
Cervicovaginal-Microbiome Analysis by 16S Sequencing and Real-Time PCR in Patients from Novosibirsk (Russia) with Cervical Lesions and Several Years after Cancer Treatment
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Material and Nucleic-Acid Extraction
2.2. PCR-Based Analyses
2.3. Preparation of a 16S rRNA Amplicon Library
2.4. 16S rRNA Gene Sequencing and Sequencing Data Analysis
2.5. Other Data Analyses
3. Results
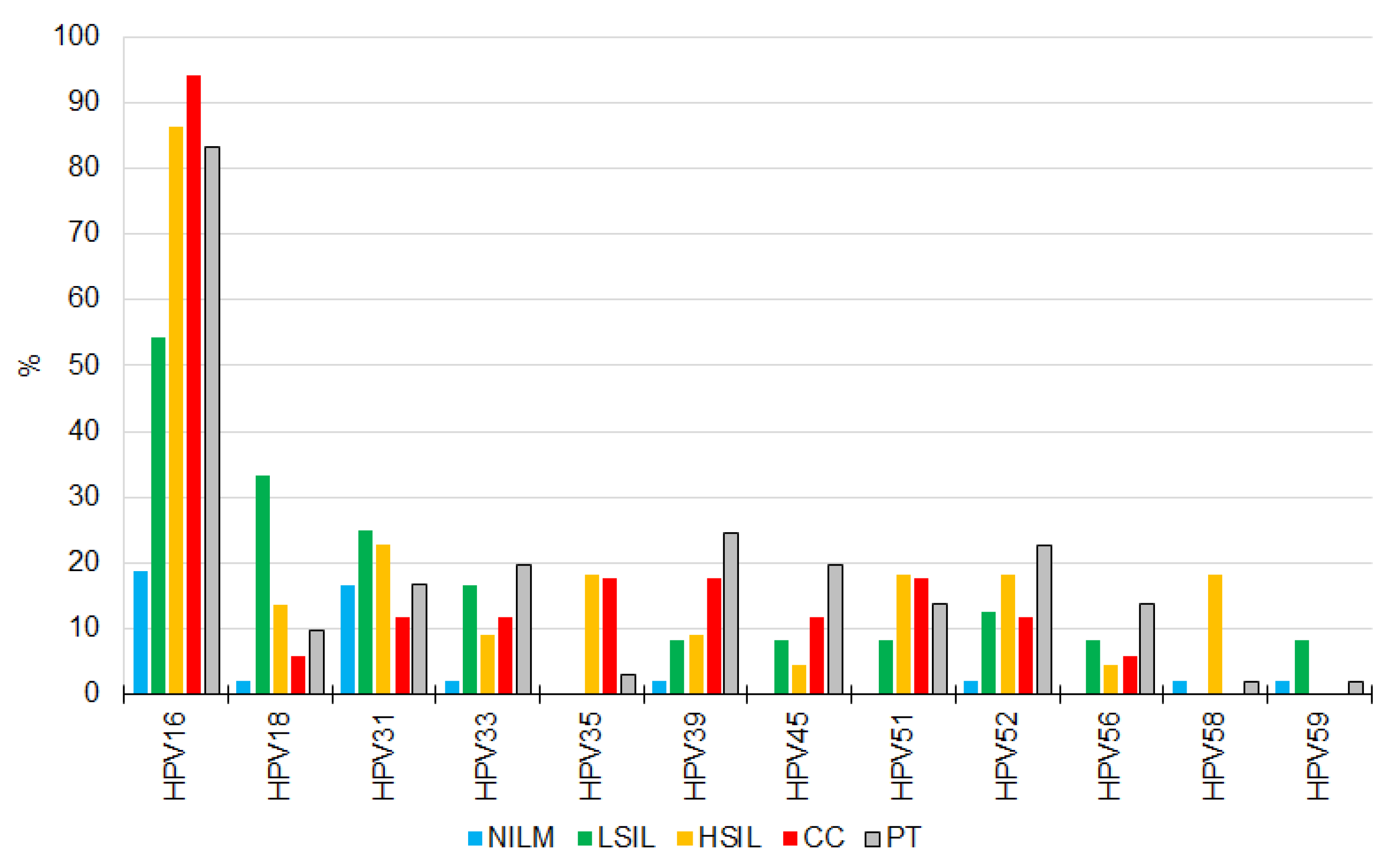
3.1. HR-HPV Detection, Genotyping, and Quantitation
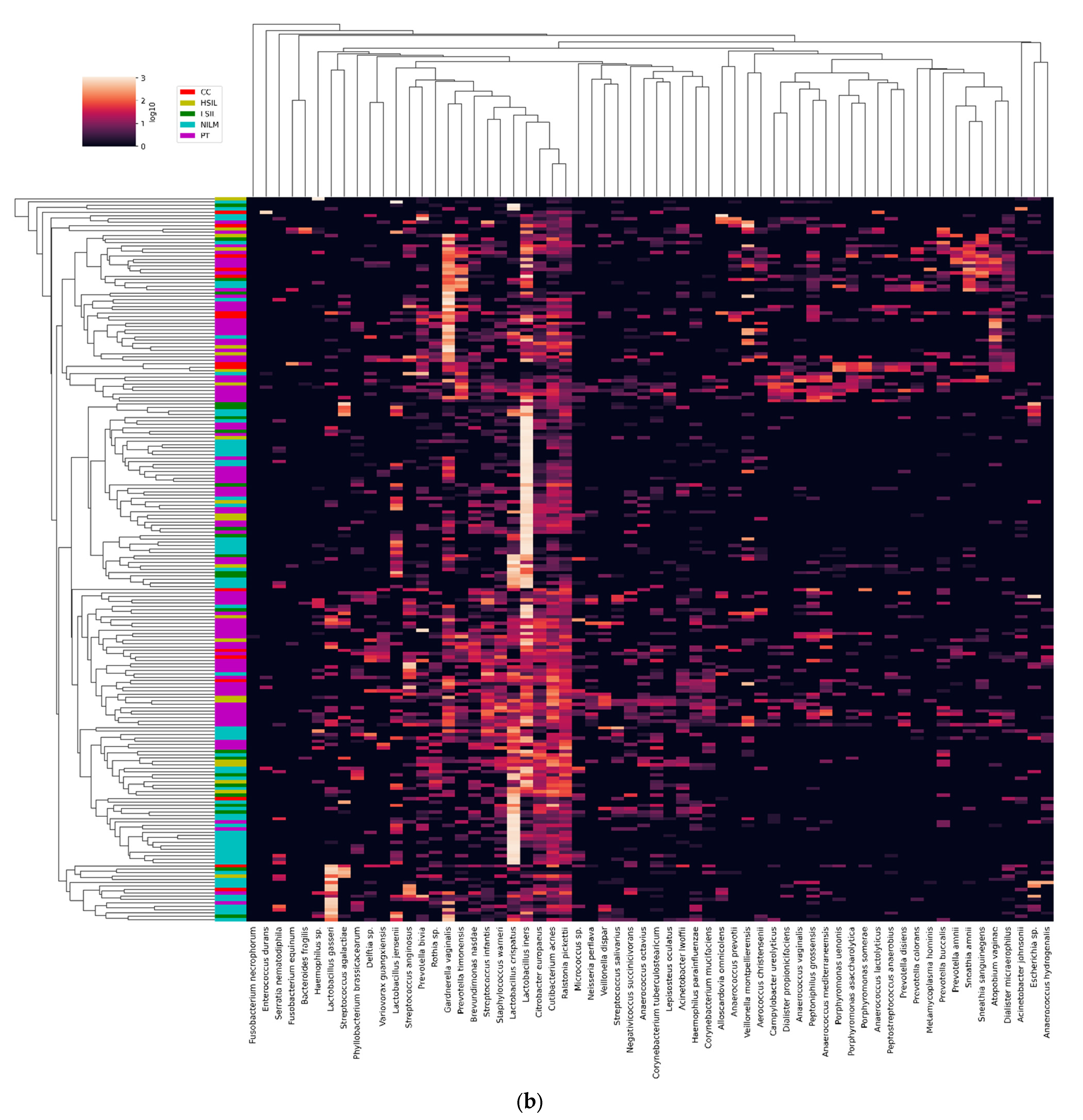
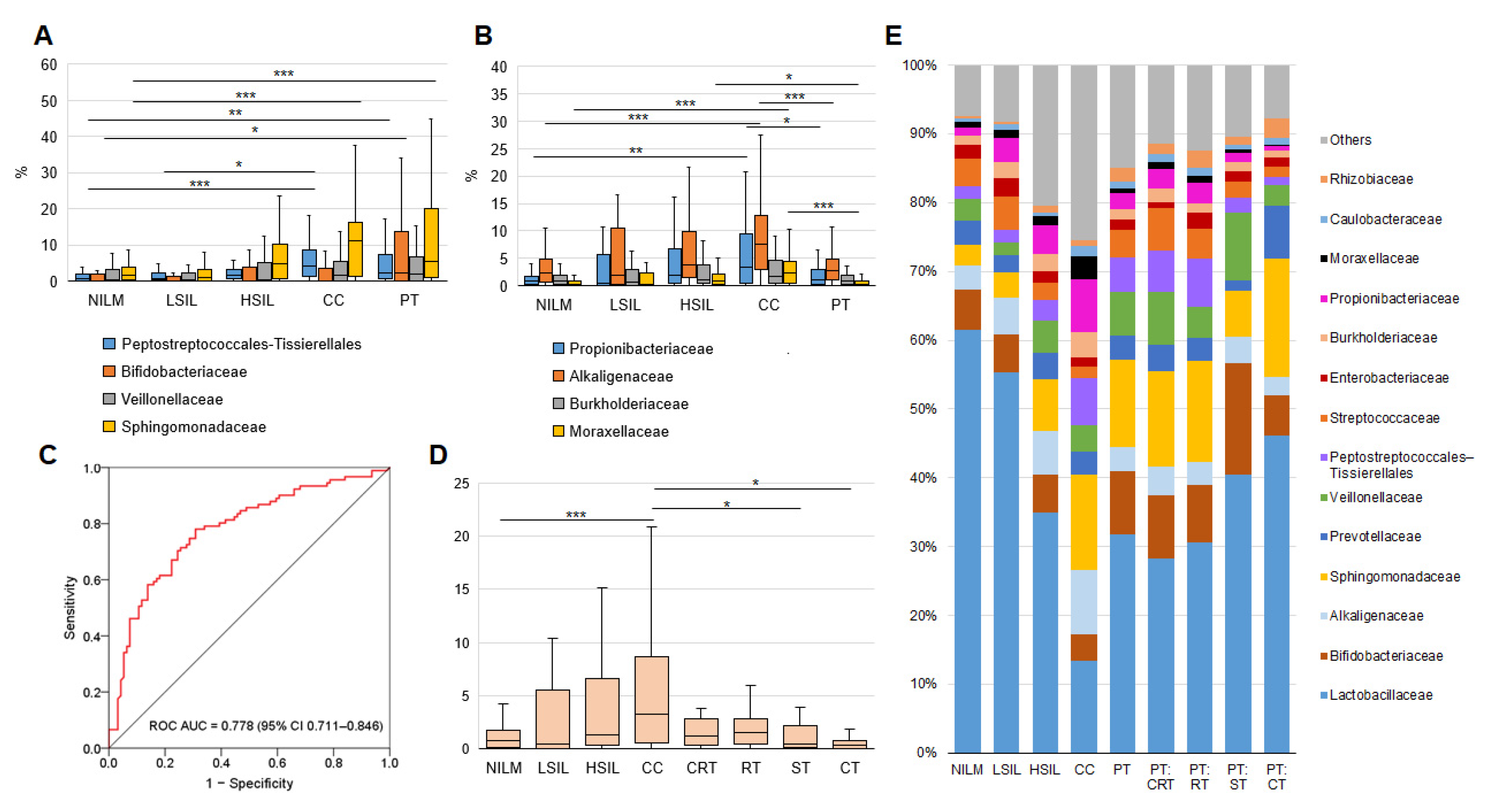
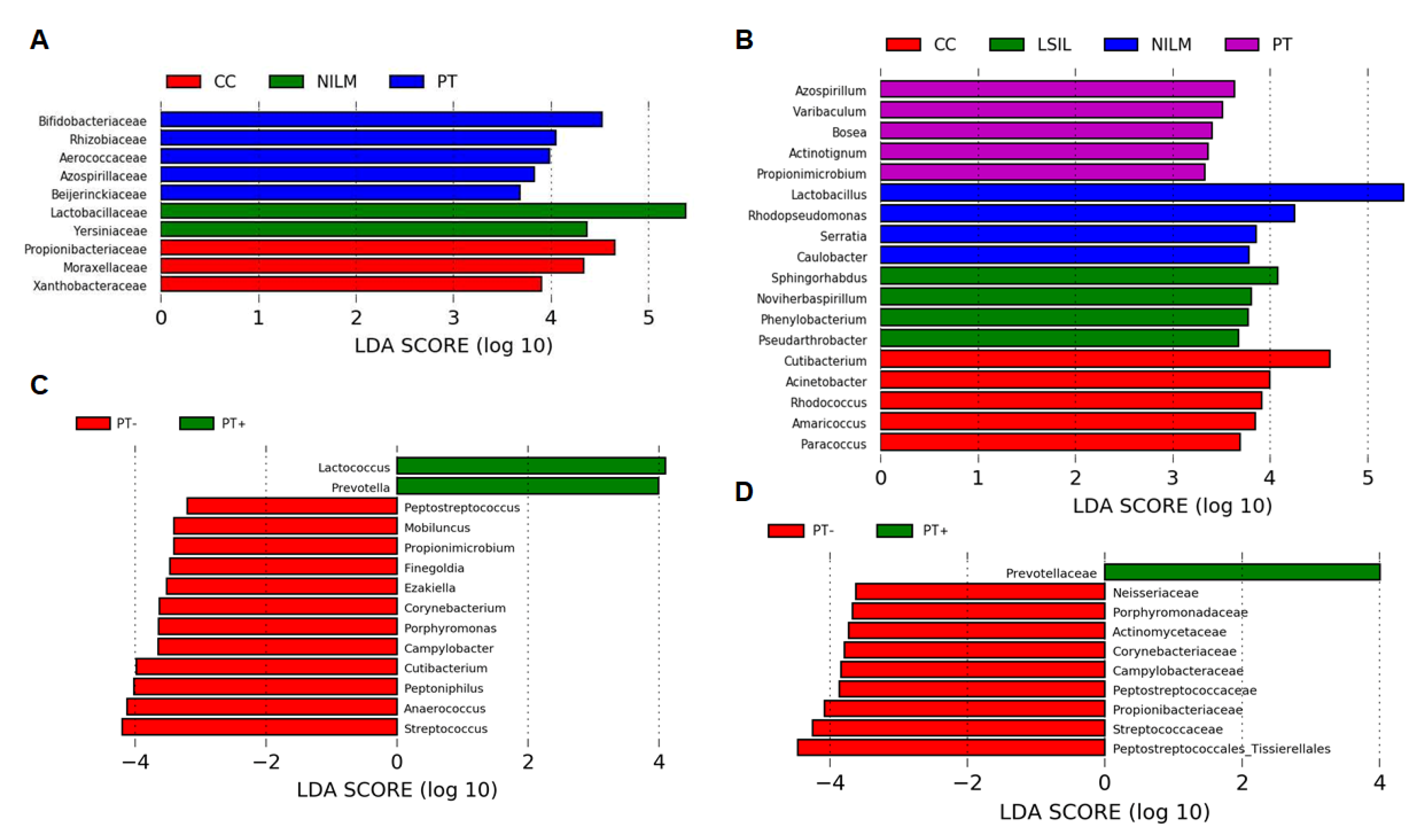
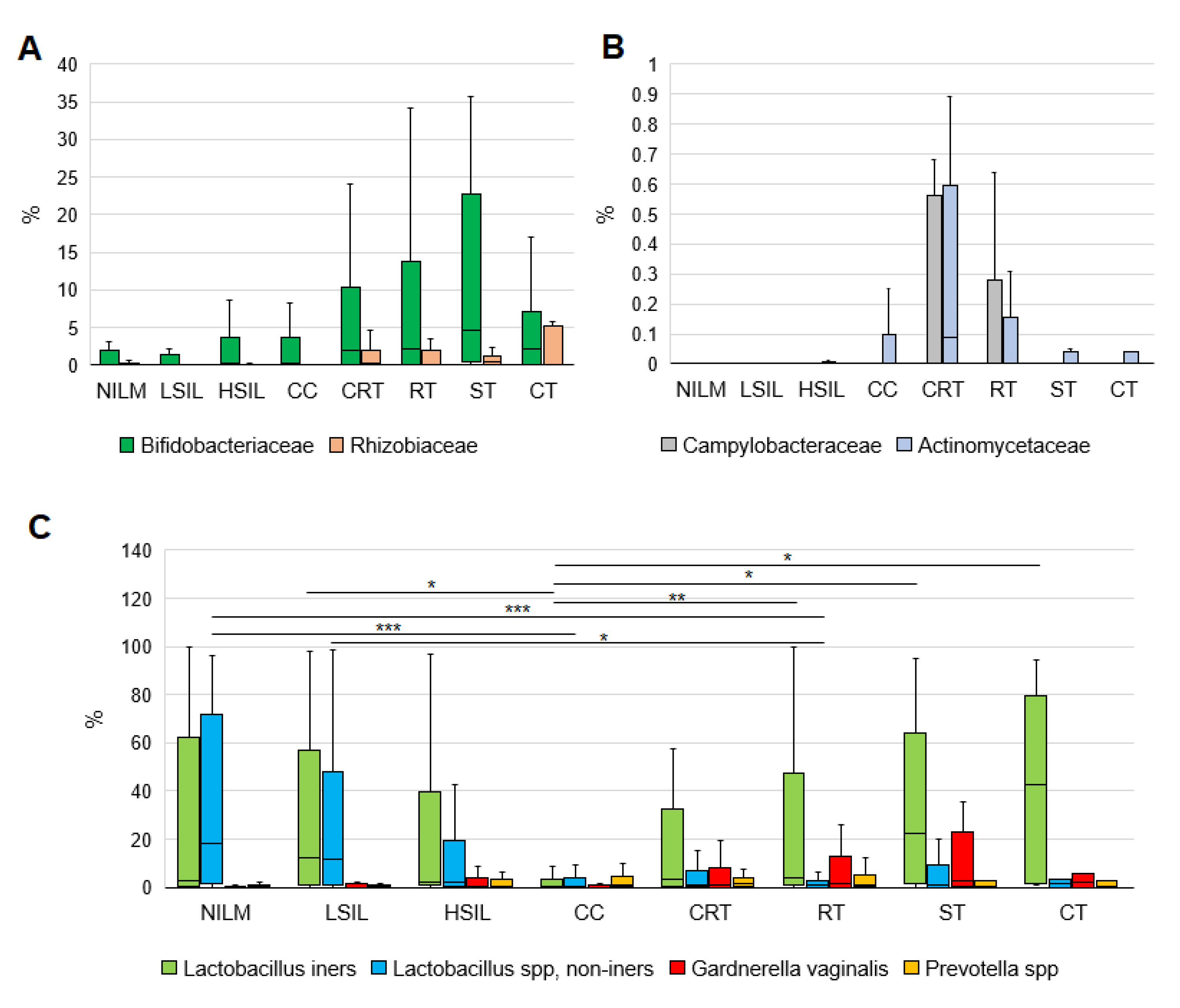
3.2. CVM Analysis by 16S rRNA Gene Sequencing
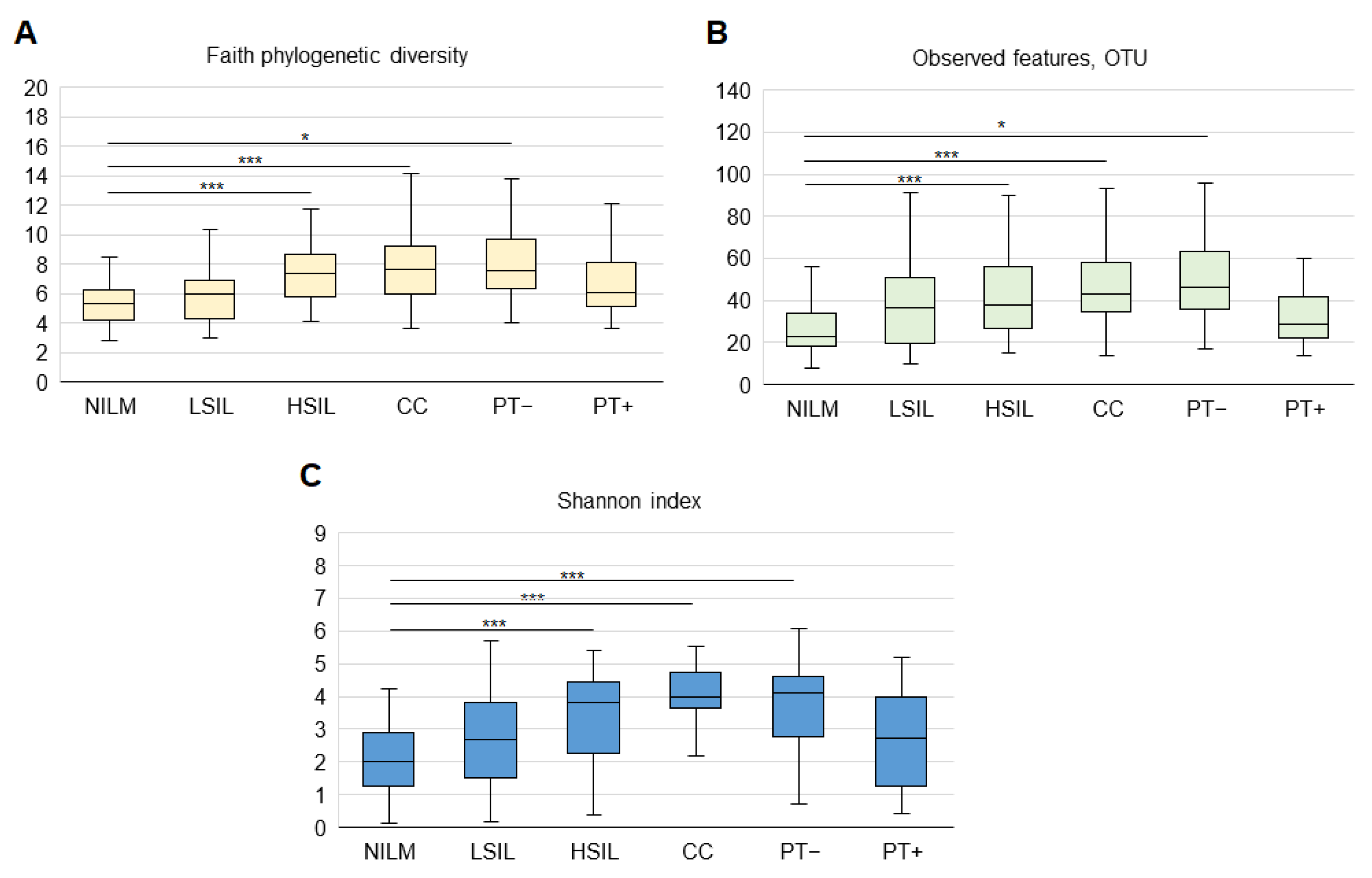
3.2.1. General Biodiversity
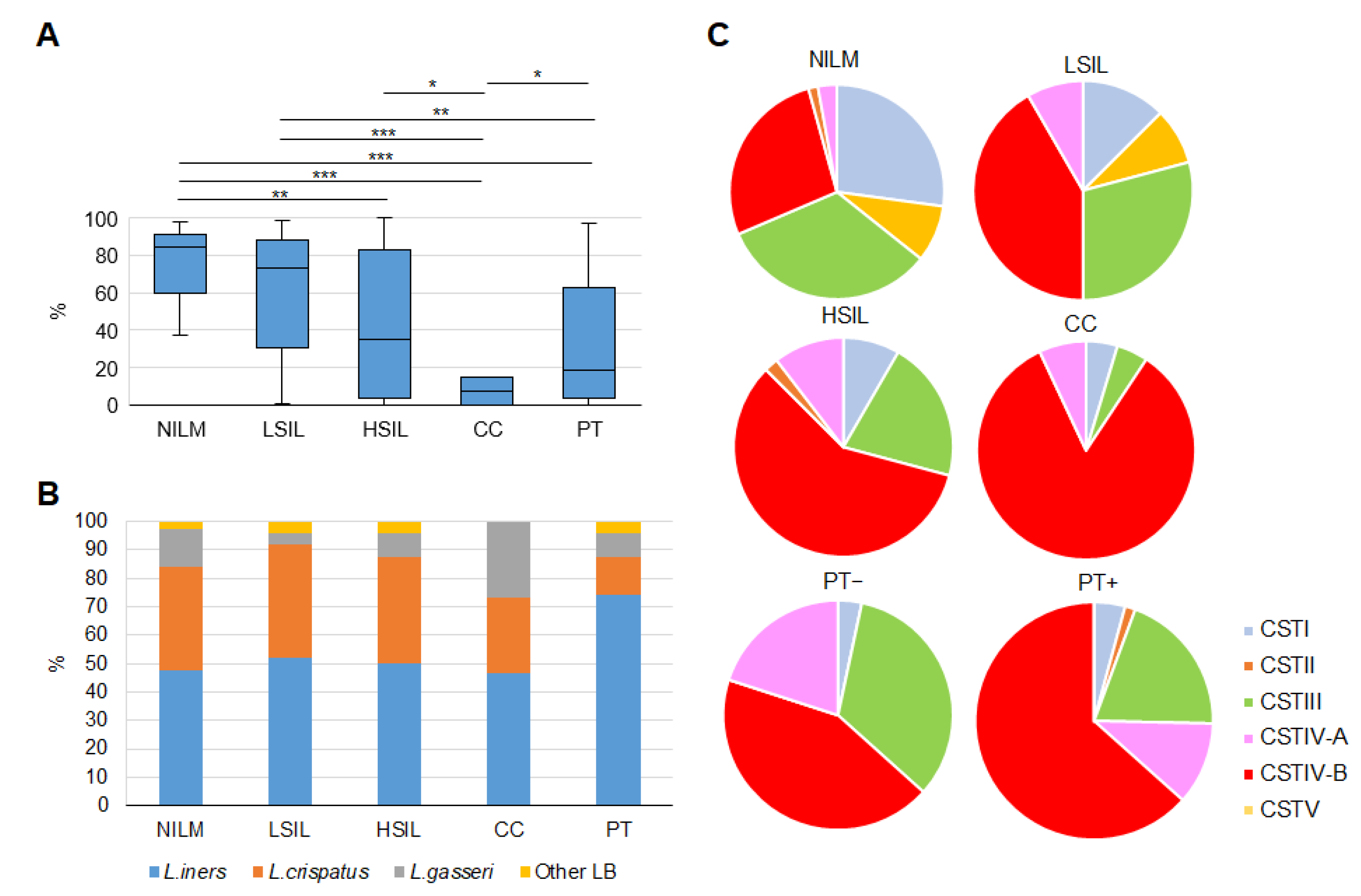
3.2.2. Enrichment with LB Species
3.2.3. Community State Types
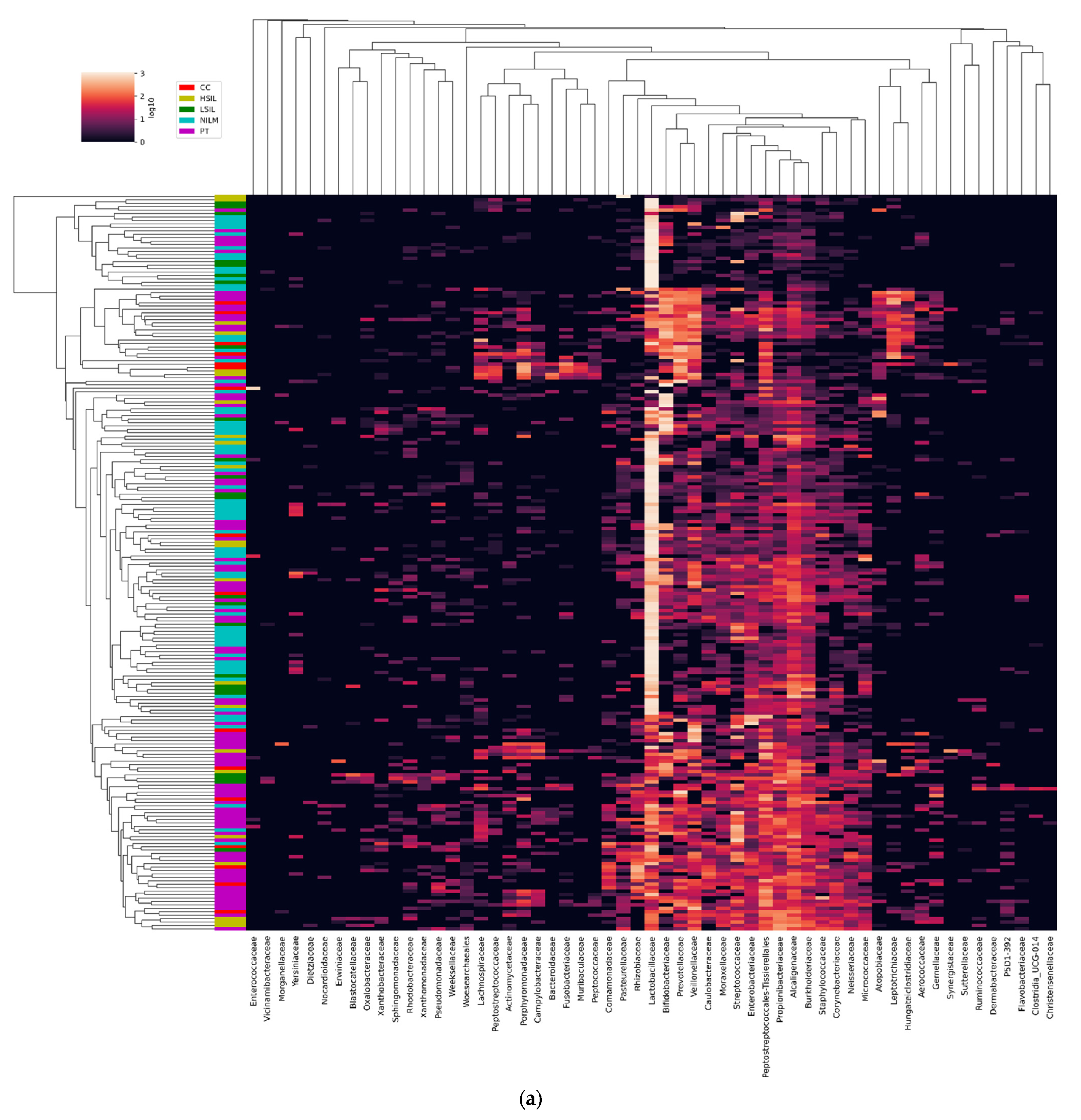
3.2.4. Non-LB Species
3.3. CVM Analysis by PCR-Based Kits
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gao, W.; Weng, J.; Gao, Y.; Chen, X. Comparison of the vaginal microbiota diversity of women with and without human papillomavirus infection: A cross-sectional study. BMC Infect. Dis. 2013, 13, 271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Da, M.; Zhang, W.; Qi, Q.; Zhang, C.; Han, S. Role of Lactobacillus in cervical cancer. Cancer Manag. Res. 2018, 10, 1219–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, W.; Du, H.; Li, S.; Wu, R. Cervicovaginal Microbiome Factors in Clearance of Human Papillomavirus Infection. Front. Oncol. 2021, 11, 722639. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; MacIntyre, D.A.; Lee, Y.S.; Smith, A.; Marchesi, J.R.; Lehne, B.; Bhatia, R.; Lyons, D.; Paraskevaidis, E.; Li, J.V.; et al. Cervical intraepithelial neoplasia disease progression is associated with increased vaginal microbiome diversity. Sci. Rep. 2015, 5, 16865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audirac-Chalifour, A.; Torres-Poveda, K.; Bahena-Román, M.; Téllez-Sosa, J.; Martínez-Barnetche, J.; Cortina-Ceballos, B.; López-Estrada, G.; Delgado-Romero, K.; Burguete-García, A.I.; Cantú, D.; et al. Cervical microbiome and cytokine profile at various stages of cervical cancer: A pilot study. PLoS ONE 2016, 11, e0153274. [Google Scholar] [CrossRef] [Green Version]
- Łaniewski, P.; Barnes, D.; Goulder, A.; Cui, H.; Roe, D.J.; Chase, D.M.; Herbst-Kralovetz, M.H. Linking cervicovaginal immune signatures, HPV and microbiota composition in cervical carcinogenesis in non-Hispanic and Hispanic women. Sci. Rep. 2018, 8, 7593. [Google Scholar] [CrossRef] [Green Version]
- Łaniewski, P.; Cui, H.; Roe, D.J.; Barnes, D.; Goulder, A.; Monk, J.B.; Greenspan, D.L.; Chase, D.M.; Herbst-Kralovetz, M.H. Features of the cervicovaginal microenvironment drive cancer biomarker signatures in patients across cervical carcinogenesis. Sci. Rep. 2019, 9, 7333. [Google Scholar] [CrossRef] [Green Version]
- Curty, G.; de Carvalho, P.S.; Soares, M.A. The Role of the Cervicovaginal Microbiome on the Genesis and as a Biomarker of Premalignant Cervical Intraepithelial Neoplasia and Invasive Cervical Cancer. Int. J. Mol. Sci. 2020, 21, 222. [Google Scholar] [CrossRef] [Green Version]
- De Sanjose, S.; Diaz, M.; Castellsague, X.; Clifford, G.; Bruni, L.; Muñoz, N.; Bosch, F.X. Worldwide prevalence and genotype distribution of cervical human papillomavirus DNA in women with normal cytology: A meta-analysis. Lancet Infect. Dis. 2007, 7, 453–459. [Google Scholar] [CrossRef]
- Huh, W.K.; Ault, K.A.; Chelmow, D.; Davey, D.D.; Goulart, R.A.; Garcia, F.A.R.; Kinney, W.K.; Massad, L.S.; Mayeaux, E.J.; Saslow, D.; et al. Use of primary high-risk human papillomavirus testing for cervical cancer screening: Interim clinical guidance. Gynecol. Oncol. 2015, 136, 178–182. [Google Scholar] [CrossRef]
- Koliopoulos, G.; Nyaga, V.N.; Santesso, N.; Bryant, A.; Martin-Hirsch, P.P.; Mustafa, R.A.; Schünemann, H.; Paraskevaidis, E.; Arbyn, M. Cytology versus HPV testing for cervical cancer screening in the general population. Cochrane Database Syst. Rev. 2017, 8, CD008587. [Google Scholar] [CrossRef]
- Sørbye, S.W.; Suhrke, P.; Revå BWBerland, J.; Maurseth, R.J.; Al-Shibli, K. Accuracy of cervical cytology: Comparison of diagnoses of 100 Pap smears read by four pathologists at three hospitals in Norway. BMC Clin. Pathol. 2017, 17, 18. [Google Scholar] [CrossRef] [Green Version]
- van den Munckhof, E.H.A.; van Sitter, R.L.; Boers, K.E.; Lamont, R.F.; te Witt, R.; le Cessie, S.; Knetsch, C.W.; van Doorn, L.-J.; Quint, W.G.V.; Molijn, A.; et al. Comparison of Amsel criteria, Nugent score, culture and two CE-IVD marked quantitative real-time PCRs with microbiota analysis for the diagnosis of bacterial vaginosis. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 959–966. [Google Scholar] [CrossRef]
- Lamont, R.F.; van den Munckhof, E.H.A.; Luef, B.M.; Vinter, C.A.; Jørgensen, J.S. Recent advances in cultivation-independent molecular-based techniques for the characterization of vaginal eubiosis and dysbiosis. Fac. Rev. 2020, 9, 21. [Google Scholar] [CrossRef]
- Chen, X.; Lu, Y.; Chen, T.; Li, R. The Female Vaginal Microbiome in Health and Bacterial Vaginosis. Front. Cell. Infect. Microbiol. 2021, 11, 631972. [Google Scholar] [CrossRef]
- Lin, D.; Kouzy, R.; Abi Jaoude, J.; Noticewala, S.S.; Delgado Medrano, A.Y.; Klopp, A.H.; Taniguchi, C.M.; Colbert, L.E. Microbiome factors in HPV-driven carcinogenesis and cancers. PLoS Pathog. 2020, 16, e1008524. [Google Scholar] [CrossRef]
- Gupta, V.K.; Paul, S.; Dutta, C. Geography, Ethnicity or Subsistence-Specific Variations in Human Microbiome Composition and Diversity. Front. Microbiol. 2017, 8, 1162. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Li, H.; Yu, H.; Yan, Y.; Wang, C.; Teng, F.; Fan, A.; Xue, F. Disturbances of Vaginal Microbiome Composition in Human Papillomavirus Infection and Cervical Carcinogenesis: A Qualitative Systematic Review. Front. Oncol. 2022, 12, 941741. [Google Scholar] [CrossRef]
- Ivanov, M.K.; Titov, S.E.; Dzyubenko, V.V.; Glushkov, S.A.; Krasilnikov, S.E.; Mansurova, A.S.; Malek, A.V.; Berlev, I.V.; Prisyazhnaya, T.S.; Kuleshova, S.V.; et al. Detection of Cervical Lesions and Cancer in Air-Dried Cytologic Smears by Combined Analysis of mRNA and miRNA Expression Levels. J. Mol. Diagn. 2021, 23, 541–554. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 15. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Walron, L.; Dirk Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic Biomarker Discovery and Explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravel, J.; Gajer, P.; Abdo, Z.; Schneider, G.M.; Koenig, S.S.K.; McCulle, S.L.; Karlebach, S.; Gorle, R.; Russell, J.; Tacket, C.O.; et al. Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. S1), 4680–4687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajer, P.; Brotman, R.M.; Bail, G.; Sakamoto, J.; Schutte, U.M.E.; Zhong, X.; Koenig, S.S.K.; Fu, L.; Ma, Z.S.; Zhou, X.; et al. Temporal dynamics of the human vaginal microbiota. Sci. Transl. Med. 2020, 4, 132ra52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrd, A.L.; Belkaid, Y.; Segre, J.A. The human skin microbiome. Nat. Rev. Microbiol. 2018, 16, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Hashim, D.; Engesæter, B.; Skare, G.B.; Castle, P.E.; Bjørge, T.; Tropé, A.; Nygård, M. Real-world data on cervical cancer risk stratification by cytology and HPV genotype to inform the management of HPV-positive women in routine cervical screening. Br. J. Cancer 2020, 122, 1715–1723. [Google Scholar] [CrossRef] [Green Version]
- Demarco, M.; Hyun, N.; Carter-Pokras, O.; Raine-Bennett, T.R.; Cheung, L.; Chen, X.; Hammer, A.; Campos, N.; Kinney, W.; Gage, J.C.; et al. A study of type-specific HPV natural history and implications for contemporary cervical cancer screening programs. EClinicalMedicine 2020, 22, 100293. [Google Scholar] [CrossRef]
- Pardini, B.; De Maria, D.; Francavilla, A.; Di Gaetano, C.; Ronco, G.; Naccarati, A. MicroRNAs as markers of progression in cervical cancer: A systematic review. BMC Cancer 2018, 18, 696. [Google Scholar] [CrossRef]
- Kori, M.; Yalcin Arga, K. Potential biomarkers and therapeutic targets in cervical cancer: Insights from the meta-analysis of transcriptomics data within network biomedicine perspective. PLoS ONE 2018, 13, e0200717. [Google Scholar] [CrossRef]
- Kelly, H.; Benavente, Y.; Pavon, M.A.; De Sanjose, S.; Mayaud, P.; Lorincz, A.T. Performance of DNA methylation assays for detection of highgrade cervical intraepithelial neoplasia (CIN2+): A systematic review and meta-analysis. Br. J. Cancer 2019, 121, 954–965. [Google Scholar] [CrossRef]
- Ravel, J.; Brotman, R.M.; Gajer, P.; Ma, B.; Nandy, M.; Fadrosh, D.W.; Sakamoto, J.; Koenig, S.S.K.; Fu, L.; Zhou, X.; et al. Daily temporal dynamics of vaginal microbiota before, during and after episodes of bacterial vaginosis. Microbiome 2013, 1, 29. [Google Scholar] [CrossRef]
- Lin, S.; Zhang, B.; Lin, Y.; Lin, Y.; Zuo, X. Dysbiosis of Cervical and Vaginal Microbiota Associated With Cervical Intraepithelial Neoplasia. Front. Cell. Infect. Microbiol. 2022, 12, 767693. [Google Scholar] [CrossRef]
- Kim, T.K.; Thomas, S.M.; Ho, M.; Sharma, S.; Reich, C.I.; Frank, J.A.; Yeater, K.M.; Biggs, D.R.; Nakamura, N.; Stumpf, R.; et al. Heterogeneity of Vaginal Microbial Communities within Individuals. J. Clin. Microbiol. 2009, 47, 1181–1189. [Google Scholar] [CrossRef] [Green Version]
- Wiik, J.; Sengpiel, V.; Kyrgiou, M.; Nilsson, S.; Mitra, A.; Tanbo, T.; Jonassen, C.M.; Tannæs, T.M.; Sjøborg, K. Cervical microbiota in women with cervical intra-epithelial neoplasia, prior to and after local excisional treatment, a Norwegian cohort study. BMC Women Health 2019, 19, 30. [Google Scholar] [CrossRef]
- Zhou, Z.-W.; Long, H.-Z.; Cheng, Y.; Luo, H.-Y.; Wen, D.-D.; Gao, L.-C. From microbiome to inflammation: The key drivers of cervical cancer (a review). Front. Microbiol. 2021, 12, 767931. [Google Scholar] [CrossRef]
- Gillet, E.; Meys, J.F.A.; Verstraelen, H.; Bosire, C.; De Sutter, P.; Temmerman, M.; Broeck, D.V. Bacterial vaginosis is associated with uterine cervical human papillomavirus infection: A meta-analysis. BMC Infect. Dis. 2011, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Brotman, R.M.; Shardell, M.D.; Gajer, P.; Tracy, K.J.; Zenilman, J.M.; Ravel, J.; Gravitt, P.E. Interplay Between the Temporal Dynamics of the Vaginal Microbiota and Human Papillomavirus Detection. J. Infect. Dis. 2014, 210, 1723–1733. [Google Scholar] [CrossRef] [Green Version]
- Di Paola, M.; Sani, C.; Clemente, A.M.; Iossa, A.; Perissi, E.; Castronovo, G.; Tanturli, M.; Rivero, D.; Cozzolino, F.; Cavalieri, D.; et al. Characterization of cervicovaginal microbiota in women developing persistent high-risk Human Papillomavirus infection. Sci. Rep. 2017, 7, 10200. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, Y.; Gao, W.; Pan, Y.; Gao, Y.; Shen, J.; Xiong, H. The direct and indirect association of cervical microbiota with the risk of cervical intraepithelial neoplasia. Cancer Med. 2018, 7, 2172–2179. [Google Scholar] [CrossRef]
- Usyk, M.; Zolnik, C.P.; Castle, P.E.; Porras, C.; Herrero, R.; Gradissimo, A.; Gonzalez, P.; Safaeian, M.; Schiffman, M.; Burk, R.D. Cervicovaginal microbiome and natural history of HPV in a longitudinal study. PLoS Pathog. 2020, 16, e1008376. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xu, X.; Yu, L.; Shi, X.; Min, M.; Xiong, L.; Pan, J.; Zhang, Y.; Liu, P.; Wu, G.; et al. Vaginal Microbiota Changes Caused by HPV Infection in Chinese Women. Front. Cell. Infect. Microbiol. 2022, 12, 814668. [Google Scholar] [CrossRef]
- Verhoeven, V.; Renard, N.; Makar, A.; Van Royen, P.; Bogers, J.-P.; Lardon, F.; Peeters, M.; Baay, M. Probiotics enhance the clearance of human papillomavirus-related cervical lesions: A prospective controlled pilot study. Eur. J. Cancer Prev. 2013, 22, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Palma, E.; Recine, N.; Domenici, L.; Giorgini, M.; Pierangeli, A.; Panici, P.B. Long-term Lactobacillus rhamnosus BMX 54 application to restore a balanced vaginal ecosystem: A promising solution against HPV-infection. BMC Infect. Dis. 2018, 18, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, Y.-C.; Fu, H.-C.; Tseng, C.-W.; Wu, C.-H.; Tsai, C.-C.; Lin, H. The influence of probiotics on genital high-risk human papilloma virus clearance and quality of cervical smear: A randomized placebo-controlled trial. BMC Women Health 2019, 19, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.-Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 2019, 10, 5029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, J.; Mun, S.; Oh, Y.; Cho, C.-S.; Yun, K.; Ahn, Y.; Chung, W.-H.; Lim, M.Y.; Lee, K.E.; Hwang, T.S.; et al. A qRT-PCR Method Capable of Quantifying Specific Microorganisms Compared to NGS-Based Metagenome Profiling Data. Microorganisms 2022, 10, 324. [Google Scholar] [CrossRef]
- Petrova, M.I.; Lievens, E.; Malik, S.; Imholz, N.; Lebeer, S. Lactobacillus species as biomarkers and agents that can promote various aspects of vaginal health. Front. Physiol. 2015, 6, 81. [Google Scholar] [CrossRef] [Green Version]
- Brotman, R.M.; Shardell, M.D.; Gajer, P.; Fadrosh, D.; Chang, K.; Silver, M.; Viscidi, R.P.; Burke, A.E.; Ravel, J.; Gravitt, P.E. Association between the vaginal microbiota, menopause status and signs of vulvovaginal atrophy. Menopause 2014, 21, 450–458. [Google Scholar] [CrossRef] [Green Version]
- Shardell, M.D.; Gravitt, P.E.; Burke, A.E.; Ravel, J.; Brotman, R.M. Association of Vaginal Microbiota With Signs and Symptoms of the Genitourinary Syndrome of Menopause Across Reproductive Stages. J. Gerontol. Biol. Sci. Med. Sci. 2021, 76, 1542–1550. [Google Scholar] [CrossRef]
- Ryan, M.P.; Pembroke, J.T.; Adley, C.C. Ralstonia pickettii: A persistent Gram-negative nosocomial infectious organism (Review). J. Hosp. Infect. 2006, 62, 278–284. [Google Scholar] [CrossRef]
- Swenson, C.E.; Sadikot, R.T. Achromobacter Respiratory Infections. Ann. Am. Thorac. Soc. 2015, 12, 252–258. [Google Scholar] [CrossRef]
- Achermann, Y.; Goldstein, E.J.C.; Coenye, T.; Shirtliff, M.E. Propionibacterium acnes: From commensal to opportunistic biofilm-associated implant pathogen. Clin. Microbiol. Rev. 2014, 27, 419–440. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Lu, J.; Lu, Y.; Cai, Q.; Liu, H.; Xu, C. Cervical microbiome is altered in cervical intraepithelial neoplasia after loop electrosurgical excision procedure in China. Sci. Rep. 2018, 8, 4923. [Google Scholar] [CrossRef] [Green Version]
- Tsakmaklis, A.; Vehreschild, M.; Farowski, F.; Trommer, M.; Kohler, C.; Herter, J.; Marnitz, S. Changes in the cervical microbiota of cervical cancer patients after primary radio-chemotherapy. Int. J. Gynecol. Cancer 2020, 30, 1326–1330. [Google Scholar] [CrossRef]
- Li, M.; Zhao, C.; Zhao, Y.; Li, J.; Wei, L. Age-Stratified Analysis of Vaginal Microbiota Dysbiosis and the Relationship with HPV Viral Load in HPV-Positive Women. J. Immunol. Res. 2022, 2022, 1372926. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivanov, M.K.; Brenner, E.V.; Hodkevich, A.A.; Dzyubenko, V.V.; Krasilnikov, S.E.; Mansurova, A.S.; Vakhturova, I.E.; Agletdinov, E.F.; Shumeikina, A.O.; Chernyshova, A.L.; et al. Cervicovaginal-Microbiome Analysis by 16S Sequencing and Real-Time PCR in Patients from Novosibirsk (Russia) with Cervical Lesions and Several Years after Cancer Treatment. Diagnostics 2023, 13, 140. https://doi.org/10.3390/diagnostics13010140
Ivanov MK, Brenner EV, Hodkevich AA, Dzyubenko VV, Krasilnikov SE, Mansurova AS, Vakhturova IE, Agletdinov EF, Shumeikina AO, Chernyshova AL, et al. Cervicovaginal-Microbiome Analysis by 16S Sequencing and Real-Time PCR in Patients from Novosibirsk (Russia) with Cervical Lesions and Several Years after Cancer Treatment. Diagnostics. 2023; 13(1):140. https://doi.org/10.3390/diagnostics13010140
Chicago/Turabian StyleIvanov, Mikhail K., Evgeny V. Brenner, Anastasia A. Hodkevich, Victoria V. Dzyubenko, Sergey E. Krasilnikov, Alphiya S. Mansurova, Irina E. Vakhturova, Eduard F. Agletdinov, Anastasia O. Shumeikina, Alyona L. Chernyshova, and et al. 2023. "Cervicovaginal-Microbiome Analysis by 16S Sequencing and Real-Time PCR in Patients from Novosibirsk (Russia) with Cervical Lesions and Several Years after Cancer Treatment" Diagnostics 13, no. 1: 140. https://doi.org/10.3390/diagnostics13010140