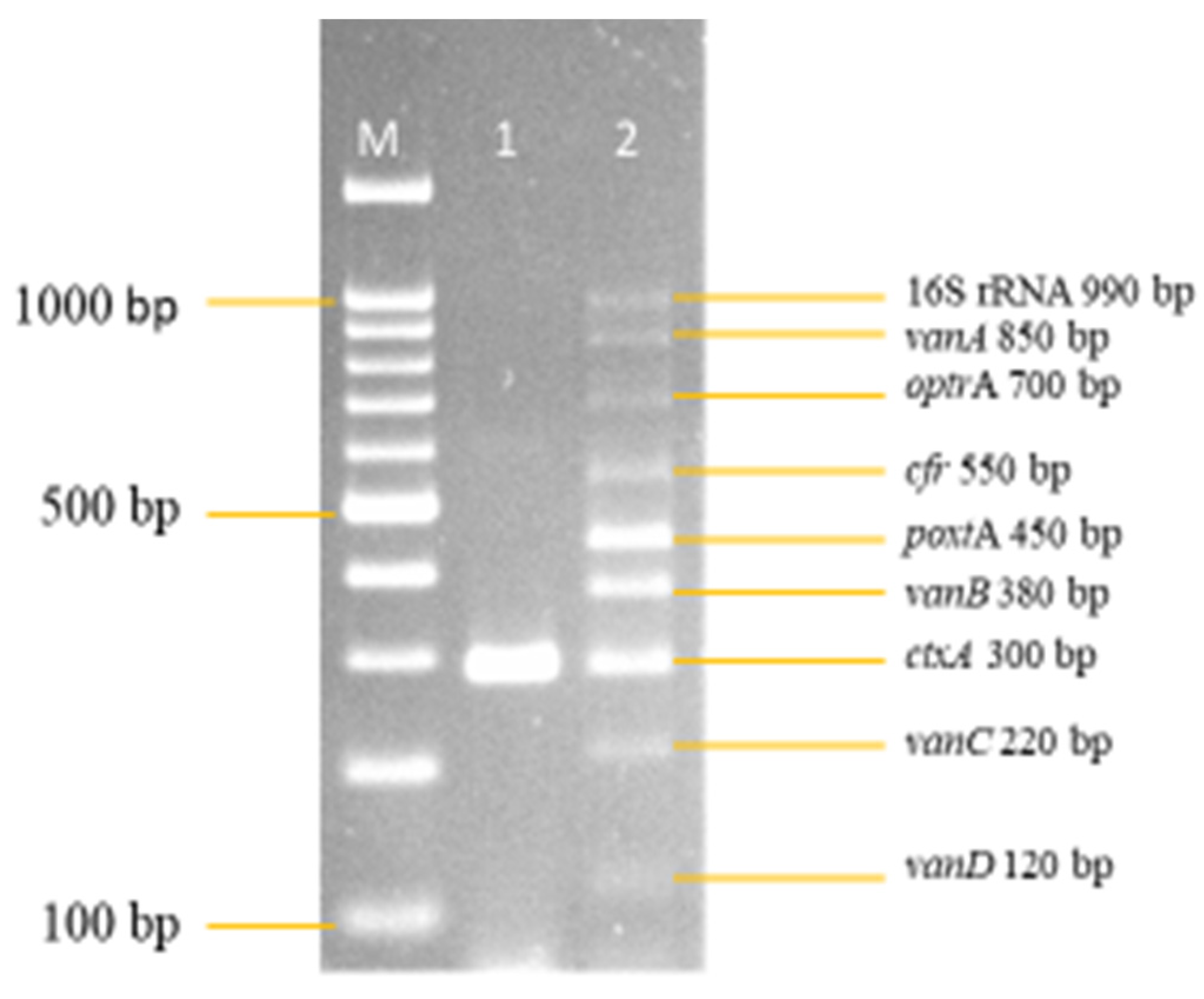
3.2. Development and Optimization of the Nanoplex PCR
Final Optimised Parameters of the Nanoplex PCR Assay
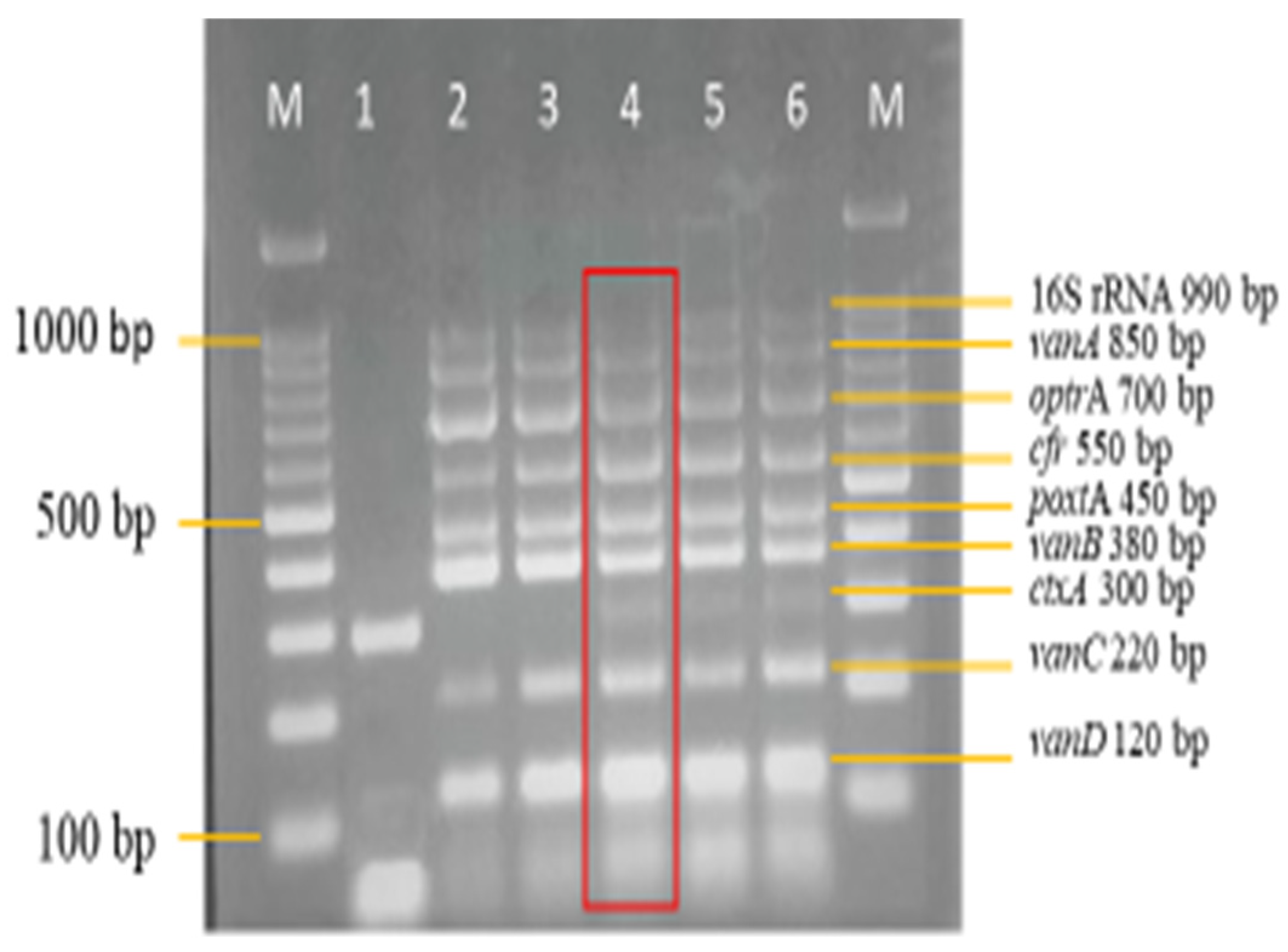
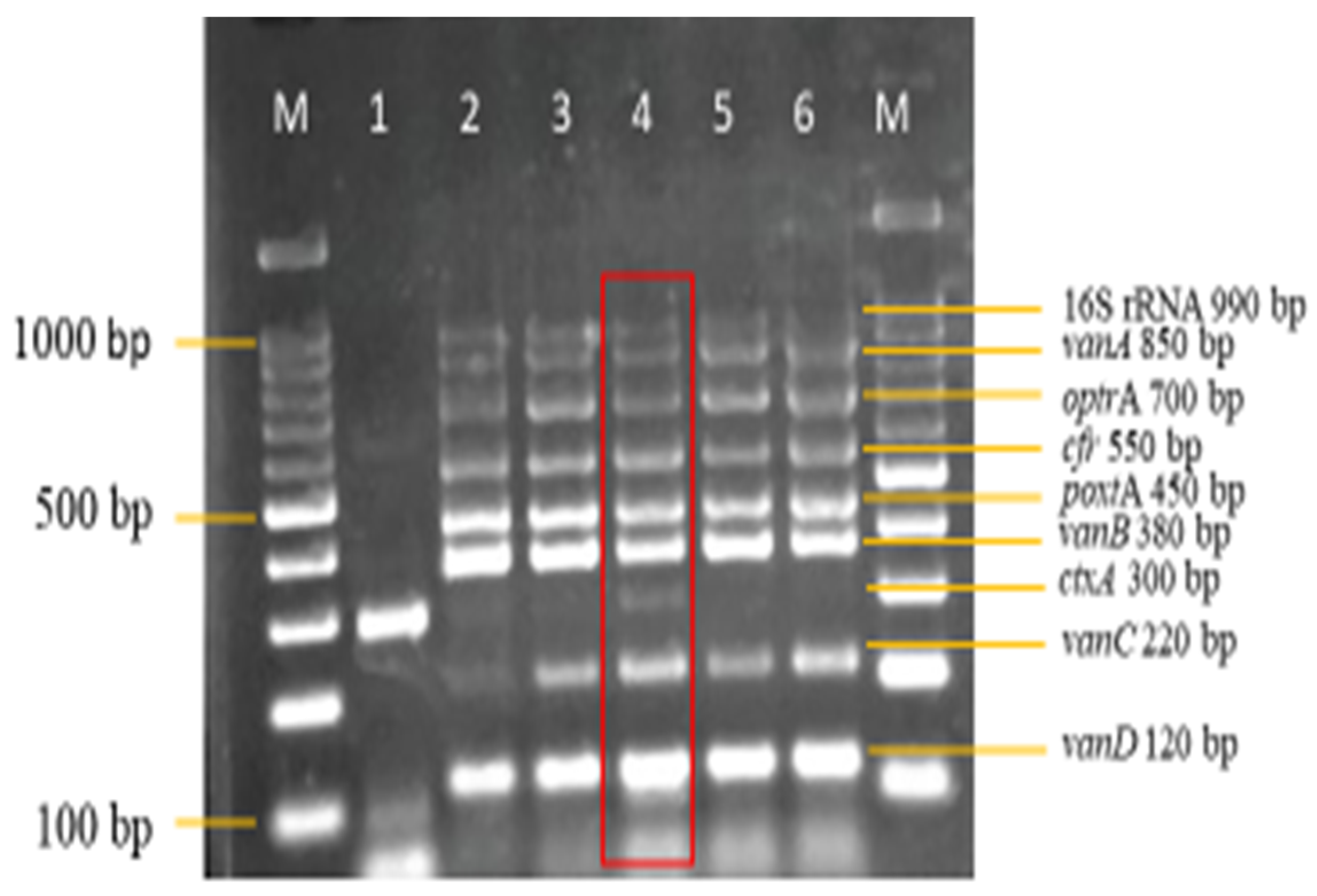
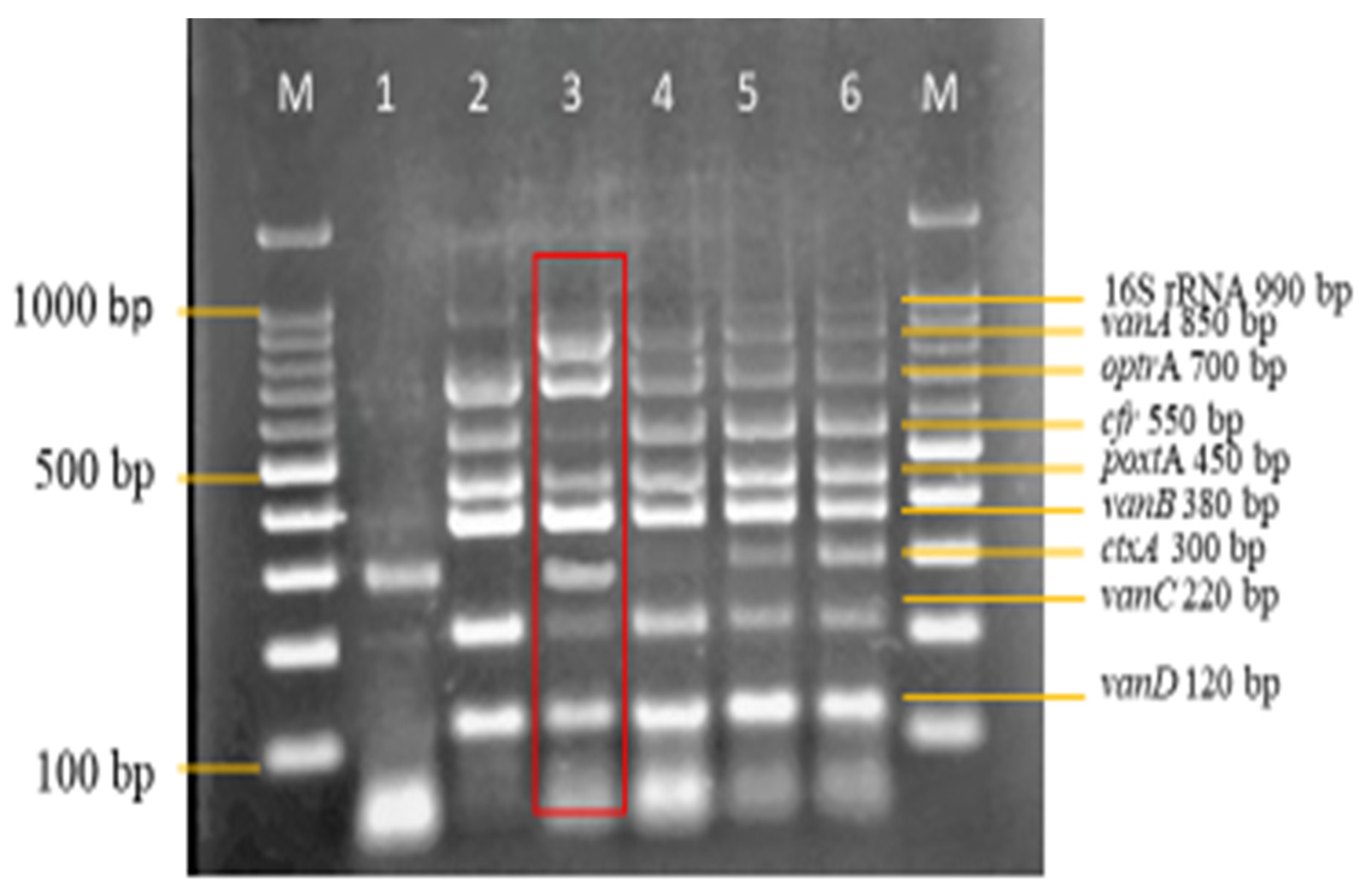
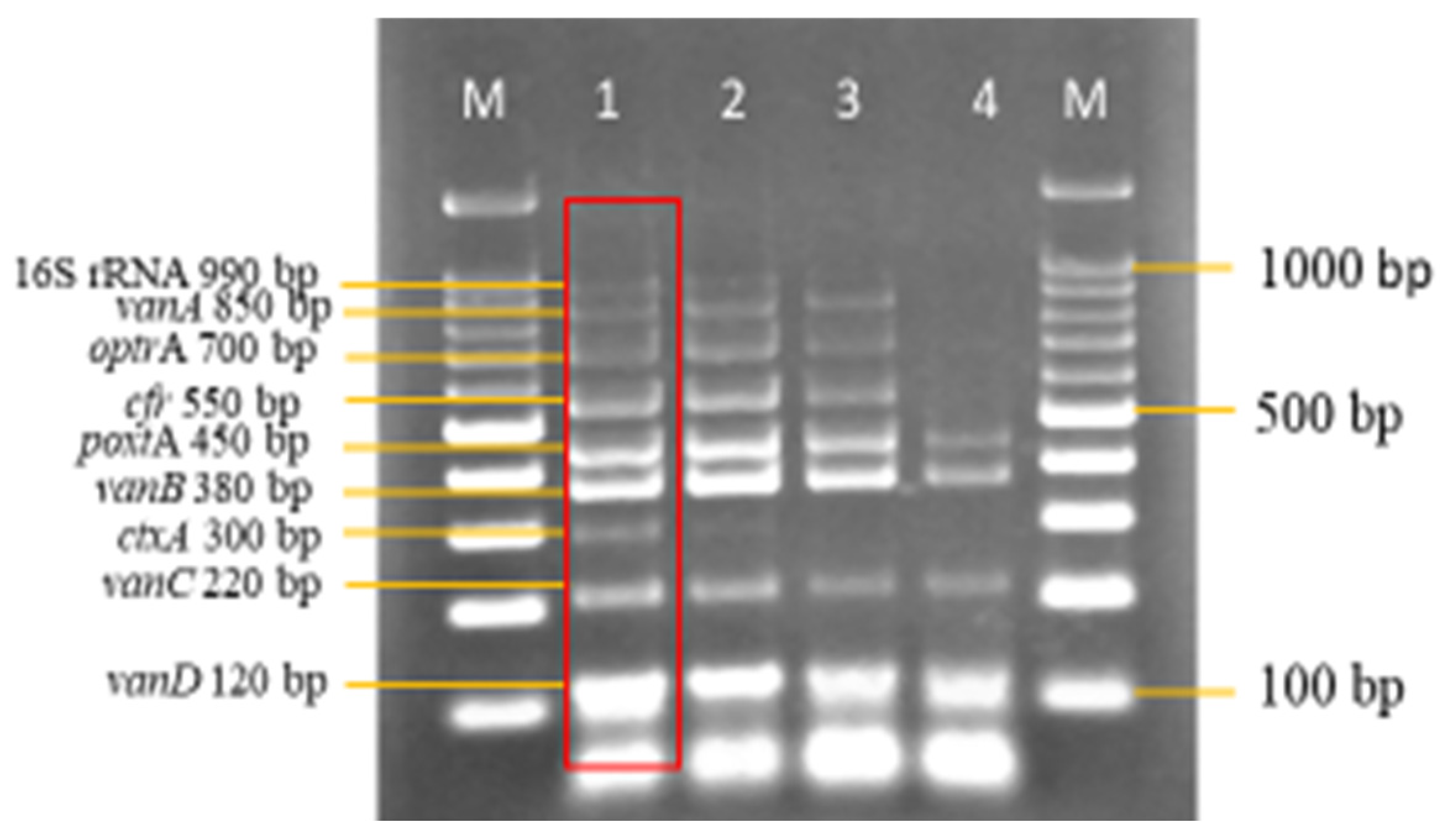
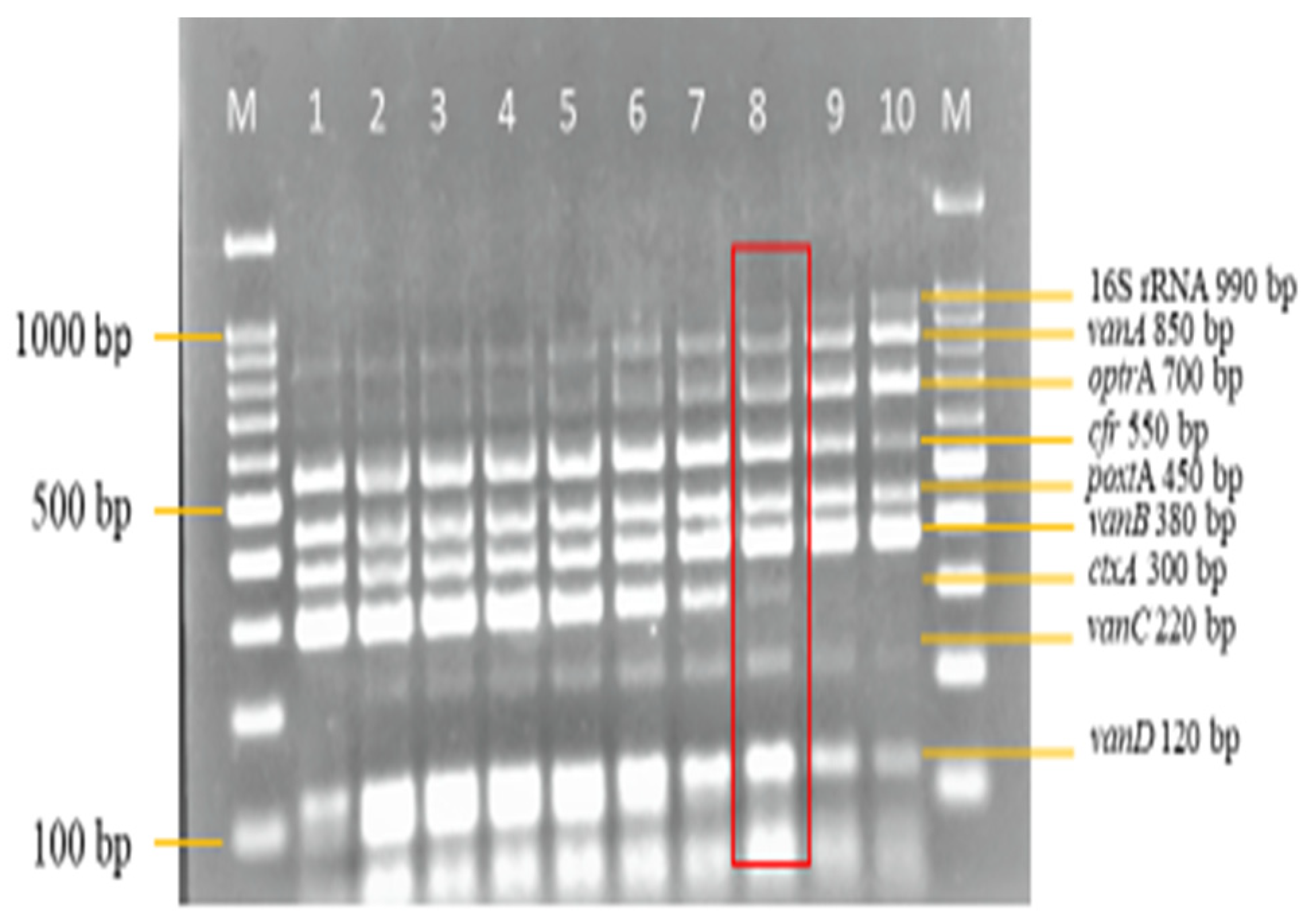
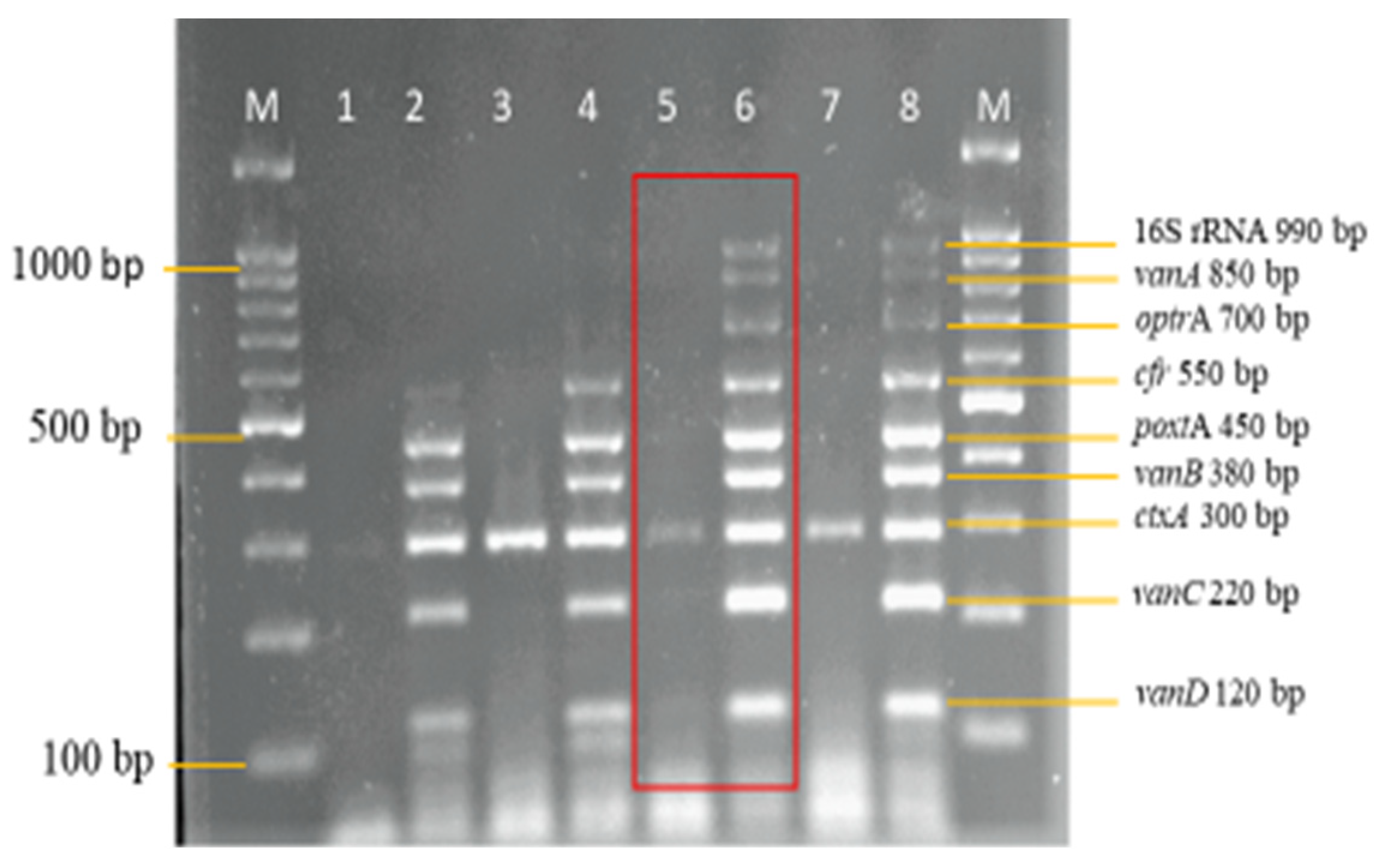
3.3. Sensitivity and Specificity Evaluation of the Nanoplex PCR Assay
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wada, Y.; Harun, A.B.; Yean, C.Y.; Zaidah, A.R. Vancomycin-Resistant Enterococcus: Issues in Human Health, Animal Health, Resistant Mechanisms and the Malaysian Paradox. Adv. Anim. Vet. Sci. 2019, 7, 1021–1034. [Google Scholar] [CrossRef]
- Antimicrobial Resistance. Available online: https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance (accessed on 19 December 2022).
- Browne, A.J.; Chipeta, M.G.; Haines-Woodhouse, G.; Kumaran, E.P.A.; Hamadani, B.H.K.; Zaraa, S.; Henry, N.J.; Deshpande, A.; Reiner, R.C.; Day, N.P.J.; et al. Global Antibiotic Consumption and Usage in Humans, 2000–2018: A Spatial Modelling Study. Lancet Planet. Health 2021, 5, e893–e904. [Google Scholar] [CrossRef] [PubMed]
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- Yean, C.Y.; Yin, L.S.; Lalitha, P.; Ravichandran, M. A Nanoplex PCR Assay for the Rapid Detection of Vancomycin and Bifunctional Aminoglycoside Resistance Genes in Enterococcus Species. BMC Microbiol. 2007, 7, 112. [Google Scholar] [CrossRef] [PubMed]
- Bender, J.K.; Fleige, C.; Klare, I.; Werner, G. Development of a Multiplex-PCR to Simultaneously Detect Acquired Linezolid Resistance Genes Cfr, OptrA and PoxtA in Enterococci of Clinical Origin. J. Microbiol. Methods 2019, 160, 101–103. [Google Scholar] [CrossRef] [PubMed]
- Zangenberg, G.; Saiki, R.K.; Reynolds, R. Multiplex PCR: Optimization Guidelines. In PCR Applications; Academic Press: Cambridge, MA, USA, 1999; pp. 73–94. [Google Scholar] [CrossRef]
- Song, H.; Bae, Y.; Jeon, E.; Kwon, Y.; Joh, S. Multiplex PCR Analysis of Virulence Genes and Their Influence on Antibiotic Resistance in Enterococcus Spp. Isolated from Broiler Chicken. J. Vet. Sci. 2019, 20, e26. [Google Scholar] [CrossRef] [PubMed]
- Mohamad Nasir, N.S.; Chan, Y.Y.; Harun, A.; Husin, A.; Kamaruzzaman, N.F.; Wada, Y.; Abdul-Rahman, Z. Malaysian Journal of Microbiology Linezolid-resistant Enterococcus casseliflavus and Enterococcus gallinarum isolated from poultry farms in Kelantan, Malaysia. Malays. J. Microbiol. 2021, 17, 361–368. [Google Scholar] [CrossRef]
- Trevethan, R. Sensitivity, Specificity, and Predictive Values: Foundations, Pliabilities, and Pitfalls in Research and Practice. Front. Public Health 2017, 5, 307. [Google Scholar] [CrossRef] [PubMed]
- Nik Zuraina, N.M.N.; Goni, M.D.; Amalina, K.N.; Hasan, H.; Mohamad, S.; Suraiya, S. Thermostable Heptaplex PCR Assay for the Detection of Six Respiratory Bacterial Pathogens. Diagnostics 2021, 11, 753. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
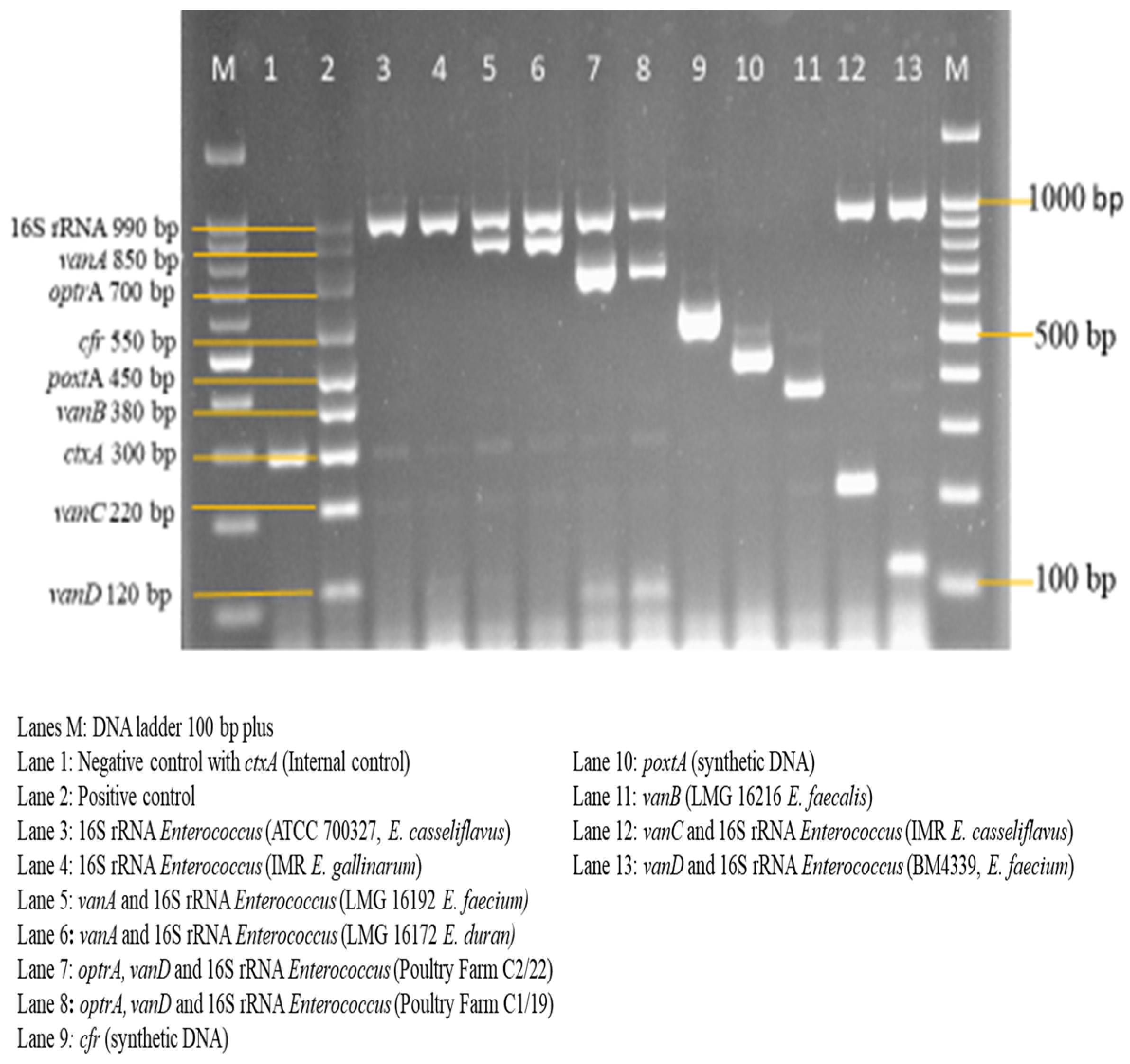{kind=link}
{kind=link}
| Species | Reference | Target Gene |
|---|---|---|
| Enterococcus casseliflavus a | ATCC 700327 | 16S rRNA Enterococcus |
| Enterococcus gallinarum b | IMR | 16S rRNA Enterococcus |
| Enterococcus raffinosus c | LMG 12172 | 16S rRNA Enterococcus |
| Enterococcus mundti c | LMG 12308 | 16S rRNA Enterococcus |
| Enterococcus faecium c | LMG 16192 | 16S rRNA Enterococcus, vanA |
| Enterococcus durans c | LMG 16172 | 16S rRNA Enterococcus, vanA, |
| Enterococcus faecalis c | LMG 16216 | vanB |
| Enterococcus casseliflavus b | IMR | 16S rRNA Enterococcus, vanC |
| Enterococcus faecium d | BM4339 | 16S rRNA Enterococcus, vanD |
| Enterococcus casseliflavus e | Poultry Farm C2/22 | 16S rRNA Enterococcus, optrA, poxtA |
| Enterococcus casseliflavus e | Poultry Farm C1/19 | 16S rRNA Enterococcus, optrA, poxtA |
| S/No | Bacteria Strains |
|---|---|
| Gram-positive | |
| 1 | Methicillin-resistant Staphylococcus aureus (MRSA) |
| 2 | Streptococcus Group A |
| 3 | Streptococcus Group B |
| 4 | Streptococcus Group G |
| 5 | Streptococcus Group F |
| 6 | Bacillus species |
| 7 | Listeria species |
| 8 | Corynebacterium species |
| 9 | Staphylococcus aureus |
| 10 | Gardnerella species |
| Gram-negative | |
| 1 | Proteus mirabilis |
| 2 | Klebsiella species |
| 3 | Shigella dysentariae |
| 4 | Plesiomonas shigelloides |
| 5 | Vibrio cholerae |
| 6 | Klebsiella pneumoniae |
| 7 | Escherichia coli (Enterohemorrhagic EHEC) |
| 8 | Escherichia coli (Enteropathogenic EPEC) |
| 9 | Vibrio parahaemolyticus |
| 10 | Shigella sonnei |
| 11 | Shigella boydii |
| 12 | Citrobacter freundii |
| 13 | Yersinia enterocolitica |
| 14 | Acinetobacter baumannii |
| 15 | Acinetobacter species |
| 16 | Pseudomonas aeruginosa |
| Synthetic dsDNA | Size (bp) |
|---|---|
| 16S rRNA Enterococcus | 993 |
| vanA | 1032 |
| optrA | 880 |
| cfr | 725 |
| poxtA | 600 |
| vanB | 1029 |
| ctxA * | 615 |
| vanC | 699 |
| vanD | 1032 |
| Target Gene | Primer Sequence (5′-3′) | Product Length (bp) |
|---|---|---|
| 16S rRNA Enterococcus | F-5′-TTC CAC CGG AGC TTG CTC C-3′ R-5′-TTT GCC CCC GAA GGG GAA G-3′ | 990 |
| vanA | F-5′-TTT GGG GGT TGC TCA GAG G-3′ R-5′-CAC ACG GGC TAG ACC TCT A-3′ | 850 |
| optrA | F-5′-TGG AAA AAC AAC CTT GCT AAA AGC-3′ R-5′-CAA GCG TGT AAT CCT TTC AAT TTC-3′ | 700 |
| cfr | F-5′-CAA AGA ATT AGT CGA TTT GAG GA-3′ R-5′-GTT CCT CAC TAT AAG GTG AGT-3′ | 550 |
| poxtA | F-5′-TGC TTT TTC TCC AGG GGA CA-3′ R-5′-GTG GAG AGC TGC AAA AGA GA-3′ | 450 |
| vanB | F-5′-AAA ACG GCG TAT GGA AGC TAT G-3′ R-5′-CGG CTT CAC AAA GAC AGG GTA G-3′ | 380 |
| ctxA * | F-5′-AAC TCA GAC GGG ATT TGT TAG GC-3′ R-5′-TCT CTG TAG CCC CTA TTA CGA TGT-3′ | 300 [5] |
| vanC | F-5′-CAG CAG CCA TTG GCG TAC A-3′ R-5′-TGT AGG AGC ACT GCG GAA C-3′ | 220 |
| vanD | F-5′-AAG CTC CGT GAT CTG CAT GG-3′ R-5′-AAA TCC TCC GTT TCC AGG C-3′ | 120 |
| Components | Initial Concentration | Per Reaction (μL) | Final Concentration |
|---|---|---|---|
| PCR-Grade dH2O | - | 2.37 | - |
| 10× Reaction Buffer | 10× | 2.0 | 1× |
| MgCl2 | 25 mM | 2.0 | 2.5 mM |
| dNTPs | 10 mM | 0.32 | 0.16 mM |
| Primers (Sense and Anti-sense) | |||
| 16S rRNA Enterococcus | 20 μM | 1.0 | 1 μM |
| vanA | 20 μM | 1.0 | 1 μM |
| optrA | 20 μM | 1.0 | 1 μM |
| cfr | 20 μM | 1.0 | 1 μM |
| poxtA | 20 μM | 0.1 | 0.1 μM |
| vanB | 20 μM | 0.08 | 0.08 μM |
| ctxA (IAC) | 20 μM | 0.07 | 0.07 μM |
| vanC | 20 μM | 0.8 | 0.8 μM |
| vanD | 20 μM | 0.1 | 0.1 μM |
| Taq DNA Polymerase | 5 units | 0.15 | 0.75 units |
| ctxA Template (IAC) | 10 ng | 1.0 | 10 pg |
| Target DNA Cocktail mix (Synthetic dsDNA) | 10 ng/μL of each target | 2.0 | 1 ng/μL of each target |
| Final Volume (μL) | 20 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wada, Y.; Harun, A.; Yean, C.Y.; Zaidah, A.R. A Nanoplex PCR Assay for the Simultaneous Detection of Vancomycin- and Linezolid-Resistant Genes in Enterococcus. Diagnostics 2023, 13, 722. https://doi.org/10.3390/diagnostics13040722
Wada Y, Harun A, Yean CY, Zaidah AR. A Nanoplex PCR Assay for the Simultaneous Detection of Vancomycin- and Linezolid-Resistant Genes in Enterococcus. Diagnostics. 2023; 13(4):722. https://doi.org/10.3390/diagnostics13040722
Chicago/Turabian StyleWada, Yusuf, Azian Harun, Chan Yean Yean, and Abdul Rahman Zaidah. 2023. "A Nanoplex PCR Assay for the Simultaneous Detection of Vancomycin- and Linezolid-Resistant Genes in Enterococcus" Diagnostics 13, no. 4: 722. https://doi.org/10.3390/diagnostics13040722