
Recent Updates on Corticosteroid-Induced Neuropsychiatric Disorders and Theranostic Advancements through Gene Editing Tools
 , ,
, ,  and
and
Abstract
:1. Introduction
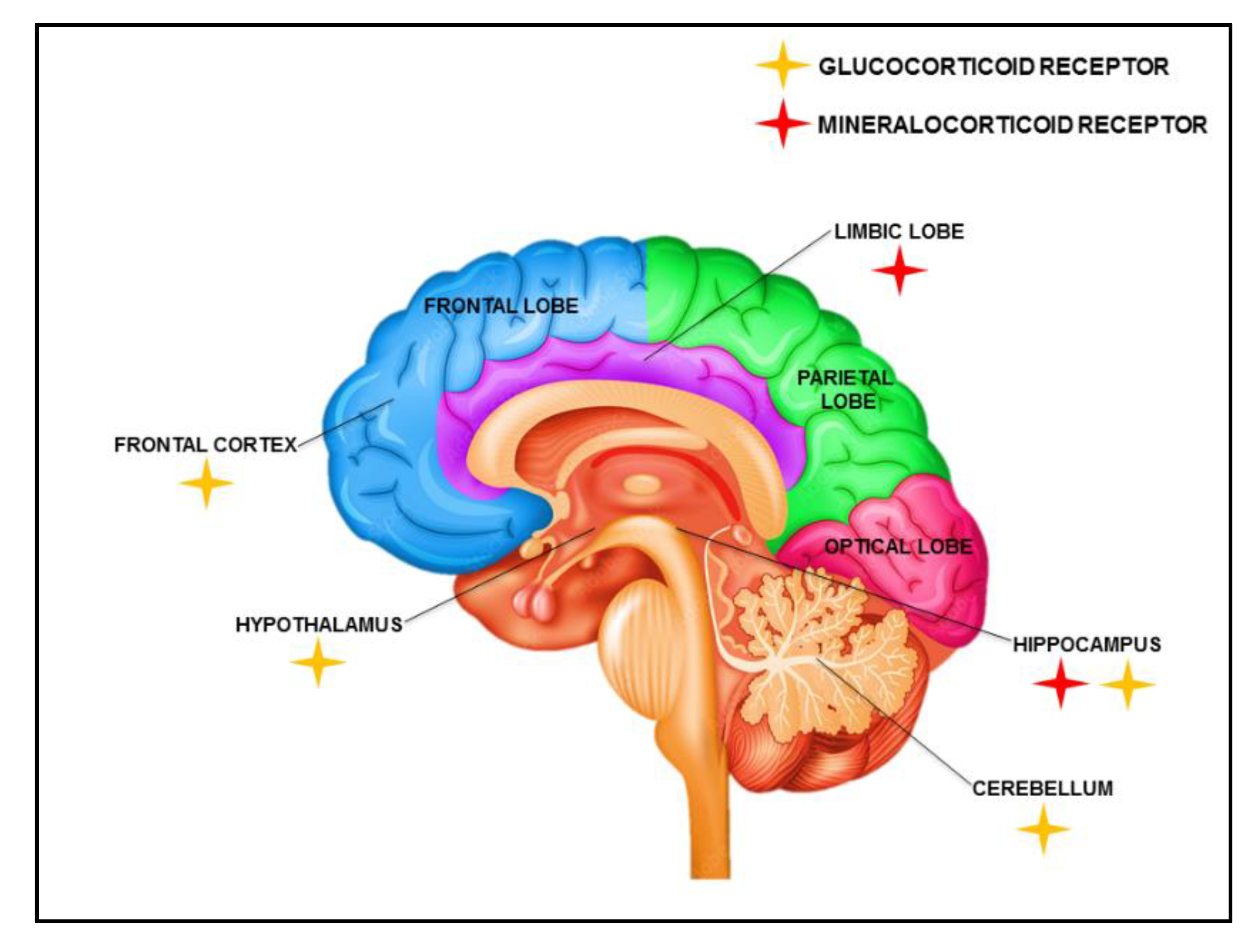
2. Biogenesis and Expression of Corticosteroids in the Brain
2.1. Gene Regulation by Glucocorticoids (GR)
2.2. Gene Regulation by Mineralo Corticosteroids (MR)
3. Non-Genomic Effects of Corticosteroids (CCSs) Related to CNS
4. Therapeutic Action of Corticosteroids
5. Types of CCS-Induced NPDs
6. Associated Concerns after CCS Administration
7. Relevance of Gene Therapies in Eliminating CCS-Induced NPD
8. Role of Non-Coding RNAs (ncRNAs)/mRNAs in NPDs
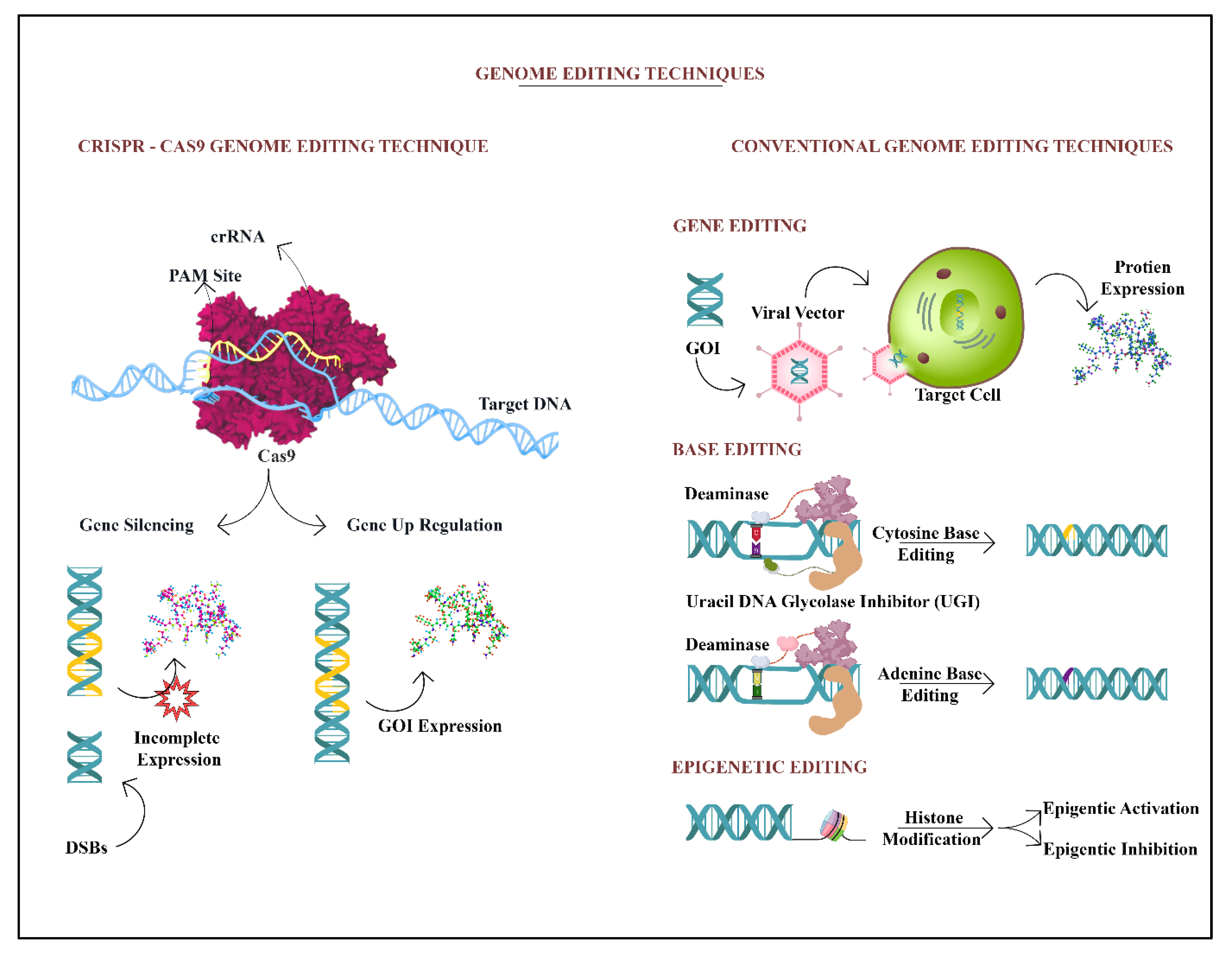
9. Gene Editing of Target DNA Locus by CRISPR/Cas 9
10. Challenges Associated with the Application of CRISPR in Neuropsychiatric Disorders
11. Other Methods of Genome Sequencing to Treat NPDs
11.1. GWAS (Genome-Wide Association Studies)
11.2. Whole Exome Sequencing (WES)
12. Discussion and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Youssef, J.; Novosad, S.A.; Winthrop, K.L. Infection Risk and Safety of Corticosteroid Use. Rheum. Dis. Clin. 2016, 42, 157–176, ix–x. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coutinho, A.E.; Chapman, K.E. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol. Cell. Endocrinol. 2011, 335, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolkowitz, O.M.; Burke, H.; Epel, E.S.; Reus, V.I. Glucocorticoids: Mood, memory, and mechanisms. Ann. N. Y. Acad. Sci. 2009, 1179, 19–40. [Google Scholar] [CrossRef] [PubMed]
- Ciriaco, M.; Ventrice, P.; Russo, G.; Scicchitano, M.; Mazzitello, G.; Scicchitano, F.; Russo, E. Corticosteroid-related central nervous system side effects. J. Pharmacol. Pharmacother. 2013, 4 (Suppl. 1), S94–S98. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Ann, L.; McCarron, R. Steroid-induced psychiatric symptoms: What you need to know. Curr. Psychiatry 2021, 20, 33–38. [Google Scholar] [CrossRef]
- Dubovsky, A.N.; Arvikar, S.; Stern, T.A.; Axelrod, L. The neuropsychiatric complications of glucocorticoid use: Steroid psychosis revisited. Psychosomatics 2012, 53, 103–115. [Google Scholar] [CrossRef]
- Roxanas, M.G.; Hunt, G.E. Rapid reversal of corticosteroid-induced mania with sodium valproate: A case series of 20 patients. Psychosomatics 2012, 53, 575–581. [Google Scholar] [CrossRef]
- Lewis, D.A.; Smith, R.E. Steroid-induced psychiatric syndromes. A report of 14 cases and a review of the literature. J. Affect. Disord. 1983, 5, 319–332. [Google Scholar] [CrossRef]
- Warren, K.N.; Katakam, J.; Espiridion, E.D. Acute-onset Mania in a Patient with Non-small Cell Lung Cancer. Cureus 2019, 11, e5436. [Google Scholar] [CrossRef]
- Morishita, R.; Aoki, M.; Kaneda, Y. Oligonucleotide-based gene therapy for cardiovascular disease: Are oligonucleotide therapeutics novel cardiovascular drugs? Curr. Drug Targets 2000, 1, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudry, E.; Vandenberghe, L.H. Therapeutic AAV Gene Transfer to the Nervous System: A Clinical Reality. Neuron 2019, 101, 839–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bromfield, E.B.; Cavazos, J.E.; Sirven, J.I. Basic Mechanisms Underlying Seizures and Epilepsy. In An Introduction to Epilepsy; American Epilepsy Society: Nashville, TN, USA, 2006. [Google Scholar]
- Desh, D.S.; Ravi, V.; Piyush, P.; Ashish, S.; Vikram, K.; Era, U.; Dharmendra, K.Y. Potential Therapeutic Relevance of CRISPR/Cas9 Guided Epigenetic Regulations for Neuropsychiatric Disorders. Curr. Top. Med. Chem. 2021, 21, 878–894. [Google Scholar]
- Trottier, G.; Srivastava, L.; Walker, C.D. Etiology of infantile autism: A review of recent advances in genetic and neurobiological research. J. Psychiatry Neurosci. Jpn. 1999, 24, 103–115. [Google Scholar] [PubMed]
- Kuehner, J.N.; Bruggeman, E.C.; Wen, Z.; Yao, B. Epigenetic Regulations in Neuropsychiatric Disorders. Front. Genet. 2019, 10, 268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhangle, S.D.; Kramer, N.; Rosenstein, E.D. Corticosteroid-induced neuropsychiatric disorders: Review and contrast with neuropsychiatric lupus. Rheumatol. Int. 2013, 33, 1923–1932. [Google Scholar] [CrossRef]
- Araujo, E.; Barbosa, M.; Silva, J.; Serodio, J.; Costelha, J. Vasculitis and Steroid Psychosis: A Case Report and Review of Literature. J. Med. Cases 2019, 10, 264–266. [Google Scholar] [CrossRef]
- Kenna, H.A.; Poon, A.W.; de los Angeles, C.P.; Koran, L.M. Psychiatric complications of treatment with corticosteroids: Review with case report. Psychiatry Clin. Neurosci. 2011, 65, 549–560. [Google Scholar] [CrossRef]
- Bladh, L.G.; Lidén, J.; Dahlman-Wright, K.; Reimers, M.; Nilsson, S.; Okret, S. Identification of endogenous glucocorticoid repressed genes differentially regulated by a glucocorticoid receptor mutant able to separate between nuclear factor-kappaB and activator protein-1 repression. Mol. Pharmacol. 2005, 67, 815–826. [Google Scholar] [CrossRef]
- Duma, D.; Jewell, C.M.; Cidlowski, J.A. Multiple glucocorticoid receptor isoforms and mechanisms of post-translational modification. J. Steroid Biochem. Mol. Biol. 2006, 102, 11–21. [Google Scholar] [CrossRef]
- Koning, A.; Buurstede, J.C.; van Weert, L.; Meijer, O.C. Glucocorticoid and Mineralocorticoid Receptors in the Brain: A Transcriptional Perspective. J. Endocr. Soc. 2019, 3, 1917–1930. [Google Scholar] [CrossRef] [PubMed]
- Zhe, D.; Fang, H.; Yuxiu, S. Expressions of hippocampal mineralocorticoid receptor (MR) and glucocorticoid receptor (GR) in the single-prolonged stress-rats. Acta Histochem. Et Cytochem. 2008, 41, 89–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitre-Aguilar, I.B.; Cabrera-Quintero, A.J.; Zentella-Dehesa, A. Genomic and non-genomic effects of glucocorticoids: Implications for breast cancer. Int. J. Clin. Exp. Pathol. 2015, 8, 1–10. [Google Scholar] [PubMed]
- Chantong, B.; Kratschmar, D.V.; Nashev, L.G.; Balazs, Z.; Odermatt, A. Mineralocorticoid and glucocorticoid receptors differentially regulate NF-kappaB activity and pro-inflammatory cytokine production in murine BV-2 microglial cells. J. Neuroinflammat. 2012, 9, 260. [Google Scholar] [CrossRef] [Green Version]
- Hudson, W.H.; Youn, C.; Ortlund, E.A. The structural basis of direct glucocorticoid-mediated transrepression. Nat. Struct. Mol. Biol. 2013, 20, 53–58. [Google Scholar] [CrossRef] [Green Version]
- Johnson, T.A.; Paakinaho, V.; Kim, S.; Hager, G.L.; Presman, D.M. Genome-wide binding potential and regulatory activity of the glucocorticoid receptor’s monomeric and dimeric forms. Nat. Commun. 2021, 12, 1987. [Google Scholar] [CrossRef]
- Oakley, R.H.; Cidlowski, J.A. The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 2013, 132, 1033–1044. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Du, H.; Li, Y.; Guan, W.; Tang, F.; Ga, Q.; Ge, R.-L. NR3C1 gene polymorphisms are associated with high-altitude pulmonary edema in Han Chinese. J. Physiol. Anthropol. 2019, 38, 4. [Google Scholar] [CrossRef] [Green Version]
- Petta, I.; Dejager, L.; Ballegeer, M.; Lievens, S.; Tavernier, J.; De Bosscher, K.; Libert, C. The Interactome of the Glucocorticoid Receptor and Its Influence on the Actions of Glucocorticoids in Combatting Inflammatory and Infectious Diseases. Microbiol. Mol. Biol. Rev. MMBR 2016, 80, 495–522. [Google Scholar] [CrossRef] [Green Version]
- Schäcke, H.; Berger, M.; Rehwinkel, H.; Asadullah, K. Selective glucocorticoid receptor agonists (SEGRAs): Novel ligands with an improved therapeutic index. Mol. Cell. Endocrinol. 2007, 275, 109–117. [Google Scholar] [CrossRef] [Green Version]
- Thibaut, F. Corticosteroid-induced psychiatric disorders: Genetic studies are needed. Eur. Arch. Psychiatry Clin. Neurosci. 2019, 269, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kampmann, M. CRISPR-based functional genomics for neurological disease. Nat. Rev. Neurol. 2020, 16, 465–480. [Google Scholar] [CrossRef]
- Nicholas, J.; Bray, M.C.O.D. The genetics of neuropsychiatric disorders. Brain Neurosci. Adv. 2018, 2, 2398212818799271. [Google Scholar]
- Barešić, A.; Nash, A.J.; Dahoun, T.; Howes, O.; Lenhard, B. Understanding the genetics of neuropsychiatric disorders: The potential role of genomic regulatory blocks. Mol. Psychiatry 2020, 25, 6–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holst, J.P.; Soldin, O.P.; Guo, T.; Soldin, S.J. Steroid hormones: Relevance and measurement in the clinical laboratory. Clin. Lab. Med. 2004, 24, 105–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joëls, M. Corticosteroids and the brain. J. Endocrinol. 2018, 238, R121–R130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higo, S.; Hojo, Y.; Ishii, H.; Komatsuzaki, Y.; Ooishi, Y.; Murakami, G.; Mukai, H.; Yamazaki, T.; Nakahara, D.; Barron, A.; et al. Endogenous Synthesis of Corticosteroids in the Hippocampus. PLoS ONE 2011, 6, e21631. [Google Scholar] [CrossRef]
- Cho, S.; Kagan, B.L.; Blackford, J.A., Jr.; Szapary, D.; Simons, S.S., Jr. Glucocorticoid receptor ligand binding domain is sufficient for the modulation of glucocorticoid induction properties by homologous receptors, coactivator transcription intermediary factor 2, and Ubc9. Mol. Endocrinol. 2005, 19, 290–311. [Google Scholar] [CrossRef] [Green Version]
- Baumgartner-Parzer, S.; Witsch-Baumgartner, M.; Hoeppner, W. EMQN best practice guidelines for molecular genetic testing and reporting of 21-hydroxylase deficiency. Eur. J. Hum. Genet. 2020, 28, 1341–1367. [Google Scholar] [CrossRef]
- Chung, B.C.; Picado-Leonard, J.; Haniu, M.; Bienkowski, M.; Hall, P.F.; Shively, J.E.; Miller, W.L. Cytochrome P450c17 (steroid 17 alpha-hydroxylase/17,20 lyase): Cloning of human adrenal and testis cDNAs indicates the same gene is expressed in both tissues. Proc. Natl. Acad. Sci. USA 1987, 84, 407–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, J.L.; Myers, R.P.; Strickler, R.C. Human placental 3 beta-hydroxy-5-ene-steroid dehydrogenase and steroid 5----4-ene-isomerase: Purification from mitochondria and kinetic profiles, biophysical characterization of the purified mitochondrial and microsomal enzymes. J. Steroid Biochem. 1989, 33, 209–217. [Google Scholar] [CrossRef]
- Lorence, M.C.; Murry, B.A.; Trant, J.M.; Mason, J.I. Human 3 beta-hydroxysteroid dehydrogenase/delta 5→4isomerase from placenta: Expression in nonsteroidogenic cells of a protein that catalyzes the dehydrogenation/isomerization of C21 and C19 steroids. Endocrinology 1990, 126, 2493–2498. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.L.; Auchus, R.J. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr. Rev. 2011, 32, 81–151. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Sanchez, E.; Gomez-Sanchez, C.E. The multifaceted mineralocorticoid receptor. Compr. Physiol. 2014, 4, 965–994. [Google Scholar] [PubMed] [Green Version]
- Sapolsky, R.M.; Romero, L.M.; Munck, A.U. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr. Rev. 2000, 21, 55–89. [Google Scholar] [PubMed] [Green Version]
- Miner, J.N.; Hong, M.H.; Negro-Vilar, A. New and improved glucocorticoid receptor ligands. Expert Opin. Investig. Drugs 2005, 14, 1527–1545. [Google Scholar] [CrossRef]
- Do Rego, J.L.; Seong, J.Y.; Burel, D.; Leprince, J.; Luu-The, V.; Tsutsui, K.; Tonon, M.-C.; Pelletier, G.; Vaudry, H. Neurosteroid biosynthesis: Enzymatic pathways and neuroendocrine regulation by neurotransmitters and neuropeptides. Front. Neuroendocrinol. 2009, 30, 259–301. [Google Scholar]
- Coirini, H.; Magariños, A.M.; De Nicola, A.F.; Rainbow, T.C.; McEwen, B.S. Further studies of brain aldosterone binding sites employing new mineralocorticoid and glucocorticoid receptor markers in vitro. Brain Res. 1985, 361, 212–216. [Google Scholar] [CrossRef]
- Coirini, H.; Marusic, E.T.; De Nicola, A.F.; Rainbow, T.C.; McEwen, B.S. Identification of Mineralocorticoid Binding Sites in Rat Brain by Competition Studies and Density Gradient Centrifugation. Neuroendocrinology 1983, 37, 354–360. [Google Scholar] [CrossRef]
- Gomez-sanchez, C.E.; Zhou, M.Y.; Cozza, E.N.; Morita, H.; Eddleman, F.C.; Gomez-sanchez, E.P. Corticosteroid synthesis in the central nervous system. Endocr. Res. 1996, 22, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Davies, E.; MacKenzie, S.M. Extra-adrenal production of corticosteroids. Clin. Exp. Pharmacol. Physiol. 2003, 30, 437–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Thompson, E.B. Transactivation Functions of the N-Terminal Domains of Nuclear Hormone Receptors: Protein Folding and Coactivator Interactions. Mol. Endocrinol. 2003, 17, 1–10. [Google Scholar] [CrossRef]
- Lavery, D.N.; McEwan, I.J. Structure and function of steroid receptor AF1 transactivation domains: Induction of active conformations. Biochem. J. 2005, 391 Pt 3, 449–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, T.E.; Pauli, F.; Sprouse, R.O.; Neff, N.F.; Newberry, K.M.; Garabedian, M.J.; Myers, R.M. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res. 2009, 19, 2163–2171. [Google Scholar] [CrossRef] [Green Version]
- Xavier, A.M.; Anunciato, A.K.O.; Rosenstock, T.R.; Glezer, I. Gene Expression Control by Glucocorticoid Receptors during Innate Immune Responses. Front. Endocrinol. 2016, 7, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Auboeuf, D.; Hönig, A.; Berget, S.M.; O’Malley, B.W. Coordinate Regulation of Transcription and Splicing by Steroid Receptor Coregulators. Science 2002, 298, 416–419. [Google Scholar] [CrossRef] [PubMed]
- Karst, H.; Berger, S.; Turiault, M.; Tronche, F.; Schütz, G.; Joëls, M. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc. Natl. Acad. Sci. USA 2005, 102, 19204–19207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual-Le Tallec, L.; Lombès, M. The mineralocorticoid receptor: A journey exploring its diversity and specificity of action. Mol. Endocrinol. 2005, 19, 2211–2221. [Google Scholar] [CrossRef] [Green Version]
- Yokota, K.; Shibata, H.; Kurihara, I.; Kobayashi, S.; Suda, N.; Murai-Takeda, A.; Saito, I.; Kitagawa, H.; Kato, S.; Saruta, T.; et al. Coactivation of the N-terminal transactivation of mineralocorticoid receptor by Ubc9. J. Biol. Chem. 2007, 282, 1998–2010. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, M.K.; Mirshahi, M. General overview of mineralocorticoid hormone action. Pharmacol. Ther. 1999, 84, 273–326. [Google Scholar] [CrossRef] [PubMed]
- Penning, T.M. Hydroxysteroid dehydrogenases and pre-receptor regulation of steroid hormone action. Hum. Reprod. Update 2003, 9, 193–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Sanchez, E.P. The mammalian mineralocorticoid receptor: Tying down a promiscuous receptor. Exp. Physiol. 2010, 95, 13–18. [Google Scholar] [CrossRef]
- Groeneweg, F.L.; Karst, H.; de Kloet, E.R.; Joëls, M. Rapid non-genomic effects of corticosteroids and their role in the central stress response. J. Endocrinol. 2011, 209, 153–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.T.; Almon, R.R.; DuBois, D.C.; Jusko, W.J.; Androulakis, I.P. Comparative analysis of acute and chronic corticosteroid pharmacogenomic effects in rat liver: Transcriptional dynamics and regulatory structures. BMC Bioinform. 2010, 11, 515. [Google Scholar] [CrossRef] [Green Version]
- Berger, S.; Wolfer, D.P.; Selbach, O.; Alter, H.; Erdmann, G.; Reichardt, H.M.; Chepkova, A.N.; Welzl, H.; Haas, H.L.; Lipp, H.-P.; et al. Loss of the limbic mineralocorticoid receptor impairs behavioral plasticity. Proc. Natl. Acad. Sci. USA 2006, 103, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Bogdan, R.; Perlis, R.H.; Fagerness, J.; Pizzagalli, D.A. The impact of mineralocorticoid receptor ISO/VAL genotype (rs5522) and stress on reward learning. Genes Brain Behav. 2010, 9, 658–667. [Google Scholar] [CrossRef] [Green Version]
- Ramamoorthy, S.; Cidlowski, J.A. Corticosteroids: Mechanisms of Action in Health and Disease. Rheum. Dis. Clin. 2016, 42, 15–31, vii. [Google Scholar] [CrossRef] [Green Version]
- Newton, R. Molecular mechanisms of glucocorticoid action: What is important? Thorax 2000, 55, 603–613. [Google Scholar] [CrossRef] [Green Version]
- Atsak, P.; Hauer, D.; Campolongo, P.; Schelling, G.; Fornari, R.V.; Roozendaal, B. Endocannabinoid signaling within the basolateral amygdala integrates multiple stress hormone effects on memory consolidation. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2015, 40, 1485–1494. [Google Scholar] [CrossRef]
- Atsak, P.; Hauer, D.; Campolongo, P.; Schelling, G.; McGaugh, J.L.; Roozendaal, B. Glucocorticoids interact with the hippocampal endocannabinoid system in impairing retrieval of contextual fear memory. Proc. Natl. Acad. Sci. USA 2012, 109, 3504–3509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campolongo, P.; Roozendaal, B.; Trezza, V.; Hauer, D.; Schelling, G.; McGaugh, J.L.; Cuomo, V. Endocannabinoids in the rat basolateral amygdala enhance memory consolidation and enable glucocorticoid modulation of memory. Proc. Natl. Acad. Sci. USA 2009, 106, 4888–4893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarante, F.F.; Vila-Verde, C.; Detoni, V.L.; Ferreira-Junior, N.C.; Guimarães, F.S.; Campos, A.C. Cannabinoid Modulation of the Stressed Hippocampus. Front. Mol. Neurosci. 2017, 10, 411. [Google Scholar] [CrossRef] [PubMed]
- Becker, D.E. Basic and Clinical Pharmacology of Glucocorticosteroids. Anesth. Prog. 2013, 60, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Bhatia, V. Corticosteroid physiology and principles of therapy. Indian J. Pediatr. 2008, 75, 1039–1044. [Google Scholar] [CrossRef]
- Claman, H.N. Corticosteroids as immunomodulators. Ann. N. Y. Acad. Sci. 1993, 685, 288–292. [Google Scholar] [CrossRef]
- De Bosscher, K.; Haegeman, G. Minireview: Latest perspectives on antiinflammatory actions of glucocorticoids. Mol. Endocrinol. 2009, 23, 281–291. [Google Scholar] [CrossRef]
- Ashwell, J.D.; Lu, F.W.; Vacchio, M.S. Glucocorticoids in T cell development and function*. Annu. Rev. Immunol. 2000, 18, 309–345. [Google Scholar] [CrossRef]
- Granger DN, S.E. Inflammation and the Microcirculation: Leukocyte–Endothelial Cell Adhesion; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2010. [Google Scholar]
- Williams, D.M. Clinical Pharmacology of Corticosteroids. Respir. Care 2018, 63, 655–670. [Google Scholar] [CrossRef] [Green Version]
- Derendorf, H.; Nave, R.; Drollmann, A.; Cerasoli, F.; Wurst, W. Relevance of pharmacokinetics and pharmacodynamics of inhaled corticosteroids to asthma. Eur. Respir. J. 2006, 28, 1042–1050. [Google Scholar] [CrossRef]
- Taylor, D.R.; Hancox, R.J. Interactions between corticosteroids and β agonists. Thorax 2000, 55, 595–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, A.O.; Lefkowitz, R.J. Regulation of beta-adrenergic receptors by steroid hormones. Annu. Rev. Physiol. 1984, 46, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Beta-adrenergic receptors and their regulation. Am. J. Respir. Crit. Care Med. 1995, 152, 838–860. [Google Scholar] [CrossRef] [PubMed]
- Scarcériaux, V.; Pélaprat, D.; Forgez, P.; Lhiaubet, A.M.; Rostène, W. Effects of dexamethasone and forskolin on neurotensin production in rat hypothalamic cultures. Endocrinology 1995, 136, 2554–2560. [Google Scholar] [CrossRef] [PubMed]
- Anna Dorai-Raj, L.S. The role of corticosteroids in rheumatology. Aust. Prescr. 1998, 21, 11–14. [Google Scholar] [CrossRef]
- van der Goes, M.C.; Jacobs, J.W.; Bijlsma, J.W. The value of glucocorticoid co-therapy in different rheumatic diseases--positive and adverse effects. Arthritis Res. Ther. 2014, 16 (Suppl. 2), S2. [Google Scholar] [CrossRef] [Green Version]
- Kirschke, E.; Goswami, D.; Southworth, D.; Griffin, P.R.; Agard, D.A. Glucocorticoid receptor function regulated by coordinated action of the Hsp90 and Hsp70 chaperone cycles. Cell 2014, 157, 1685–1697. [Google Scholar] [CrossRef] [Green Version]
- Auphan, N.; DiDonato, J.A.; Rosette, C.; Helmberg, A.; Karin, M. Immunosuppression by glucocorticoids: Inhibition of NF-kappa B activity through induction of I kappa B synthesis. Science 1995, 270, 286–290. [Google Scholar] [CrossRef]
- Savas, M.; Vinkers, C.H.; Rosmalen, J.G.M.; Hartman, C.A.; Wester, V.L.; van den Akker, E.L.T.; Iyer, A.M.; McEwen, B.S.; van Rossum, E.F.C. Systemic and Local Corticosteroid Use Is Associated with Reduced Executive Cognition, and Mood and Anxiety Disorders. Neuroendocrinology 2020, 110, 282–291. [Google Scholar] [CrossRef]
- Zorumski, C.F.; Paul, S.M.; Covey, D.F.; Mennerick, S. Neurosteroids as novel antidepressants and anxiolytics: GABA-A receptors and beyond. Neurobiol. Stress 2019, 11, 100196. [Google Scholar] [CrossRef]
- Jasani, R.; Deacon, J.W.; Sertich, A. Corticosteroid-Induced Mania After Previous Tolerance of Higher Doses. Cureus 2021, 13, e17719. [Google Scholar] [CrossRef] [PubMed]
- Yasir, M.; Goyal, A.; Bansal, P.; Sonthalia, S. Corticosteroid Adverse Effects; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Boston Collaborative Drug Surveillance Program. Acute adverse reactions to prednisone in relation to dosage. Clin. Pharmacol. Ther. 1972, 13, 694–698. [Google Scholar]
- Corvol, H.; Nathan, N.; Charlier, C.; Chadelat, K.; Le Rouzic, P.; Tabary, O.; Fauroux, B.; Henrion-Caude, A.; Feingold, J.; Boelle, P.-Y.; et al. Glucocorticoid receptor gene polymorphisms associated with progression of lung disease in young patients with cystic fibrosis. Respir. Res. 2007, 8, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pommepuy, N.; Januel, D. Catatonia: Resurgence of a concept. A review of the international literature. Encephale 2002, 28 6 Pt 1, 481–492. [Google Scholar]
- Janssens, H.J.; Janssen, M.; van de Lisdonk, E.H.; van Riel, P.L.; van Weel, C. Use of oral prednisolone or naproxen for the treatment of gout arthritis: A double-blind, randomised equivalence trial. Lancet 2008, 371, 1854–1860. [Google Scholar] [CrossRef]
- Keller, L.-A.; Merkel, O.; Popp, A. Intranasal drug delivery: Opportunities and toxicologic challenges during drug development. Drug Deliv. Transl. Res. 2022, 12, 735–757. [Google Scholar] [CrossRef] [PubMed]
- Szefler, S.; Weiss, S.; Tonascia, J.; Adkinson, N.F.; Bender, B.; Cherniack, R.; Donithan, M.; Kelly, H.W.; Reisman, J.; Shapiro, G.G.; et al. Long-term effects of budesonide or nedocromil in children with asthma. N. Engl. J. Med. 2000, 343, 1054–1063. [Google Scholar]
- Mrakotsky, C.; Forbes, P.W.; Bernstein, J.H.; Grand, R.J.; Bousvaros, A.; Szigethy, E.; Waber, D.P. Acute cognitive and behavioral effects of systemic corticosteroids in children treated for inflammatory bowel disease. J. Int. Neuropsychol. Soc. JINS 2013, 19, 96–109. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Pu, J.; Shen, X.D.; He, B. Prednisone induced two-way myocardial development in a patient with systemic lupus erythematosus. BMJ Case Rep. 2014, 2014, bcr2013203046. [Google Scholar] [CrossRef]
- Pines, A.; Ehrenfeld, M.; Fisman, E.Z.; Kaplinsky, N.; Samra, Y.; Ronnen, M.; Kellermann, J.J. Mitral valve prolapse in psoriatic arthritis. Arch. Intern. Med. 1986, 146, 1371–1373. [Google Scholar] [CrossRef]
- Hall, R.C.; Popkin, M.K.; Stickney, S.K.; Gardner, E.R. Presentation of the steroid psychoses. J. Nerv. Ment. Dis. 1979, 167, 229–236. [Google Scholar] [CrossRef]
- Stephens, M.A.; Wand, G. Stress and the HPA axis: Role of glucocorticoids in alcohol dependence. Alcohol Res. Curr. Rev. 2012, 34, 468–483. [Google Scholar]
- Soria, V.; González-Rodríguez, A.; Huerta-Ramos, E.; Usall, J.; Cobo, J.; Bioque, M.; Barbero, J.D.; García-Rizo, C.; Tost, M.; Monreal, J.A.; et al. Targeting hypothalamic-pituitary-adrenal axis hormones and sex steroids for improving cognition in major mood disorders and schizophrenia: A systematic review and narrative synthesis. Psychoneuroendocrinology 2018, 93, 8–19. [Google Scholar] [CrossRef]
- Sergent, J.S.; Lockshin, M.D.; Klempner, M.S.; Lipsky, B.A. Central nervous system disease in systemic lupus erythematosus. Therapy and prognosis. Am. J. Med. 1975, 58, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Stiefel, F.C.; Breitbart, W.S.; Holland, J.C. Corticosteroids in cancer: Neuropsychiatric complications. Cancer Investig. 1989, 7, 479–491. [Google Scholar] [CrossRef]
- Mani, S.; Jindal, D.; Singh, M. Gene Therapy, A potential Therapeutic Tool for Neurological and Neuropsychiatric Disorders: Applications, Challenges and Future Prospective. Curr. Gene Ther. 2022. [Google Scholar] [CrossRef] [PubMed]
- Goverdhana, S.; Puntel, M.; Xiong, W.; Zirger, J.M.; Barcia, C.; Curtin, J.F.; Soffer, E.B.; Mondkar, S.; King, G.D.; Hu, J.; et al. Regulatable gene expression systems for gene therapy applications: Progress and future challenges. Mol. Ther. J. Am. Soc. Gene Ther. 2005, 12, 189–211. [Google Scholar] [CrossRef]
- Shahcheraghi, S.H.; Ayatollahi, J.; Lotfi, M.; Aljabali, A.A.A.; Al-Zoubi, M.S.; Panda, P.K.; Mishra, V.; Satija, S.; Charbe, N.B.; Serrano-Aroca, Á.; et al. Gene Therapy for Neuropsychiatric Disorders: Potential Targets and Tools. CNS Neurol. Disord. Drug Targets 2022. [Google Scholar] [CrossRef]
- Li, Q.; Qin, Z.; Wang, Q.; Xu, T.; Yang, Y.; He, Z. Applications of Genome Editing Technology in Animal Disease Modeling and Gene Therapy. Comput. Struct. Biotechnol. J. 2019, 17, 689–698. [Google Scholar] [CrossRef]
- Gu, W.; Gurguis, C.I.; Zhou, J.J.; Zhu, Y.; Ko, E.A.; Ko, J.H.; Wang, T.; Zhou, T. Functional and Structural Consequence of Rare Exonic Single Nucleotide Polymorphisms: One Story, Two Tales. Genome Biol. Evol. 2015, 7, 2929–2940. [Google Scholar] [CrossRef] [Green Version]
- Kher, G.; Trehan, S.; Misra, A. Antisense Oligonucleotides and RNA Interference. In Challenges in Delivery of Therapeutic Genomics and Proteomics; Elsevier: Amsterdam, The Netherlands, 2011. [Google Scholar]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Mimmack, M.L.; Ryan, M.; Baba, H.; Navarro-Ruiz, J.; Iritani, S.; Faull, R.L.; McKenna, P.J.; Jones, P.B.; Arai, H.; Starkey, M.; et al. Gene expression analysis in schizophrenia: Reproducible up-regulation of several members of the apolipoprotein L family located in a high-susceptibility locus for schizophrenia on chromosome 22. Proc. Natl. Acad. Sci. USA 2002, 99, 4680–4685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cacabelos, R.; Martínez-Bouza, R. Genomics and pharmacogenomics of schizophrenia. CNS Neurosci. Ther. 2011, 17, 541–565. [Google Scholar] [CrossRef] [PubMed]
- Sekar, A.; Bialas, A.R.; de Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; Van Doren, V.; et al. Schizophrenia risk from complex variation of complement component 4. Nature 2016, 530, 177–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Austin, J. Schizophrenia: An update and review. J. Genet. Couns. 2005, 14, 329–340. [Google Scholar] [CrossRef]
- Lai, M.C.; Lombardo, M.V.; Baron-Cohen, S. Autism. Lancet 2014, 383, 896–910. [Google Scholar] [CrossRef]
- Levy, S.E.; Mandell, D.S.; Schultz, R.T. Autism. Lancet 2009, 374, 1627–1638. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, M.; Nomura, J.; Ji, X.; Tamada, K.; Arai, T.; Takahashi, E.; Bućan, M.; Takumi, T. Functional significance of rare neuroligin 1 variants found in autism. PLoS Genet. 2017, 13, e1006940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhle, R.; Trentacoste, S.V.; Rapin, I. The genetics of autism. Pediatrics 2004, 113, e472–e486. [Google Scholar] [CrossRef] [Green Version]
- Morrow, E.M.; Yoo, S.Y.; Flavell, S.W.; Kim, T.K.; Lin, Y.; Hill, R.S.; Mukaddes, N.M.; Balkhy, S.; Gascon, G.; Hashmi, A.; et al. Identifying autism loci and genes by tracing recent shared ancestry. Science 2008, 321, 218–223. [Google Scholar] [CrossRef] [Green Version]
- Leehey, M.A.; Berry-Kravis, E.; Goetz, C.G.; Zhang, L.; Hall, D.A.; Li, L.; Rice, C.D.; Lara, R.; Cogswell, J.; Reynolds, A.; et al. FMR1 CGG repeat length predicts motor dysfunction in premutation carriers. Neurology 2008, 70 16 Pt 2, 1397–1402. [Google Scholar] [CrossRef]
- Niu, Y.Q.; Yang, J.C.; Hall, D.A.; Leehey, M.A.; Tassone, F.; Olichney, J.M.; Hagerman, R.J.; Zhang, L. Parkinsonism in fragile X-associated tremor/ataxia syndrome (FXTAS): Revisited. Parkinsonism Relat. Disord. 2014, 20, 456–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dibbens, L.M.; Feng, H.J.; Richards, M.C.; Harkin, L.A.; Hodgson, B.L.; Scott, D.; Jenkins, M.; Petrou, S.; Sutherland, G.R.; Scheffer, I.E.; et al. GABRD encoding a protein for extra- or peri-synaptic GABAA receptors is a susceptibility locus for generalized epilepsies. Hum. Mol. Genet. 2004, 13, 1315–1319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marini, C.; Scheffer, I.E.; Crossland, K.M.; Grinton, B.E.; Phillips, F.L.; McMahon, J.M.; Turner, S.J.; Dean, J.T.; Kivity, S.; Mazarib, A.; et al. Genetic architecture of idiopathic generalized epilepsy: Clinical genetic analysis of 55 multiplex families. Epilepsia 2004, 45, 467–478. [Google Scholar] [CrossRef] [PubMed]
- Heron, S.E.; Phillips, H.A.; Mulley, J.C.; Mazarib, A.; Neufeld, M.Y.; Berkovic, S.F.; Scheffer, I.E. Genetic variation of CACNA1H in idiopathic generalized epilepsy. Ann. Neurol. 2004, 55, 595–596. [Google Scholar] [CrossRef]
- Berg, A.T.; Berkovic, S.F.; Brodie, M.J.; Buchhalter, J.; Cross, J.H.; van Emde Boas, W.; Engel, J.; French, J.; Glauser, T.A.; Mathern, G.W.; et al. Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 2010, 51, 676–685. [Google Scholar] [CrossRef]
- Gitler, A.D.; Chesi, A.; Geddie, M.L.; Strathearn, K.E.; Hamamichi, S.; Hill, K.J.; Caldwell, K.A.; Caldwell, G.A.; Cooper, A.A.; Rochet, J.C.; et al. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat. Genet. 2009, 41, 308–315. [Google Scholar] [CrossRef]
- Klein, C.; Lohmann-Hedrich, K.; Rogaeva, E.; Schlossmacher, M.G.; Lang, A.E. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol. 2007, 6, 652–662. [Google Scholar] [CrossRef]
- Hardy, J.; Lewis, P.; Revesz, T.; Lees, A.; Paisan-Ruiz, C. The genetics of Parkinson’s syndromes: A critical review. Curr. Opin. Genet. Dev. 2009, 19, 254–265. [Google Scholar] [CrossRef]
- Lesage, S.; Drouet, V.; Majounie, E.; Deramecourt, V.; Jacoupy, M.; Nicolas, A.; Cormier-Dequaire, F.; Hassoun, S.M.; Pujol, C.; Ciura, S.; et al. Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy. Am. J. Hum. Genet. 2016, 98, 500–513. [Google Scholar] [CrossRef] [Green Version]
- Le, W.D.; Xu, P.; Jankovic, J.; Jiang, H.; Appel, S.H.; Smith, R.G.; Vassilatis, D.K. Mutations in NR4A2 associated with familial Parkinson disease. Nat. Genet. 2003, 33, 85–89. [Google Scholar] [CrossRef]
- Taft, R.J.; Simons, C.; Nahkuri, S.; Oey, H.; Korbie, D.J.; Mercer, T.R.; Holst, J.; Ritchie, W.; Wong, J.J.; Rasko, J.E.; et al. Nuclear-localized tiny RNAs are associated with transcription initiation and splice sites in metazoans. Nat. Struct. Mol. Biol. 2010, 17, 1030–1034. [Google Scholar] [CrossRef] [PubMed]
- Mehler, M.F.; Mattick, J.S. Noncoding RNAs and RNA editing in brain development, functional diversification, and neurological disease. Physiol. Rev. 2007, 87, 799–823. [Google Scholar] [CrossRef] [Green Version]
- Salta, E.; De Strooper, B. Non-coding RNAs with essential roles in neurodegenerative disorders. Lancet. Neurol. 2012, 11, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Webb, A.; Papp, A.C.; Curtis, A.; Newman, L.C.; Pietrzak, M.; Seweryn, M.; Handelman, S.K.; Rempala, G.A.; Wang, D.; Graziosa, E.; et al. RNA sequencing of transcriptomes in human brain regions: Protein-coding and non-coding RNAs, isoforms and alleles. BMC Genom. 2015, 16, 990. [Google Scholar] [CrossRef] [Green Version]
- Lau, P.; Verrier, J.D.; Nielsen, J.A.; Johnson, K.R.; Notterpek, L.; Hudson, L.D. Identification of Dynamically Regulated MicroRNA and mRNA Networks in Developing Oligodendrocytes. J. Neurosci. 2008, 28, 11720–11730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, S.Y.; Johnson, R.; Stanton, L.W. Human long non-coding RNAs promote pluripotency and neuronal differentiation by association with chromatin modifiers and transcription factors. EMBO J. 2012, 31, 522–533. [Google Scholar] [CrossRef]
- Onoguchi, M.; Hirabayashi, Y.; Koseki, H.; Gotoh, Y. A noncoding RNA regulates the neurogenin1 gene locus during mouse neocortical development. Proc. Natl. Acad. Sci. USA 2012, 109, 16939–16944. [Google Scholar] [CrossRef] [Green Version]
- Qureshi, I.A.; Mehler, M.F. Long non-coding RNAs: Novel targets for nervous system disease diagnosis and therapy. NeuroTherapeutics 2013, 10, 632–646. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; La Russa, M.; Qi, L.S. CRISPR/Cas9 in Genome Editing and Beyond. Annu. Rev. Biochem. 2016, 85, 227–264. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolotin, A.; Quinquis, B.; Sorokin, A.; Ehrlich, S.D. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology 2005, 151 Pt 8, 2551–2561. [Google Scholar] [CrossRef] [Green Version]
- Lo, A.; Qi, L. Genetic and epigenetic control of gene expression by CRISPR-Cas systems. F1000Research 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Dissing-Olesen, L.; MacVicar, B.A.; Stevens, B. Microglia: Dynamic Mediators of Synapse Development and Plasticity. Trends Immunol. 2015, 36, 605–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, A.A.; Webb, W.M.; Lubin, F.D. Regulatory RNAs and control of epigenetic mechanisms: Expectations for cognition and cognitive dysfunction. Epigenomics 2016, 8, 135–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doherty, J.L.; Owen, M.J. Genomic insights into the overlap between psychiatric disorders: Implications for research and clinical practice. Genome Med. 2014, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Rieusset, A.; Schaller, F.; Unmehopa, U.; Matarazzo, V.; Watrin, F.; Linke, M.; Georges, B.; Bischof, J.; Dijkstra, F.; Bloemsma, M.; et al. Stochastic loss of silencing of the imprinted Ndn/NDN allele, in a mouse model and humans with prader-willi syndrome, has functional consequences. PLoS Genet. 2013, 9, e1003752. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zhou, Y.; Campbell, S.L.; Le, T.; Li, E.; Sweatt, J.D.; Silva, A.J.; Fan, G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 2010, 13, 423–430. [Google Scholar] [CrossRef] [Green Version]
- Miller, N.L.; Wevrick, R.; Mellon, P.L. Necdin, a Prader-Willi syndrome candidate gene, regulates gonadotropin-releasing hormone neurons during development. Hum. Mol. Genet. 2009, 18, 248–260. [Google Scholar] [CrossRef] [Green Version]
- Ehrenhofer-Murray, A.E. Cross-Talk between Dnmt2-Dependent tRNA Methylation and Queuosine Modification. Biomolecules 2017, 7, 14. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Wang, C.; Shen, J.; Tong, P.; O’Keefe, R. Ablation of Dnmt3b in chondrocytes suppresses cell maturation during embryonic development. J. Cell. Biochem. 2018, 119, 5852–5863. [Google Scholar] [CrossRef] [PubMed]
- Driessen, E.; Hollon, S.D. Cognitive behavioral therapy for mood disorders: Efficacy, moderators and mediators. Psychiatr. Clin. 2010, 33, 537–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Hu, G.; Li, Z.; Wang, Q.; Wang, X.; Yuan, C.; Wang, Z.; Hong, W.; Lu, W.; Cao, L.; et al. Differential gene expression in patients with subsyndromal symptomatic depression and major depressive disorder. PLoS ONE 2017, 12, e0172692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garafola, C.S.; Henn, F.A. A change in hippocampal protocadherin gamma expression in a learned helpless rat. Brain Res. 2014, 1593, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Grayson, D.R.; Guidotti, A. The dynamics of DNA methylation in schizophrenia and related psychiatric disorders. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2013, 38, 138–166. [Google Scholar] [CrossRef] [Green Version]
- Zhuo, C.; Hou, W.; Hu, L.; Lin, C.; Chen, C.; Lin, X. Genomic Editing of Non-Coding RNA Genes with CRISPR/Cas9 Ushers in a Potential Novel Approach to Study and Treat Schizophrenia. Front. Mol. Neurosci. 2017, 10, 28. [Google Scholar] [CrossRef]
- Dahoun, T.; Trossbach, S.V.; Brandon, N.J.; Korth, C.; Howes, O.D. The impact of Disrupted-in-Schizophrenia 1 (DISC1) on the dopaminergic system: A systematic review. Transl. Psychiatry 2017, 7, e1015. [Google Scholar] [CrossRef] [Green Version]
- Srikanth, P.; Han, K.; Callahan, D.G.; Makovkina, E.; Muratore, C.R.; Lalli, M.A.; Zhou, H.; Boyd, J.D.; Kosik, K.S.; Selkoe, D.J.; et al. Genomic DISC1 Disruption in hiPSCs Alters Wnt Signaling and Neural Cell Fate. Cell Rep. 2015, 12, 1414–1429. [Google Scholar] [CrossRef] [Green Version]
- Alotaibi, M.; Ramzan, K. A de novo variant of CHD8 in a patient with autism spectrum disorder. Discoveries 2020, 8, e107. [Google Scholar] [CrossRef]
- Wang, P.; Lin, M.; Pedrosa, E.; Hrabovsky, A.; Zhang, Z.; Guo, W.; Lachman, H.M.; Zheng, D. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in neurodevelopment. Mol. Autism 2015, 6, 55. [Google Scholar] [CrossRef] [Green Version]
- Wolter, J.M.; Mao, H.; Fragola, G.; Simon, J.M.; Krantz, J.L.; Bazick, H.O.; Oztemiz, B.; Stein, J.L.; Zylka, M.J. Cas9 gene therapy for Angelman syndrome traps Ube3a-ATS long non-coding RNA. Nature 2020, 587, 281–284. [Google Scholar] [CrossRef] [PubMed]
- González-Romero, E.; Martínez-Valiente, C.; García-Ruiz, C.; Vázquez-Manrique, R.P.; Cervera, J.; Sanjuan-Pla, A. CRISPR to fix bad blood: A new tool in basic and clinical hematology. Haematologica 2019, 104, 881–893. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.H.; Tee, L.Y.; Wang, X.G.; Huang, Q.S.; Yang, S.H. Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol. Ther.-Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Olivieri, M.; Petris, G.; Montagna, C.; Reginato, G.; Maule, G.; Lorenzin, F.; Prandi, D.; Romanel, A.; Demichelis, F.; et al. A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nat. Biotechnol. 2018, 36, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Vakulskas, C.A.; Dever, D.P.; Rettig, G.R.; Turk, R.; Jacobi, A.M.; Collingwood, M.A.; Bode, N.M.; McNeill, M.S.; Yan, S.; Camarena, J.; et al. A high-fidelity Cas9 mutant delivered as a ribonucleoprotein complex enables efficient gene editing in human hematopoietic stem and progenitor cells. Nat. Med. 2018, 24, 1216–1224. [Google Scholar] [CrossRef] [Green Version]
- Lo, C.L.; Choudhury, S.R.; Irudayaraj, J.; Zhou, F.C. Epigenetic Editing of Ascl1 Gene in Neural Stem Cells by Optogenetics. Sci. Rep. 2017, 7, 42047. [Google Scholar] [CrossRef]
- Wagnon, J.L. Promoting CRISPRa for Targeted Treatment of Epilepsy. Epilepsy Curr. 2020, 20, 227–229. [Google Scholar] [CrossRef]
- Khajanchi, N.; Saha, K. Controlling CRISPR with small molecule regulation for somatic cell genome editing. Mol. Ther. 2022, 30, 17–31. [Google Scholar] [CrossRef]
- Zou, Y.; Sun, X.; Yang, Q.; Zheng, M.; Shimoni, O.; Ruan, W.; Wang, Y.; Zhang, D.; Yin, J.; Huang, X.; et al. Blood-brain barrier-penetrating single CRISPR-Cas9 nanocapsules for effective and safe glioblastoma gene therapy. Sci. Adv. 2022, 8, eabm8011. [Google Scholar] [CrossRef]
- Kaushik, A.; Yndart, A.; Atluri, V.; Tiwari, S.; Tomitaka, A.; Gupta, P.; Jayant, R.D.; Alvarez-Carbonell, D.; Khalili, K.; Nair, M. Magnetically guided non-invasive CRISPR-Cas9/gRNA delivery across blood-brain barrier to eradicate latent HIV-1 infection. Sci. Rep. 2019, 9, 3928. [Google Scholar] [CrossRef] [Green Version]
- Ceña, V.; Játiva, P. Nanoparticle crossing of blood–brain barrier: A road to new therapeutic approaches to central nervous system diseases. Nanomedicine 2018, 13, 1513–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaFramboise, T. Single nucleotide polymorphism arrays: A decade of biological, computational and technological advances. Nucleic Acids Res. 2009, 37, 4181–4193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jannot, A.-S.; Ehret, G.; Perneger, T. P < 5 ∗ 10−8 has emerged as a standard of statistical significance for genome-wide association studies. J. Clin. Epidemiol. 2015, 68, 460–465. [Google Scholar] [PubMed]
- Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [CrossRef] [PubMed] [Green Version]
- Schmidt-Kastner, R.; Guloksuz, S.; Kietzmann, T.; van Os, J.; Rutten, B.P.F. Analysis of GWAS-Derived Schizophrenia Genes for Links to Ischemia-Hypoxia Response of the Brain. Front. Psychiatry 2020, 11, 393. [Google Scholar] [CrossRef]
- Levinson, D.; Shi, J.; Wang, K.; Oh, S.; Riley, B.; Pulver, A.; Wildenauer, D.; Laurent, C.; Mowry, B.; Gejman, P.; et al. Genome-Wide Association Study of Multiplex Schizophrenia Pedigrees. Am. J. Psychiatry 2012, 169, 963–973. [Google Scholar] [CrossRef]
- Retterer, K.; Juusola, J.; Cho, M.T.; Vitazka, P.; Millan, F.; Gibellini, F.; Vertino-Bell, A.; Smaoui, N.; Neidich, J.; Monaghan, K.G.; et al. Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 2016, 18, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Rabbani, B.; Tekin, M.; Mahdieh, N. The promise of whole-exome sequencing in medical genetics. J. Hum. Genet. 2014, 59, 5–15. [Google Scholar] [CrossRef]
- Cukier, H.N.; Dueker, N.D.; Slifer, S.H.; Lee, J.M.; Whitehead, P.L.; Lalanne, E.; Leyva, N.; Konidari, I.; Gentry, R.C.; Hulme, W.F.; et al. Exome sequencing of extended families with autism reveals genes shared across neurodevelopmental and neuropsychiatric disorders. Mol. Autism 2014, 5, 1. [Google Scholar] [CrossRef] [Green Version]
- Shmelkov, S.V.; Hormigo, A.; Jing, D.; Proenca, C.C.; Bath, K.G.; Milde, T.; Shmelkov, E.; Kushner, J.S.; Baljevic, M.; Dincheva, I.; et al. Slitrk5 deficiency impairs corticostriatal circuitry and leads to obsessive-compulsive–like behaviors in mice. Nat. Med. 2010, 16, 598–602. [Google Scholar] [CrossRef] [Green Version]
- Halvorsen, M.; Samuels, J.; Wang, Y.; Greenberg, B.D.; Fyer, A.J.; McCracken, J.T.; Geller, D.A.; Knowles, J.A.; Zoghbi, A.W.; Pottinger, T.D.; et al. Exome sequencing in obsessive–compulsive disorder reveals a burden of rare damaging coding variants. Nat. Neurosci. 2021, 24, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Ganesh, S.; Ahmed, P.H.; Nadella, R.K.; More, R.P.; Seshadri, M.; Viswanath, B.; Rao, M.; Jain, S.; Mukherjee, O.; The ADBS Consortium. Exome sequencing in families with severe mental illness identifies novel and rare variants in genes implicated in Mendelian neuropsychiatric syndromes. Psychiatry Clin. Neurosci. 2019, 73, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreiber, M.; Dorschner, M.; Tsuang, D. Next-generation sequencing in schizophrenia and other neuropsychiatric disorders. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2013, 162, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct. Target. Ther. 2020, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luther, D.C.; Lee, Y.W.; Nagaraj, H.; Scaletti, F.; Rotello, V.M. Delivery approaches for CRISPR/Cas9 therapeutics in vivo: Advances and challenges. Expert Opin. Drug Deliv. 2018, 15, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Hana, S.; Peterson, M.; McLaughlin, H.; Marshall, E.; Fabian, A.J.; McKissick, O.; Koszka, K.; Marsh, G.; Craft, M.; Xu, S.; et al. Highly efficient neuronal gene knockout in vivo by CRISPR-Cas9 via neonatal intracerebroventricular injection of AAV in mice. Gene Ther. 2021, 28, 646–658. [Google Scholar] [CrossRef]
- Caligiuri, S.P.; Kenny, P.J. The Promise of Genome Editing for Modeling Psychiatric Disorders. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2018, 43, 223–224. [Google Scholar] [CrossRef] [Green Version]
- Naeem, M.; Majeed, S.; Hoque, M.Z.; Ahmad, I. Latest Developed Strategies to Minimize the Off-Target Effects in CRISPR-Cas-Mediated Genome Editing. Cells 2020, 9, 1608. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| S.No. | Name of the NPD | Genes/Genetic Locus Associated | References |
|---|---|---|---|
| 1 | Schizophrenia | MTHFR, CGI3L1, DISC1. DISC2, SYN2, DRD3, RTN4R, DAOA, HTR2A, AKT1, C4A, APOL2. APOL4, PRODH, NRG, SHANK3, NRXN1, SLC1A1, RBM12 | [16,115,116,117,118] |
| 2 | Autism | CNTNAP2, SLC9A9, SHANK2, CHD8, EIF4E, BLGN1, NLGN3, NLGN4X, MECP2, PTCHD1, RPL10, TMLHE | [16,119,120,121,122,123] |
| 3 | Fragile X Syndrome | FMR1 | [124,125] |
| 4 | Epilepsy/Seizures | CACNA1H, CASR, CACNB4, GABRD, CLCN2, SLC2A1, GABRA1, SLC12A5, RORB, KCNMA1 | [126,127,128,129] |
| 5 | Parkinson’s disease (PD) | SNCA, Parkin, UCHL1, PINK1, DJ1, LRRK2, ATP13A2, GIGYF2, HTRA2, PLA2G6, FBX07, VPS35, EIF4G1, DNAJC6, CHCHD2, VPS13C, PSAP, NR4A2, MAPT, PARK2, PARK6, PARK8 | [130,131,132,133,134] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, M.; Agarwal, V.; Jindal, D.; Pancham, P.; Agarwal, S.; Mani, S.; Tiwari, R.K.; Das, K.; Alghamdi, B.S.; Abujamel, T.S.; et al. Recent Updates on Corticosteroid-Induced Neuropsychiatric Disorders and Theranostic Advancements through Gene Editing Tools. Diagnostics 2023, 13, 337. https://doi.org/10.3390/diagnostics13030337
Singh M, Agarwal V, Jindal D, Pancham P, Agarwal S, Mani S, Tiwari RK, Das K, Alghamdi BS, Abujamel TS, et al. Recent Updates on Corticosteroid-Induced Neuropsychiatric Disorders and Theranostic Advancements through Gene Editing Tools. Diagnostics. 2023; 13(3):337. https://doi.org/10.3390/diagnostics13030337
Chicago/Turabian StyleSingh, Manisha, Vinayak Agarwal, Divya Jindal, Pranav Pancham, Shriya Agarwal, Shalini Mani, Raj Kumar Tiwari, Koushik Das, Badrah S. Alghamdi, Tukri S. Abujamel, and et al. 2023. "Recent Updates on Corticosteroid-Induced Neuropsychiatric Disorders and Theranostic Advancements through Gene Editing Tools" Diagnostics 13, no. 3: 337. https://doi.org/10.3390/diagnostics13030337