
Imaging More than Skin-Deep: Radiologic and Dermatologic Presentations of Systemic Disorders

 ,
,  , and
, and
Abstract
:1. Introduction
2. Autoimmune/Inflammatory Disorders and Vasculitides
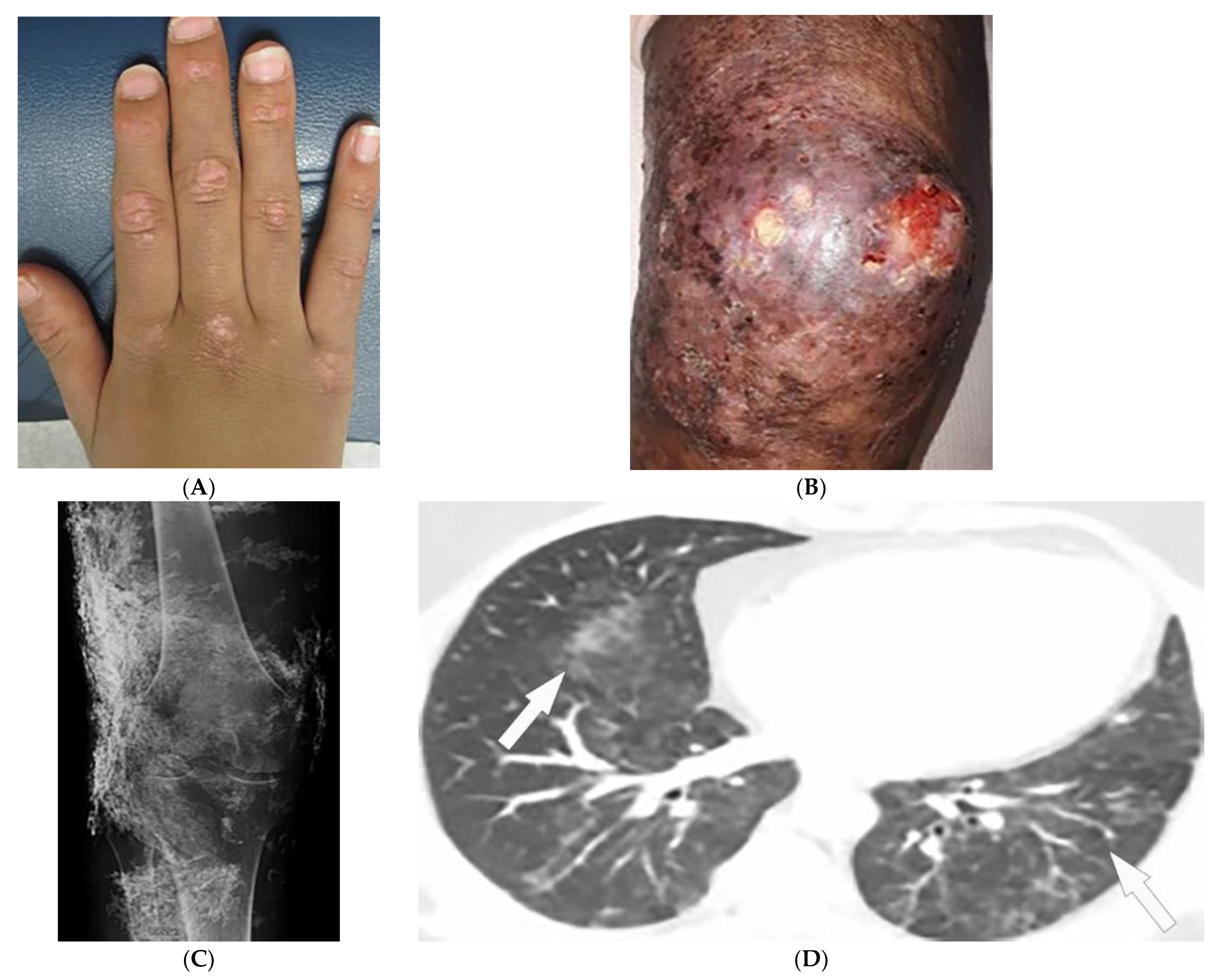
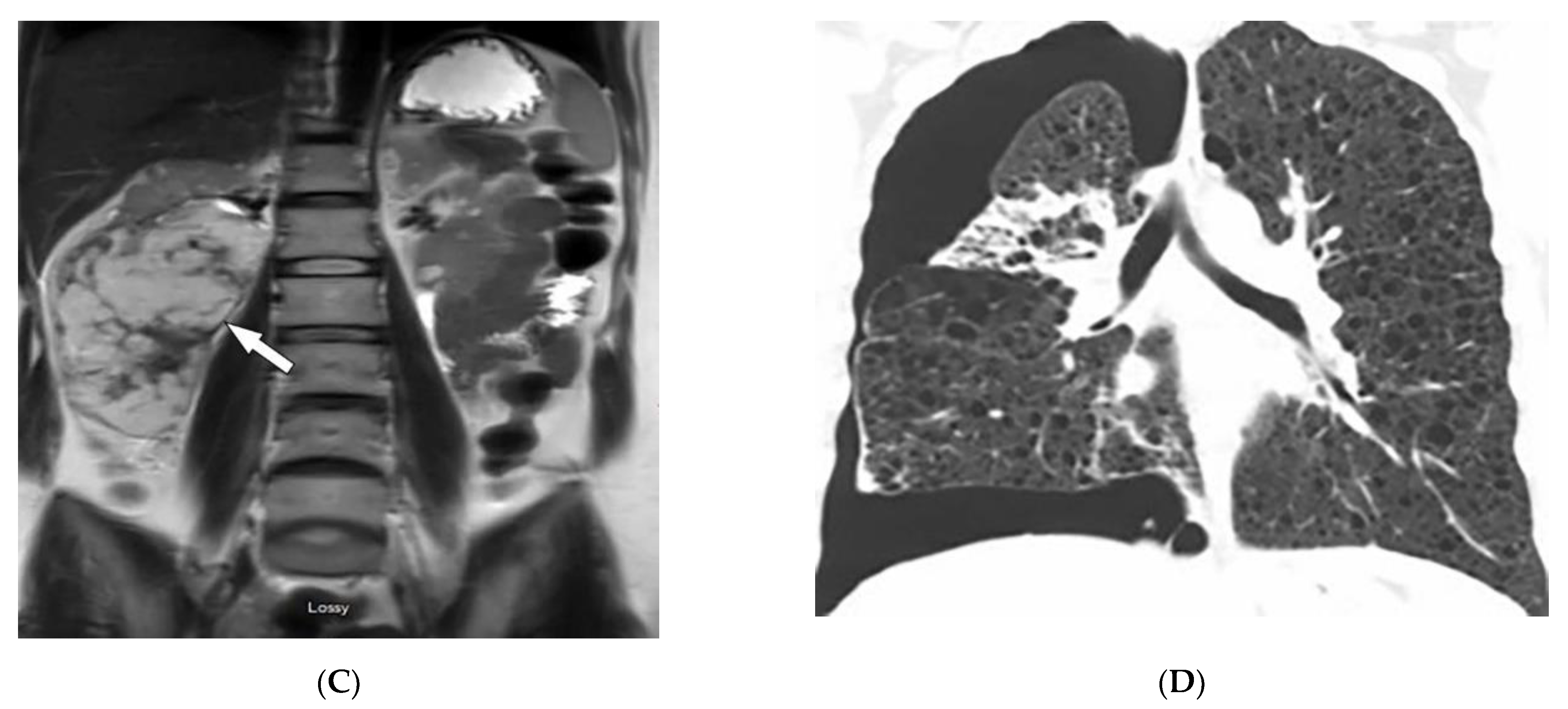
2.1. Dermatomyositis
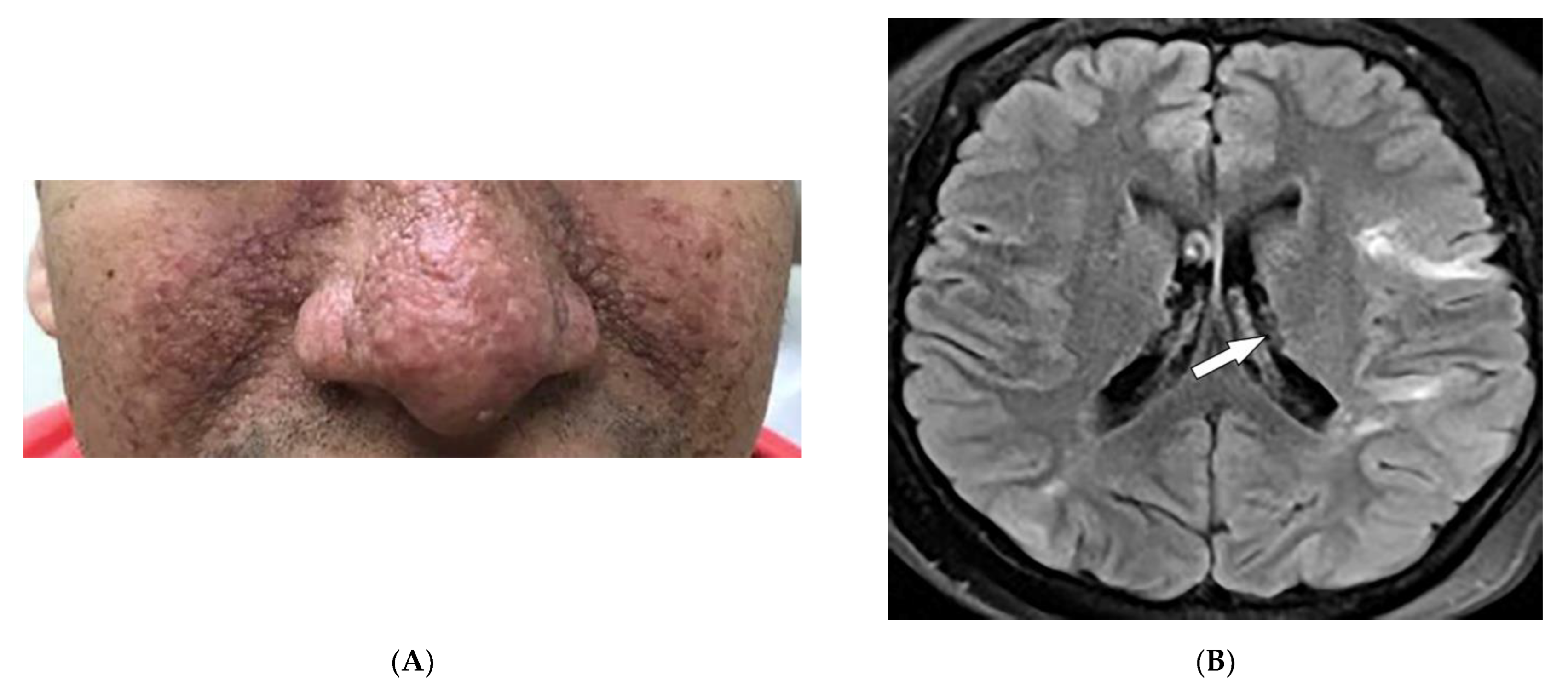
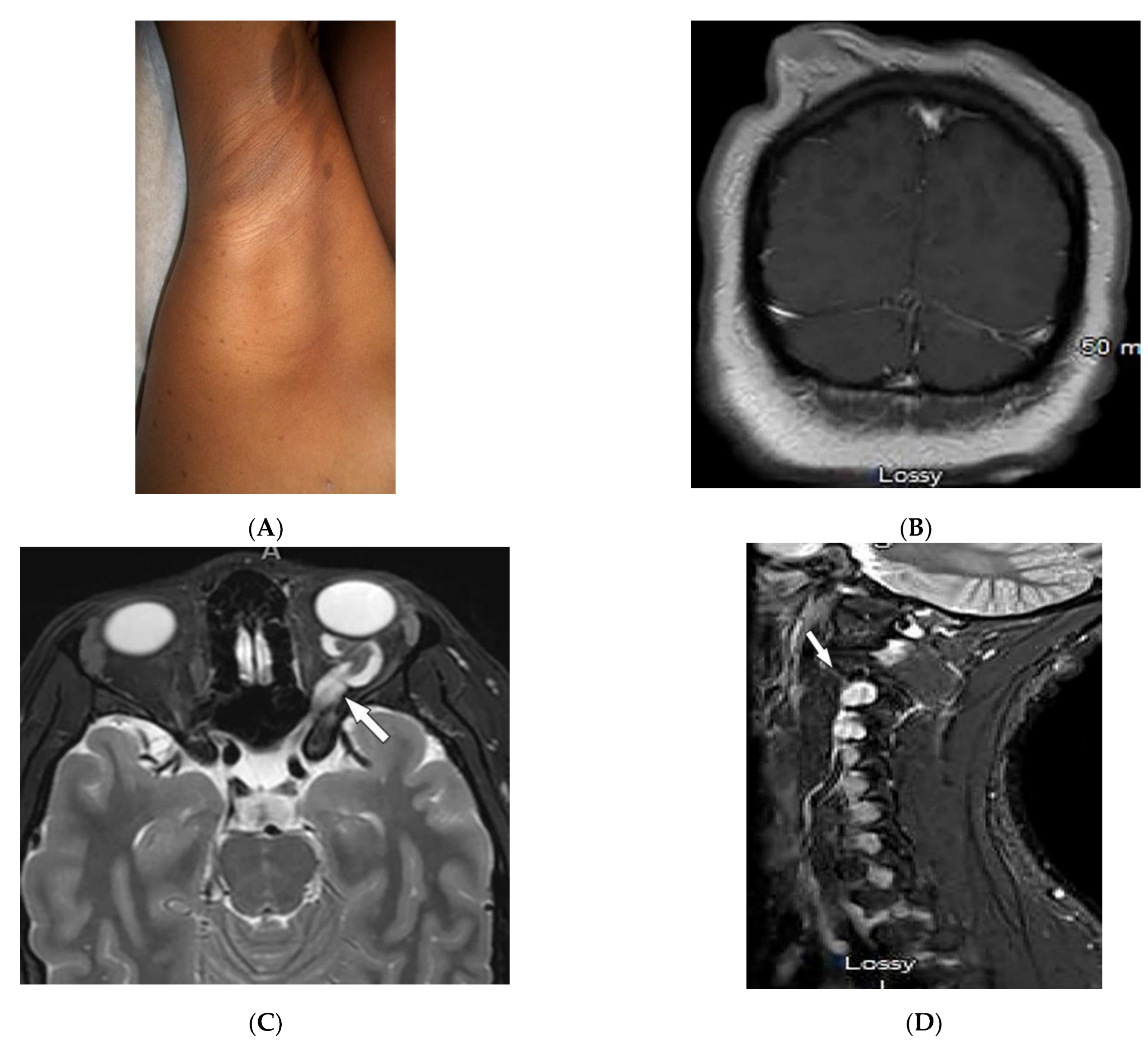
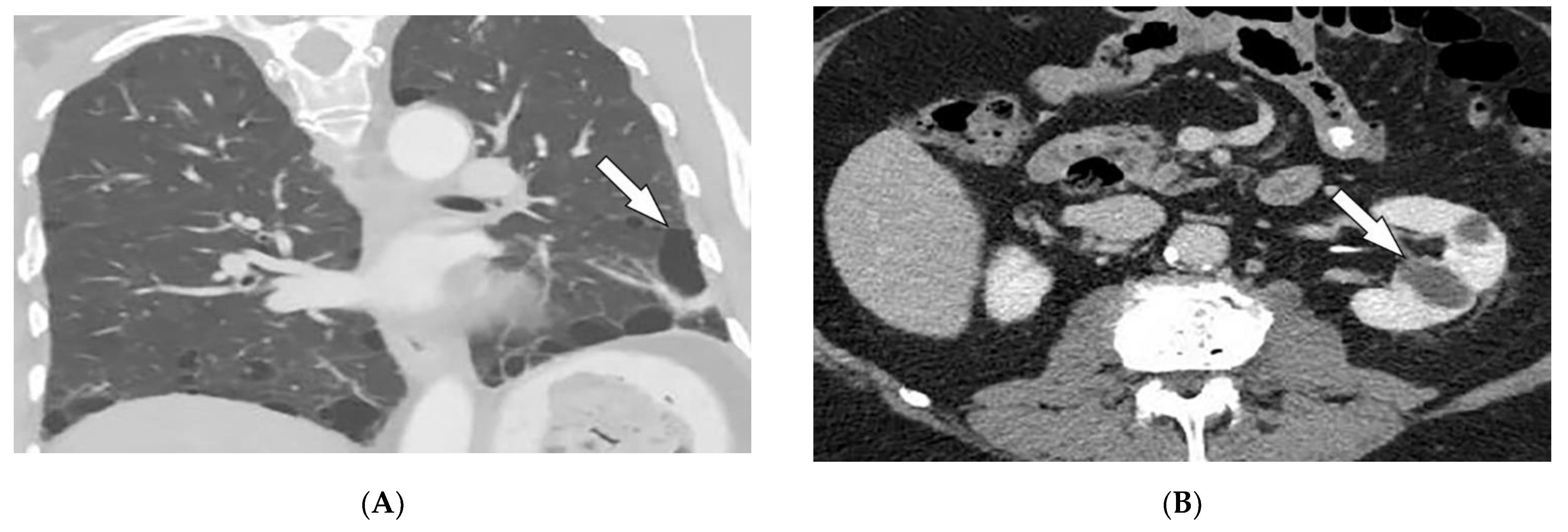
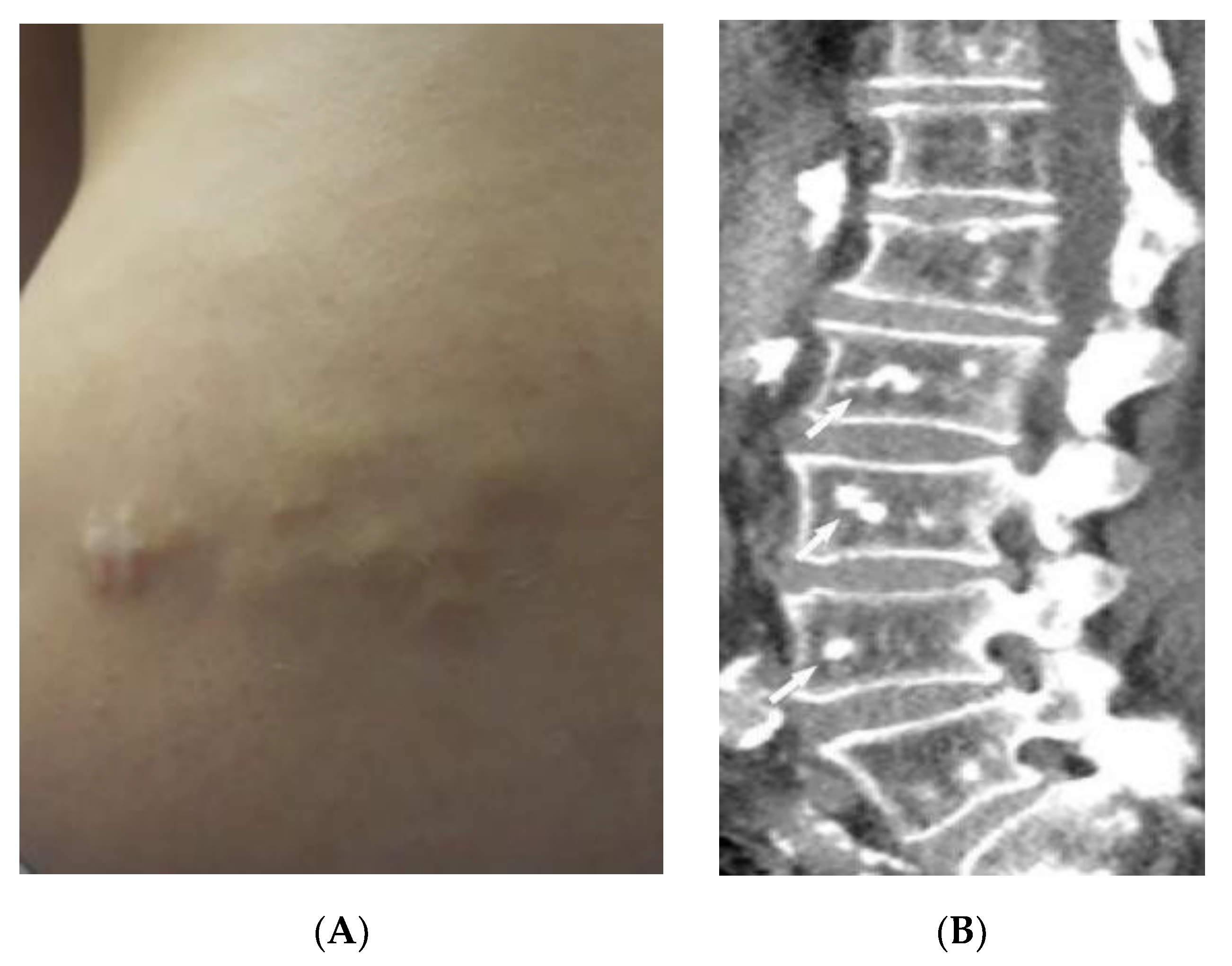
2.2. Sarcoidosis
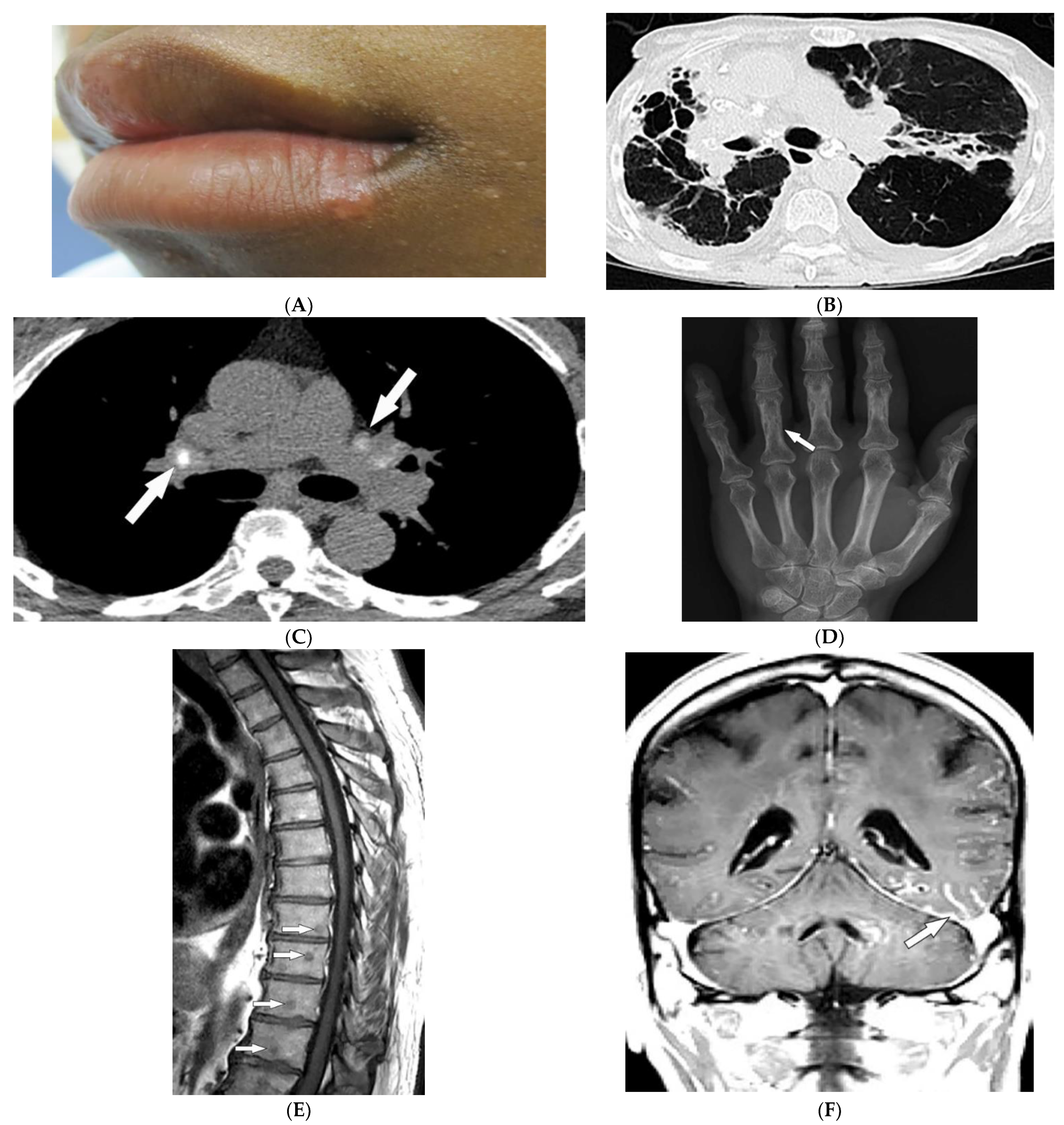
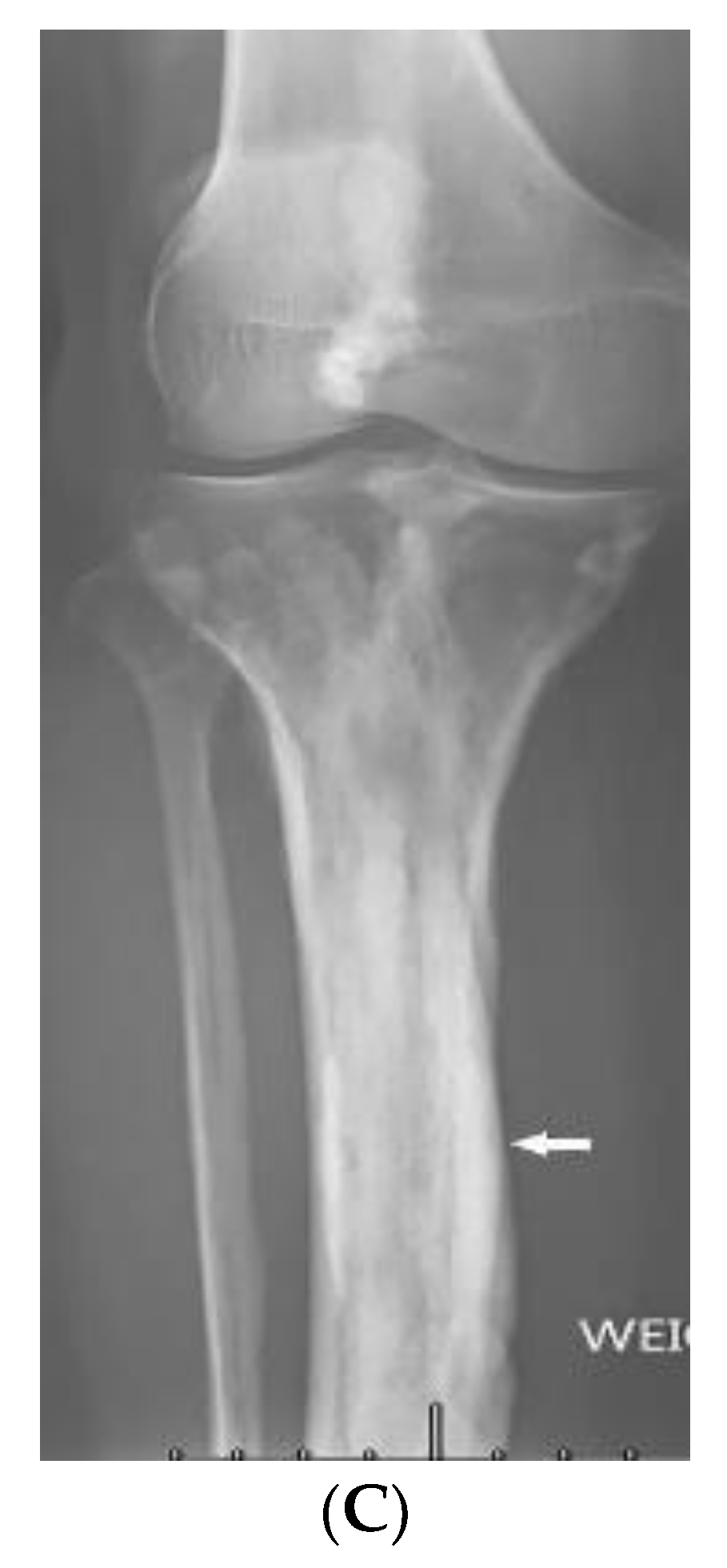
2.3. Scleroderma
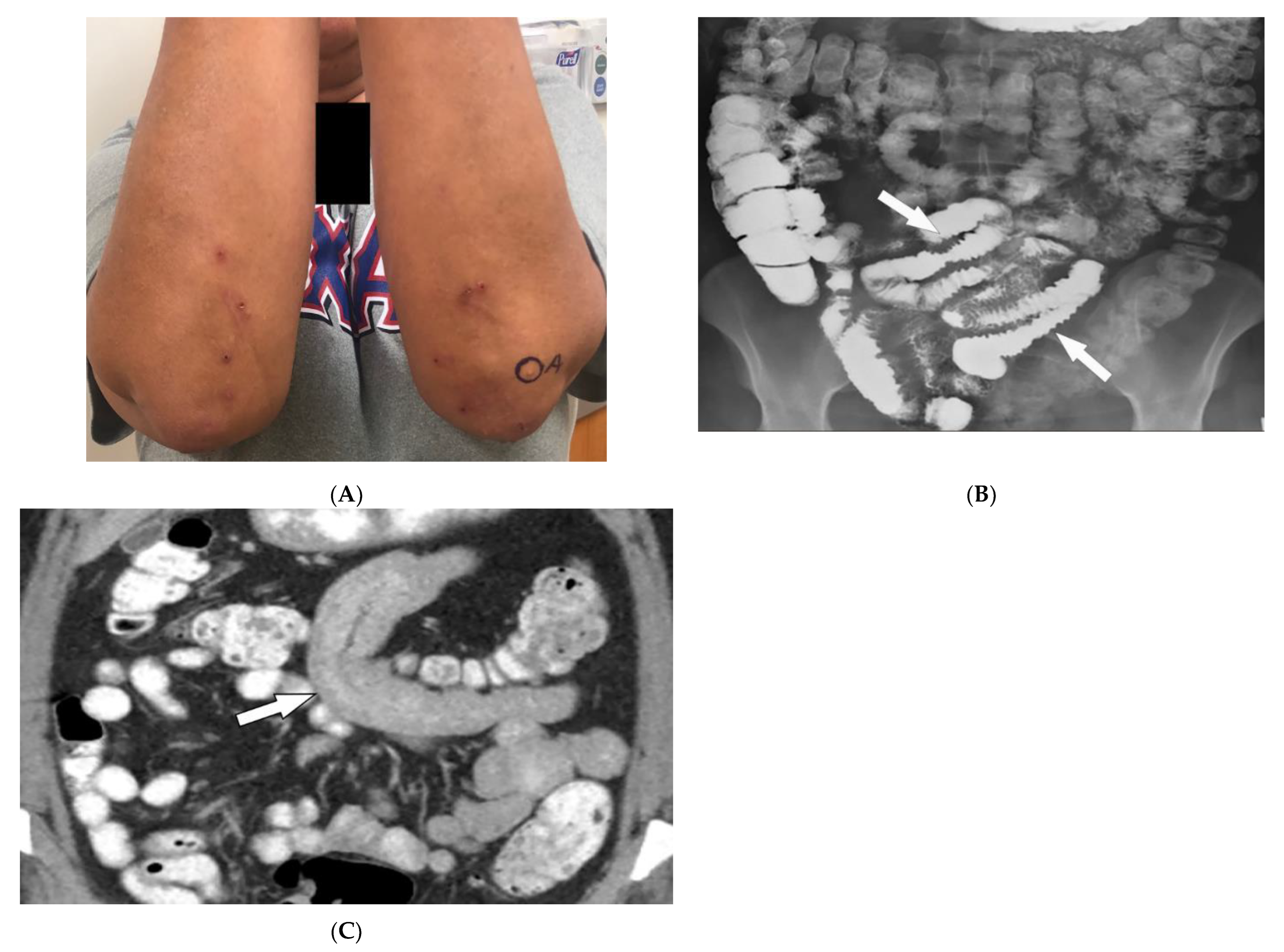
2.4. Celiac Disease
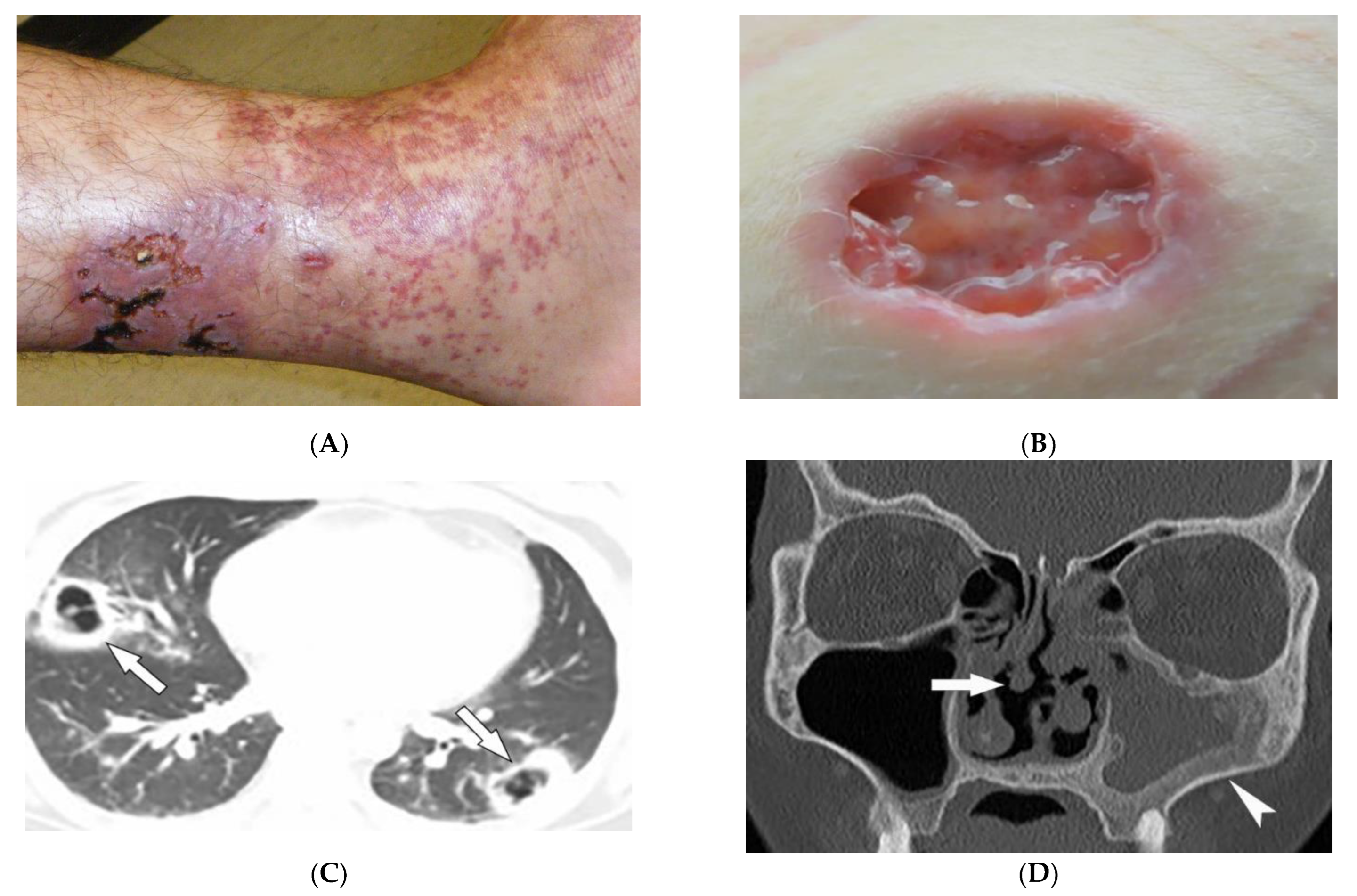
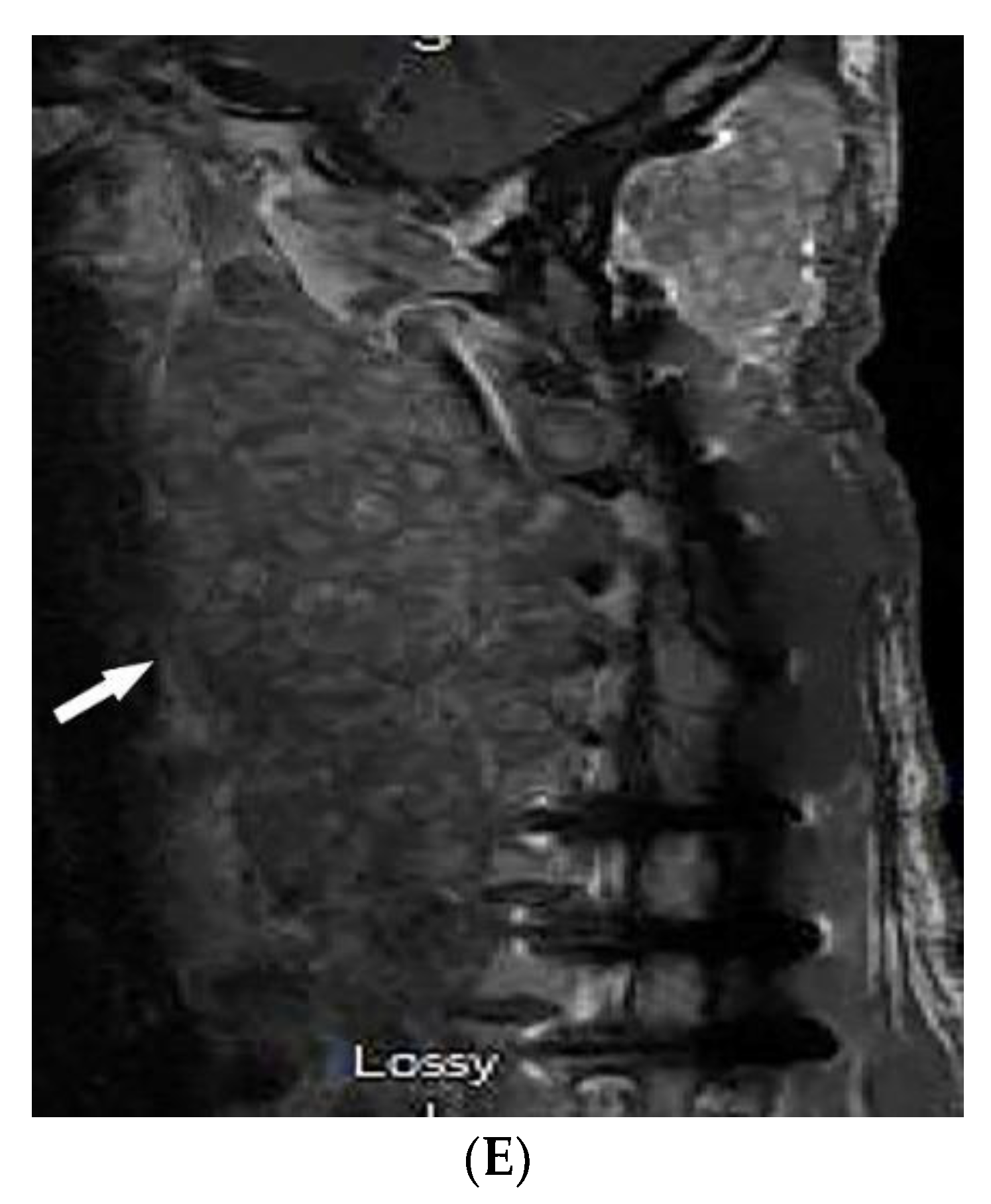
2.5. Granulomatosis with Polyangiitis
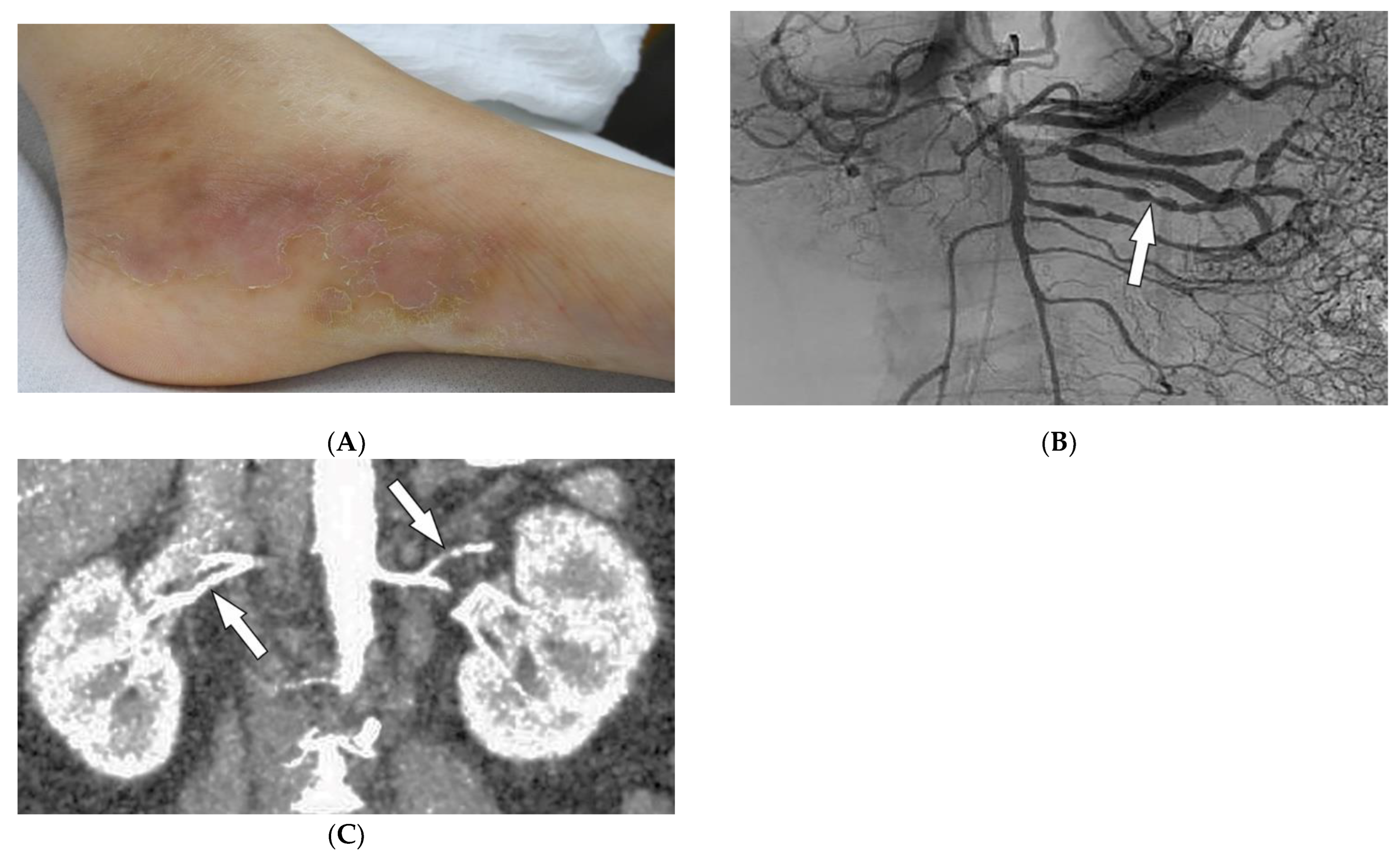
2.6. Polyarteritis Nodosa
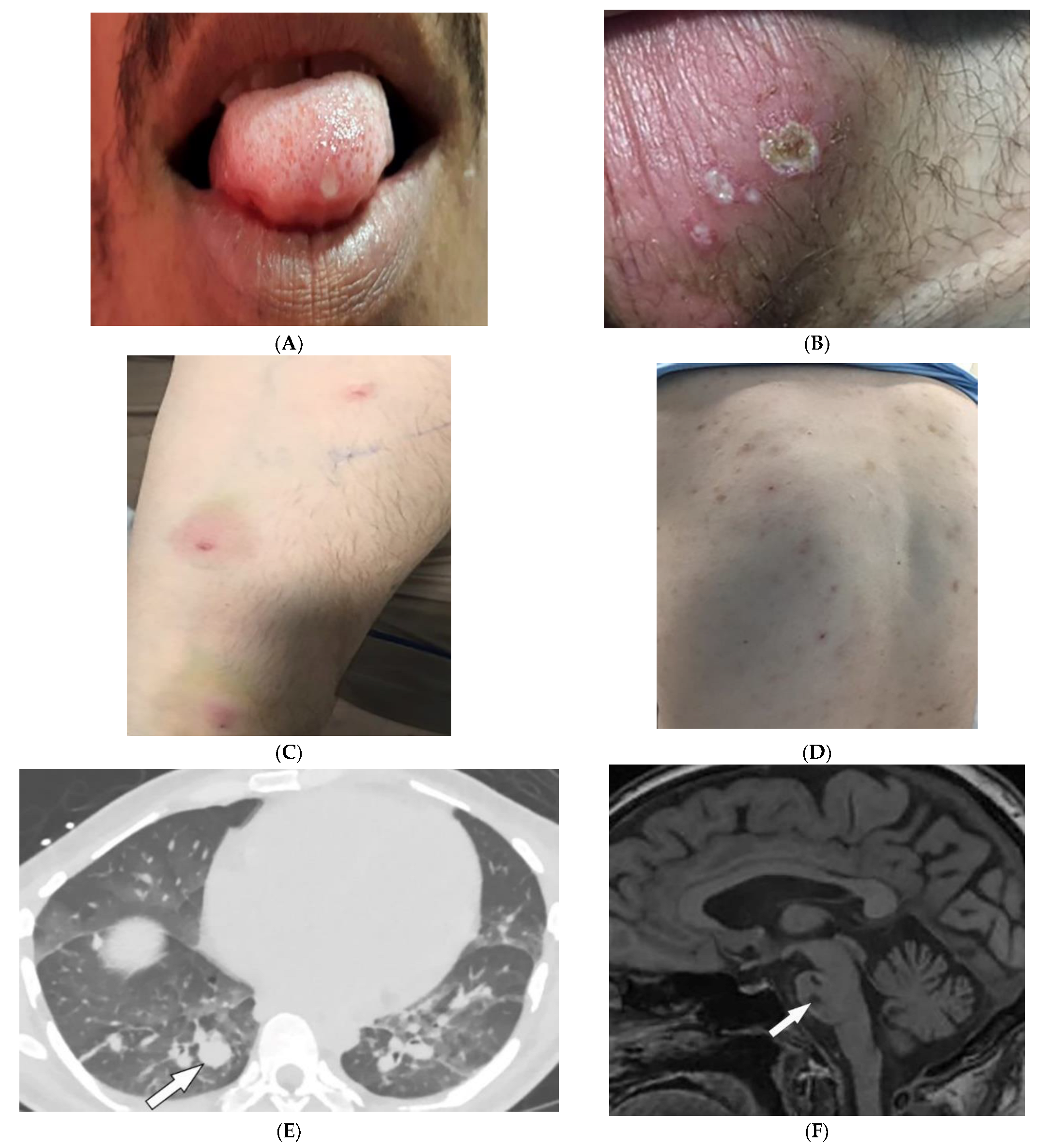
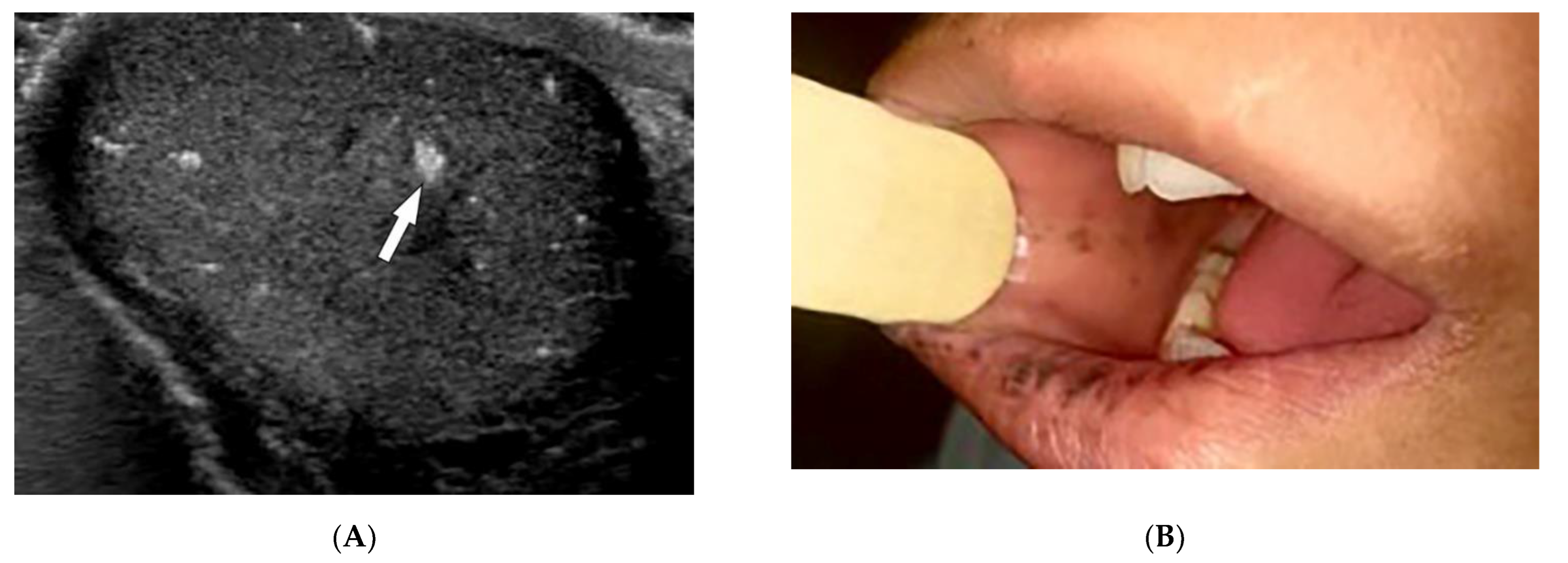
2.7. Behcet’s Disease
3. Genetic/Congenital Disorders
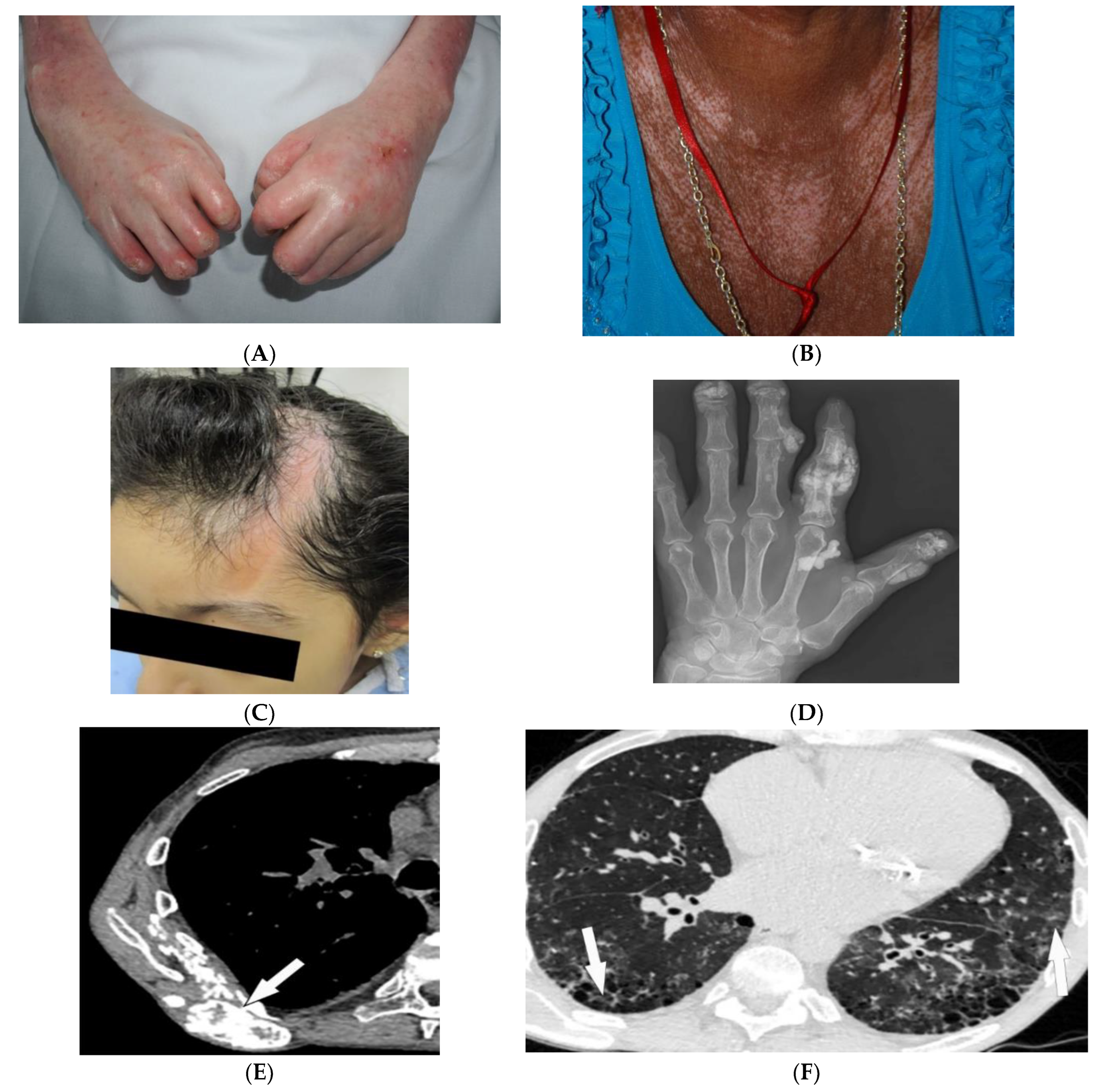
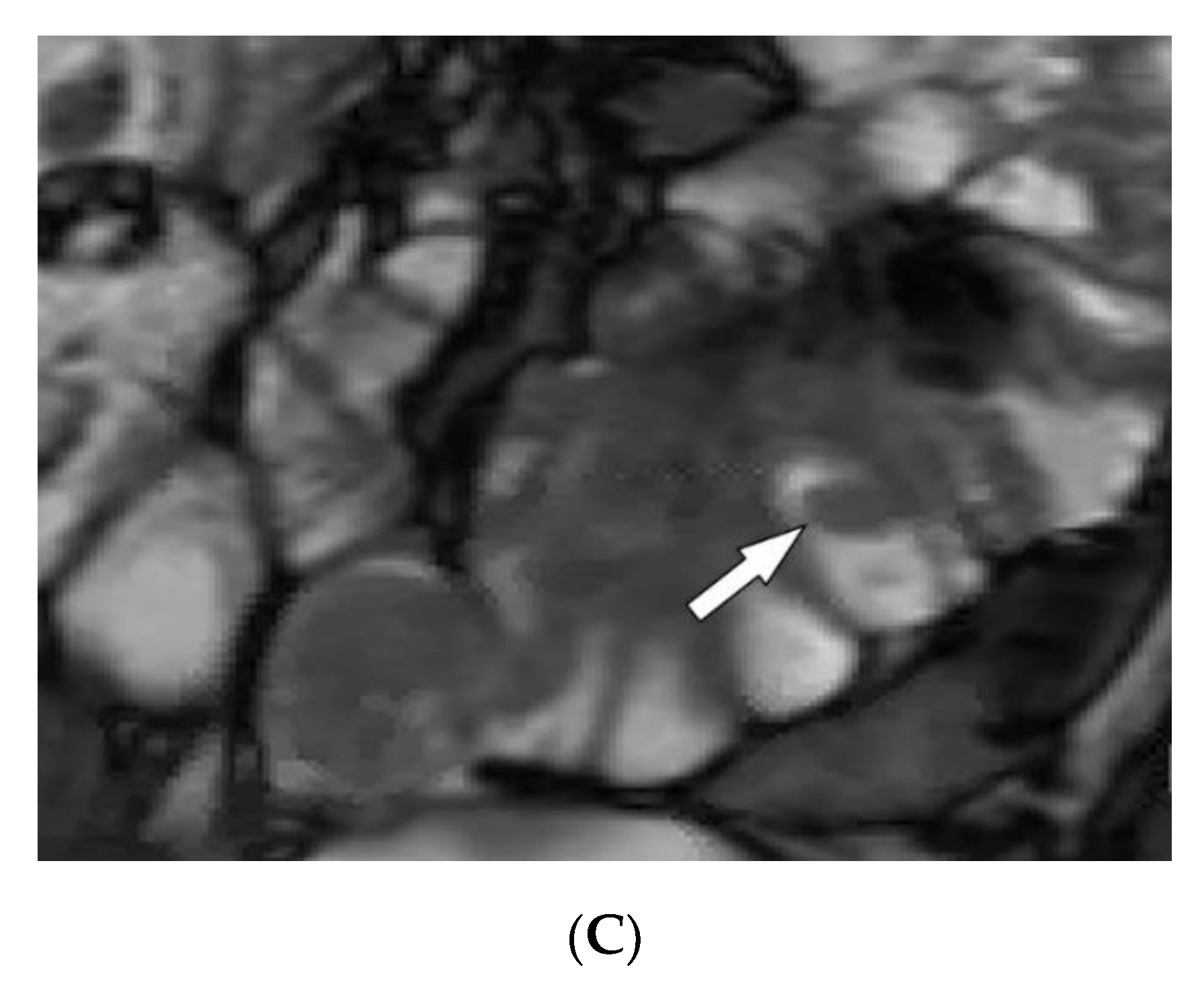
3.1. Tuberous Sclerosis Complex
3.2. Neurofibromatosis Type 1
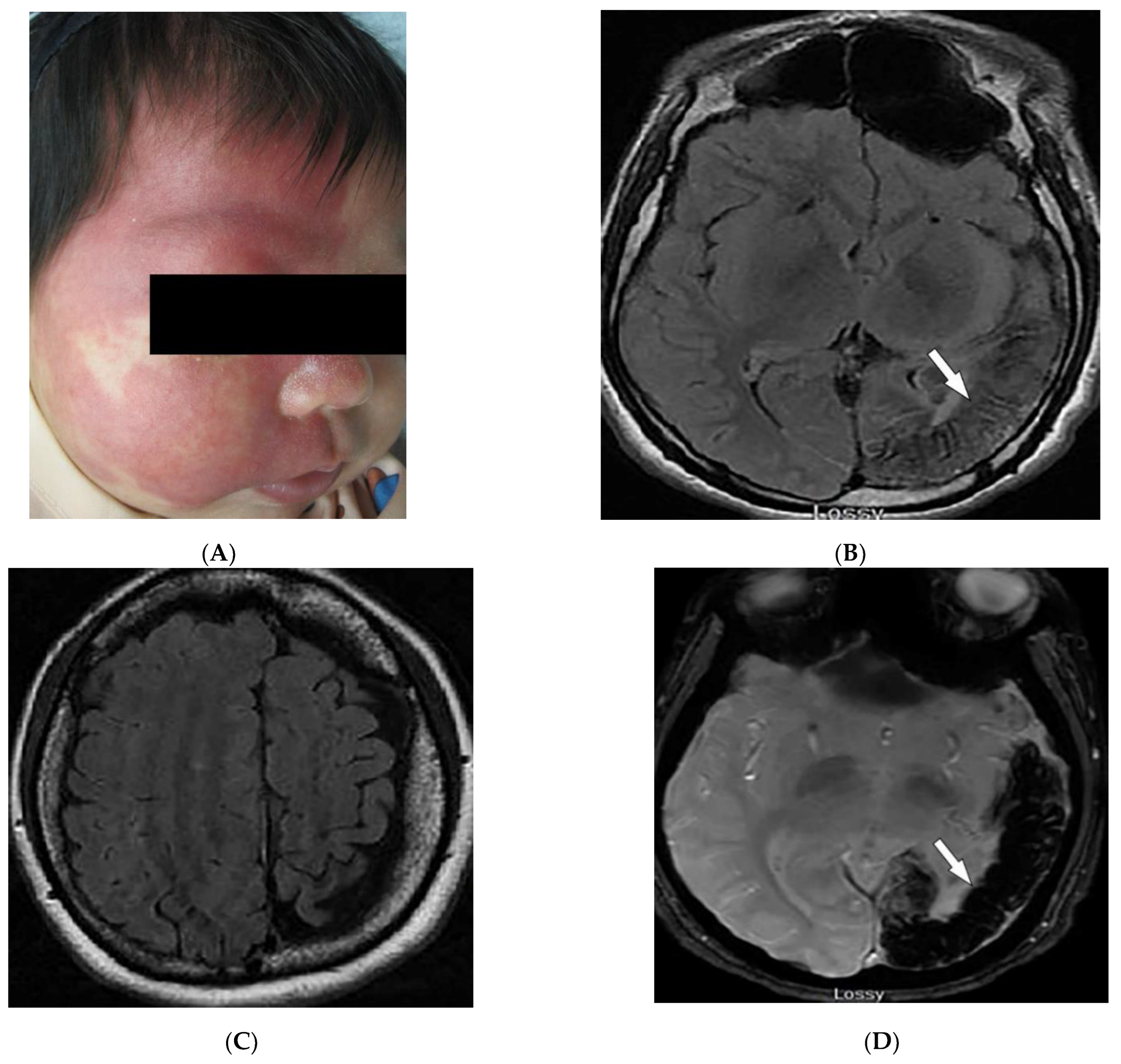
3.3. Sturge–Weber Syndrome
3.4. PHACES Syndrome
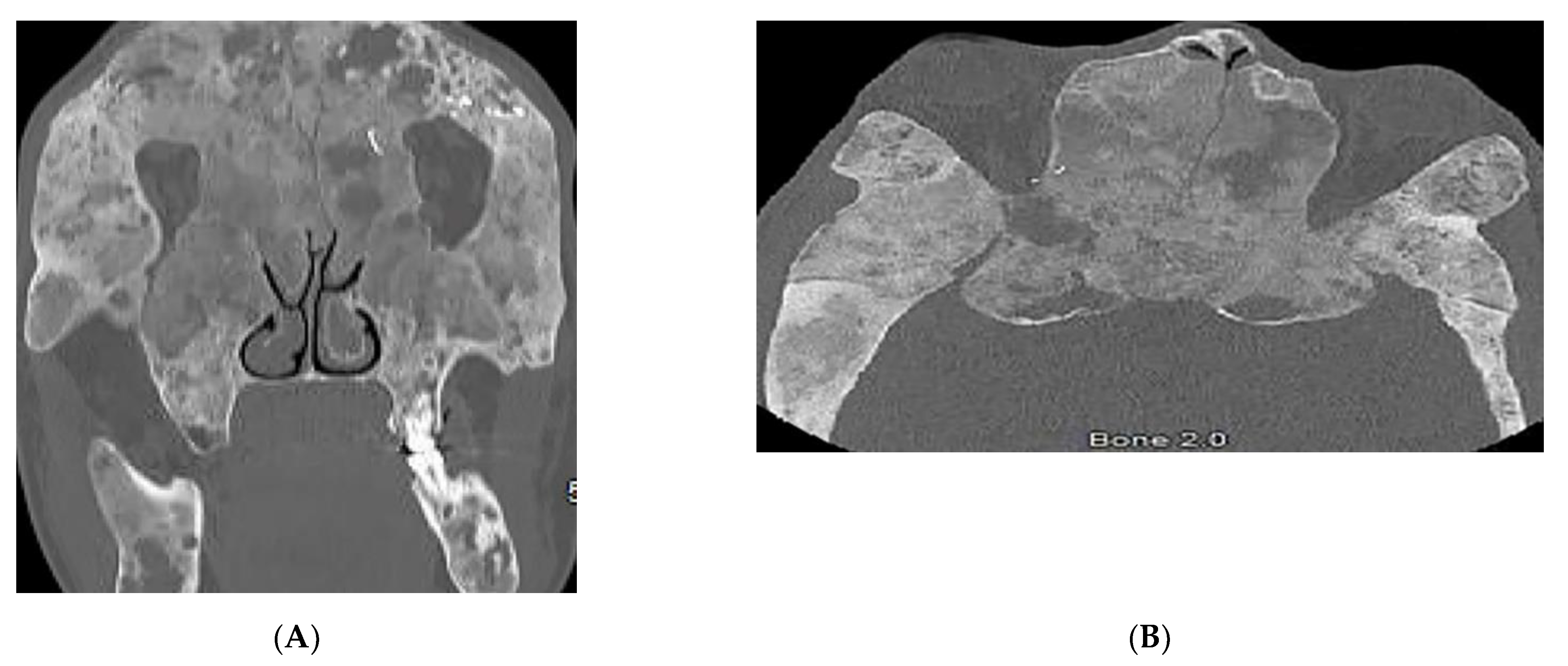
3.5. Nevoid BCC Syndrome (NBCCS)
3.6. Hereditary Hemorrhagic Telangiectasia
3.7. Birt–Hogg–Dube’ Syndrome
3.8. McCune–Albright Syndrome
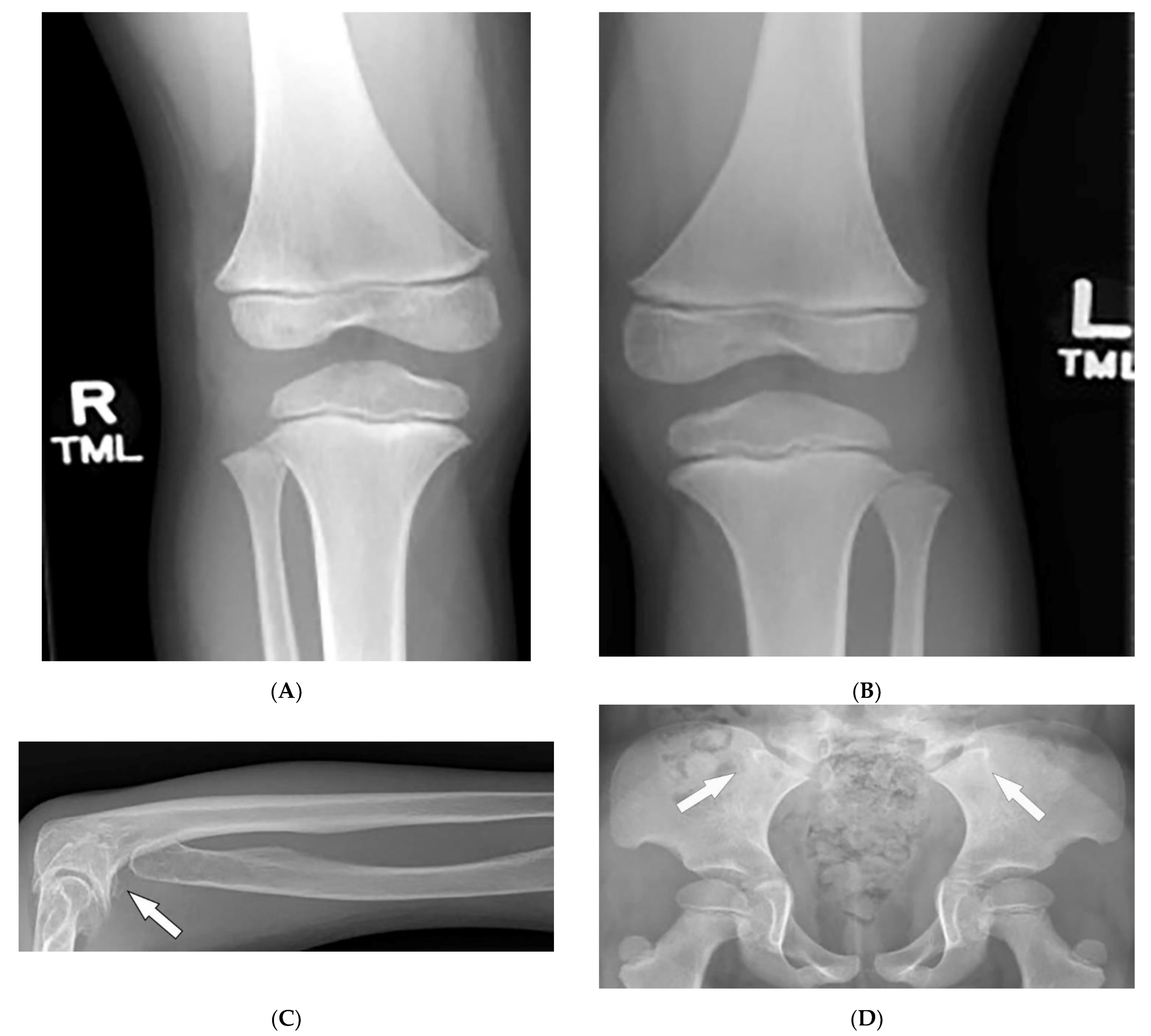
3.9. Fong Disease
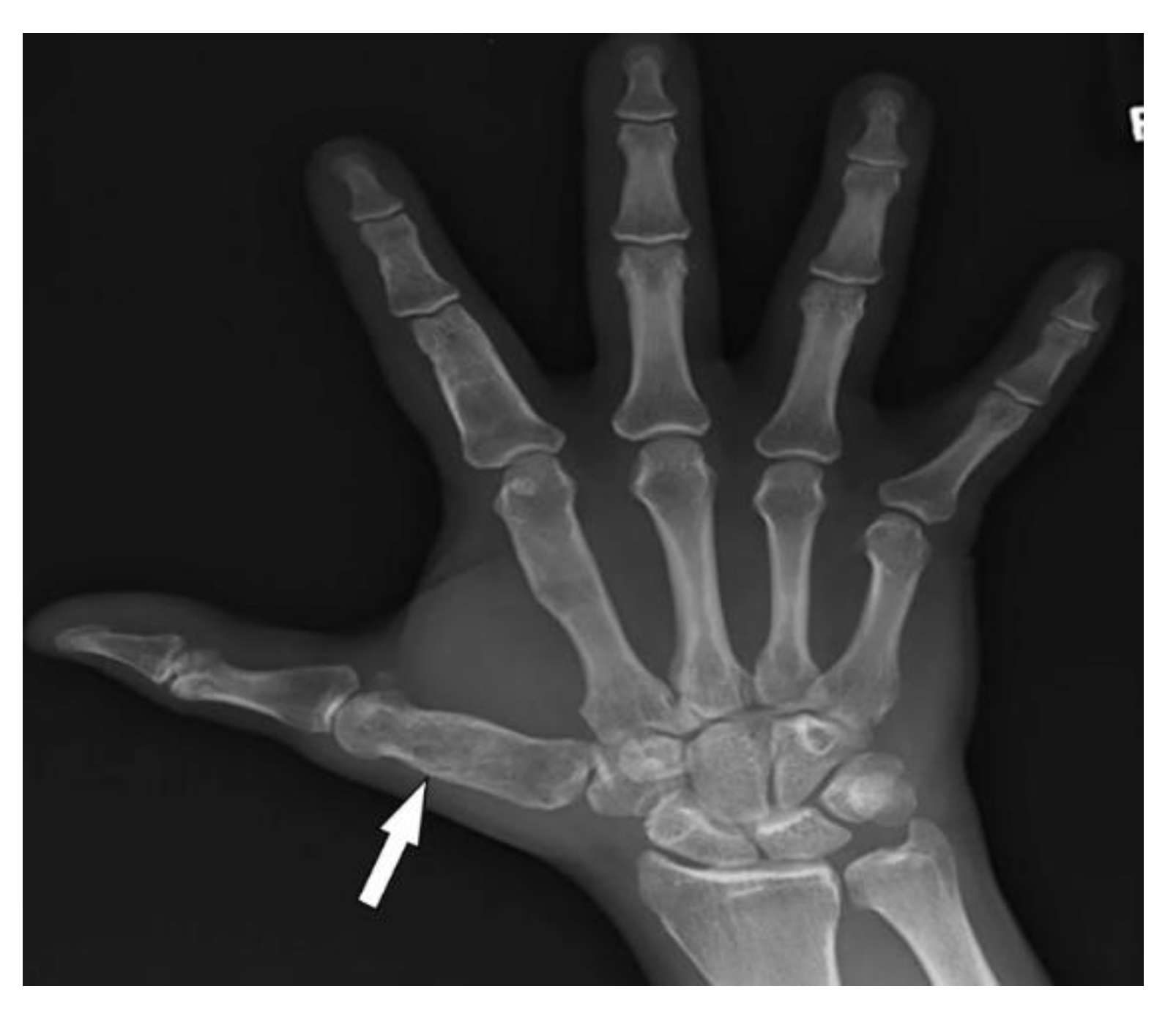
3.10. Maffucci Syndrome
3.11. Buschke–Ollendorff Syndrome
3.12. Peutz–Jeghers Syndrome (PJS)
4. Neoplasms
4.1. Melanotic Melanoma
4.2. Kaposi Sarcoma
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wei, W.; Ehlerding, E.B.; Lan, X.; Luo, Q.; Cai, W. PET and SPECT imaging of melanoma: The state of the art. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 132–150. [Google Scholar] [CrossRef] [PubMed]
- Fink, A.Z.; Gittler, J.K.; Nakrani, R.N.; Alis, J.; Blumfield, E.; Levin, T.L. Imaging findings in systemic childhood diseases presenting with dermatologic manifestations. Clin. Imaging 2018, 49, 17–36. [Google Scholar] [CrossRef] [PubMed]
- Kanne, J.P.; Donald, R.; Yandow, I.; Haemel, A.K.; Meyer, C.A. Beyond Skin Deep: Thoracic Manifestations of Systemic Disorders Affecting the Skin. RadioGraphics 2011, 31, 1651–1668. [Google Scholar] [CrossRef] [PubMed]
- Kolasinski, S.L.; Chi, A.S.; Lopez-Garib, A.J. Current Perspectives on Imaging for Systemic Lupus Erythematosus, Systemic Sclerosis, and Dermatomyositis/Polymyositis. Rheum. Dis. Clin. N. Am. 2016, 42, 711–732. [Google Scholar] [CrossRef] [PubMed]
- Mainetti, C.; Terziroli Beretta-Piccoli, B.; Selmi, C. Cutaneous Manifestations of Dermatomyositis: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2017, 53, 337–356. [Google Scholar] [CrossRef]
- Ahuja, J.; Arora, D.; Kanne, J.P.; Henry, T.S.; Godwin, J.D. Imaging of Pulmonary Manifestations of Connective Tissue Diseases. Radiol. Clin. N. Am. 2016, 54, 1015–1031. [Google Scholar] [CrossRef]
- Kamperman, R.G.; van der Kooi, A.J.; de Visser, M.; Aronica, E.; Raaphorst, J. Pathophysiological Mechanisms and Treatment of Dermatomyositis and Immune Mediated Necrotizing Myopathies: A Focused Review. Int. J. Mol. Sci. 2022, 23, 4301. [Google Scholar] [CrossRef]
- Ran, J.; Dai, B.; Liu, C.; Zhang, H.; Li, Y.; Hou, B.; Li, X. The diagnostic value of T2 map, diffusion tensor imaging, and diffusion kurtosis imaging in differentiating dermatomyositis from muscular dystrophy. Acta Radiol. 2022, 63, 467–473. [Google Scholar] [CrossRef]
- Kubinova, K.; Dejthevaporn, R.; Mann, H.; Machado, P.M.; Vencovsky, J. The role of imaging in evaluating patients with idiopathic inflammatory myopathies. Clin. Exp. Rheumatol. 2018, 36 (Suppl. S114), 74–81. [Google Scholar]
- Schulze, M.; Kötter, I.; Ernemann, U.; Fenchel, M.; Tzaribatchev, N.; Claussen, C.D.; Horger, M. MRI Findings in Inflammatory Muscle Diseases and Their Noninflammatory Mimics. Am. J. Roentgenol. 2009, 192, 1708–1716. [Google Scholar] [CrossRef]
- Smitaman, E.; Flores, D.V.; Gómez, C.M.; Pathria, M.N. MR Imaging of Atraumatic Muscle Disorders. Radiogr. A Rev. Publ. Radiol. Soc. N. Am. Inc. 2018, 38, 500–522. [Google Scholar] [CrossRef] [PubMed]
- Belperio, J.A.; Shaikh, F.; Abtin, F.G.; Fishbein, M.C.; Weigt, S.S.; Saggar, R.; Lynch, J.P. Diagnosis and treatment of pulmonary sarcoidosis: A review. JAMA 2022, 327, 856–867. [Google Scholar] [CrossRef] [PubMed]
- Guidry, C.; Fricke, R.G.; Ram, R.; Pandey, T.; Jambhekar, K. Imaging of Sarcoidosis: A Contemporary Review. Radiol. Clin. N. Am. 2016, 54, 519–534. [Google Scholar] [CrossRef] [PubMed]
- Ganeshan, D.; Menias, C.O.; Lubner, M.G.; Pickhardt, P.J.; Sandrasegaran, K.; Bhalla, S. Sarcoidosis from Head to Toe: What the Radiologist Needs to Know. Radiogr. Rev. Publ. Radiol. Soc. N. Am. Inc. 2018, 38, 1180–1200. [Google Scholar] [CrossRef]
- Lew, P.P.; Ngai, S.S.; Hamidi, R.; Cho, J.K.; Birnbaum, R.A.; Peng, D.H.; Varma, R.K. Imaging of Disorders Affecting the Bone and Skin. Radiogr. Rev. Publ. Radiol. Soc. N. Am. Inc. 2014, 34, 197–216. [Google Scholar] [CrossRef]
- Redissi, A.; Penmetsa, G.K.; Litaiem, N. Lupus pernio. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Noe, M.H.; Rosenbach, M. Cutaneous sarcoidosis. Curr. Opin. Pulm. Med. 2017, 23, 482–486. [Google Scholar] [CrossRef]
- Koyama, T.; Ueda, H.; Togashi, K.; Umeoka, S.; Kataoka, M.; Nagai, S. Radiologic Manifestations of Sarcoidosis in Various Organs. Radiogr. Rev. Publ. Radiol. Soc. N. Am. Inc. 2004, 24, 87–104. [Google Scholar] [CrossRef]
- Sève, P.; Pacheco, Y.; Durupt, F.; Jamilloux, Y.; Gerfaud-Valentin, M.; Isaac, S.; Boussel, L.; Calender, A.; Androdias, G.; Valeyre, D.; et al. Sarcoidosis: A clinical overview from symptoms to diagnosis. Cells 2021, 10, 766. [Google Scholar] [CrossRef]
- Ruaro, B.; Confalonieri, P.; Santagiuliana, M.; Wade, B.; Baratella, E.; Kodric, M.; Berria, M.; Jaber, M.; Torregiani, C.; Bruni, C.; et al. Correlation between Potential Risk Factors and Pulmonary Embolism in Sarcoidosis Patients Timely Treated. J. Clin. Med. 2021, 10, 2462. [Google Scholar] [CrossRef]
- Barreras, P.; Stern, B.J. Clinical features and diagnosis of neurosarcoidosis–review article. J. Neuroimmunol. 2022, 368, 577871. [Google Scholar] [CrossRef]
- Chapin, R.; Hant, F.N. Imaging of scleroderma. Rheum. Dis. Clin. N. Am. 2013, 39, 515–546. [Google Scholar] [CrossRef] [PubMed]
- Kowalska-Kępczyńska, A. Systemic Scleroderma—Definition, Clinical Picture and Laboratory Diagnostics. J. Clin. Med. 2022, 11, 2299. [Google Scholar] [CrossRef] [PubMed]
- Abbas, L.F.; O’Brien, J.C.; Goldman, S.; Pezeshk, P.; Chalian, M.; Chhabra, A.; Jacobe, H.T. A Cross-sectional Comparison of Magnetic Resonance Imaging Findings and Clinical Assessment in Patients With Morphea. JAMA Dermatol. 2020, 156, 590–592. [Google Scholar] [CrossRef] [PubMed]
- Ruaro, B.; Sulli, A.; Pizzorni, C.; Paolino, S.; Smith, V.; Alessandri, E.; Trombetta, A.C.; Alsheyyab, J.; Cutolo, M. Correlations between blood perfusion and dermal thickness in different skin areas of systemic sclerosis patients. Microvasc. Res. 2018, 115, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Sulli, A.; Ruaro, B.; Cutolo, M. Evaluation of blood perfusion by laser speckle contrast analysis in different areas of hands and face in patients with systemic sclerosis. Ann. Rheum. Dis. 2014, 73, 2059–2061. [Google Scholar] [CrossRef]
- Dzwigala, M.; Sobolewski, P.; Maslinska, M.; Yurtsever, I.; Szymanska, E.; Walecka, I. High-resolution ultrasound imaging of skin involvement in systemic sclerosis: A systematic review. Rheumatol. Int. 2021, 41, 285–295. [Google Scholar] [CrossRef]
- Hughes, M.; Bruni, C.; Cuomo, G.; Delle Sedie, A.; Gargani, L.; Gutierrez, M.; Lepri, G.; Ruaro, B.; Santiago, T.; Suliman, Y.; et al. The role of ultrasound in systemic sclerosis: On the cutting edge to foster clinical and research advancement. J. Scleroderma Relat. Disord. 2021, 6, 123–132. [Google Scholar] [CrossRef]
- Chatzinikolaou, S.L.; Quirk, B.; Murray, C.; Planche, K. Radiological findings in gastrointestinal scleroderma. J. Scleroderma Relat. Disord. 2020, 5, 21–32. [Google Scholar] [CrossRef]
- Abenavoli, L.; Dastoli, S.; Bennardo, L.; Boccuto, L.; Passante, M.; Silvestri, M.; Proietti, I.; Potenza, C.; Luzza, F.; Nisticò, S.P. The Skin in Celiac Disease Patients: The Other Side of the Coin. Medicina 2019, 55, 578. [Google Scholar] [CrossRef]
- Tennyson, C.A.; Semrad, C.E. Small bowel imaging in celiac disease. Gastrointest. Endosc. Clin. N. Am. 2012, 22, 735–746. [Google Scholar] [CrossRef]
- Sheedy, S.P.; Barlow, J.M.; Fletcher, J.G.; Smyrk, T.C.; Scholz, F.J.; Codipilly, D.C.; Al Bawardy, B.F.; Fidler, J.L. Beyond moulage sign and TTG levels: The role of cross-sectional imaging in celiac sprue. Abdom. Radiol. 2017, 42, 361–388. [Google Scholar] [CrossRef] [PubMed]
- Paolantonio, P.; Tomei, E.; Rengo, M.; Ferrari, R.; Lucchesi, P.; Laghi, A. Adult celiac disease: MRI findings. Abdom. Imaging 2007, 32, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Marzano, A.V.; Raimondo, M.G.; Berti, E.; Meroni, P.L.; Ingegnoli, F. Cutaneous Manifestations of ANCA-Associated Small Vessels Vasculitis. Clin. Rev. Allergy Immunol. 2017, 53, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Singhal, M.; Gupta, P.; Sharma, A. Imaging in small and medium vessel vasculitis. Int. J. Rheum. Dis. 2019, 22 (Suppl. S1), 78–85. [Google Scholar] [CrossRef]
- Schmidt, W.A. Imaging in vasculitis. Best Pract. Res. Clin. Rheumatol. 2013, 27, 107–118. [Google Scholar] [CrossRef]
- Howard, T.; Ahmad, K.; Swanson, J.A.; Misra, S. Polyarteritis nodosa. Tech. Vasc. Interv. Radiol. 2014, 17, 247–251. [Google Scholar] [CrossRef]
- Chasset, F.; Frances, C. Cutaneous Manifestations of Medium- and Large-Vessel Vasculitis. Clin. Rev. Allergy Immunol. 2017, 53, 452–468. [Google Scholar] [CrossRef]
- Diaz-Perez, J.L.; De Lagran, Z.M.; Diaz-Ramon, J.L.; Winkelmann, R.K. Cutaneous polyarteritis nodosa. Semin. Cutan. Med. Surg. 2007, 26, 77–86. [Google Scholar] [CrossRef]
- Mehdipoor, G.; Davatchi, F.; Ghoreishian, H.; Arjmand Shabestari, A. Imaging manifestations of Behcet’s disease: Key considerations and major features. Eur. J. Radiol. 2018, 98, 214–225. [Google Scholar] [CrossRef]
- Elbendary, A.; Abdel-Halim, M.R.; Ragab, G. Updates in cutaneous manifestations of systemic vasculitis. Curr. Opin. Rheumatol. 2022, 34, 25–32. [Google Scholar] [CrossRef]
- Chae, E.J.; Do, K.-H.; Seo, J.B.; Park, S.H.; Kang, J.-W.; Jang, Y.M.; Lee, J.S.; Song, J.-W.; Song, K.-S.; Lee, J.H.; et al. Radiologic and Clinical Findings of Behçet Disease: Comprehensive Review of Multisystemic Involvement. Radiogr. Rev. Publ. Radiol. Soc. N. Am. Inc. 2008, 28, e31. [Google Scholar] [CrossRef] [PubMed]
- Vural, S.; Boyvat, A. The skin in Behçet’s disease: Mucocutaneous findings and differential diagnosis. JEADV Clin. Pract. 2022, 1, 11–20. [Google Scholar] [CrossRef]
- Krishnan, A.; Kaza, R.K.; Vummidi, D.R. Cross-sectional Imaging Review of Tuberous Sclerosis. Radiol. Clin. N. Am. 2016, 54, 423–440. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.D.; DarConte, M.D.; Hebert, A.A. The cutaneous manifestations of tuberous sclerosis complex. Am. J. Med. Genet. Part C Semin. Med. Genet. 2018, 178, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Umeoka, S.; Koyama, T.; Miki, Y.; Akai, M.; Tsutsui, K.; Togashi, K. Pictorial review of tuberous sclerosis in various organs. Radiogr. A Rev. Publ. Radiol. Soc. N. Am. Inc. 2008, 28, e32. [Google Scholar] [CrossRef]
- Vézina, G. Neuroimaging of phakomatoses: Overview and advances. Pediatric Radiol. 2015, 45 (Suppl. S3), S433–S442. [Google Scholar] [CrossRef]
- Razek, A. MR imaging of neoplastic and non-neoplastic lesions of the brain and spine in neurofibromatosis type I. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2018, 39, 821–827. [Google Scholar] [CrossRef]
- Salamon, J.; Mautner, V.F.; Adam, G.; Derlin, T. Multimodal Imaging in Neurofibromatosis Type 1-associated Nerve Sheath Tumors. RoFo Fortschr. Auf Geb. Rontgenstrahlen Nukl. 2015, 187, 1084–1092. [Google Scholar] [CrossRef]
- Hernández-Martín, A.; Duat-Rodríguez, A. An Update on Neurofibromatosis Type 1: Not Just Café-au-Lait Spots, Freckling, and Neurofibromas. An Update. Part I. Dermatological Clinical Criteria Diagnostic of the Disease. Actas Dermo-Sifiliogr. 2016, 107, 454–464. [Google Scholar] [CrossRef]
- Abdel Razek, A.A. Vascular neurocutaneous disorders: Neurospinal and craniofacial imaging findings. Jpn. J. Radiol. 2014, 32, 519–528. [Google Scholar] [CrossRef]
- Puttgen, K.B.; Lin, D.D. Neurocutaneous vascular syndromes. Child’s nervous system. ChNS Off. J. Int. Soc. Pediatric Neurosurg. 2010, 26, 1407–1415. [Google Scholar] [CrossRef]
- Rotter, A.; Samorano, L.P.; Rivitti-Machado, M.C.; Oliveira, Z.N.P.; Gontijo, B. PHACE syndrome: Clinical manifestations, diagnostic criteria, and management. An. Bras. Dermatol. 2018, 93, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Barros, F.S.; Marussi, V.H.R.; Amaral, L.L.F.; Da Rocha, A.J.; Campos, C.M.; Freitas, L.F.; Huisman, T.A.G.M.; Soares, B.P. The Rare Neurocutaneous Disorders: Update on Clinical, Molecular, and Neuroimaging Features. Top. Magn. Reson. Imaging TMRI 2018, 27, 433–462. [Google Scholar] [CrossRef] [PubMed]
- Subramanyam, S.B.; Sujata, D.N.; Sridhar, K.; Pushpanjali, M. Nevoid Basal cell carcinoma syndrome: A case report and review. J. Maxillofac. Oral Surg. 2015, 14, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Dupuis-Girod, S.; Cottin, V.; Shovlin, C. The lung in hereditary hemorrhagic telangiectasia. Respiration 2017, 94, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Azma, R.; Dmytriw, A.A.; Biswas, A.; Pollak, M.; Ratjen, F.; Amirabadi, A.; Branson, H.M.; Kulkarni, A.V.; Dirks, P.; Muthusami, P. Neurovascular Manifestations in Pediatric Patients With Hereditary Haemorrhagic Telangiectasia. Pediatric Neurol. 2022, 129, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Guttmacher, A.E.; Marchuk, D.A.; White, R.I., Jr. Hereditary hemorrhagic telangiectasia. N. Engl. J. Med. 1995, 333, 918–924. [Google Scholar] [CrossRef]
- Contegiacomo, A.; Del Ciello, A.; Rella, R.; Attempati, N.; Coppolino, D.; Larici, A.R.; Di Stasi, C.; Marano, G.; Manfredi, R. Pulmonary arteriovenous malformations: What the interventional radiologist needs to know. Radiol. Med. 2019, 124, 973–988. [Google Scholar] [CrossRef]
- Tong, Y.; Schneider, J.A.; Coda, A.B.; Hata, T.R.; Cohen, P.R. Birt–Hogg–Dubé syndrome: A review of dermatological manifestations and other symptoms. Am. J. Clin. Dermatol. 2018, 19, 87–101. [Google Scholar] [CrossRef]
- Agarwal, P.P.; Gross, B.H.; Holloway, B.J.; Seely, J.; Stark, P.; Kazerooni, E.A. Thoracic CT findings in birt-hogg-dube syndrome. Am. J. Roentgenol. 2011, 196, 349–352. [Google Scholar] [CrossRef]
- Lee, J.E.; Cha, Y.K.; Kim, J.S.; Choi, J.-H. Birt-Hogg-Dubé syndrome: Characteristic CT findings differentiating it from other diffuse cystic lung diseases. Diagn. Interv. Radiol. 2017, 23, 354. [Google Scholar] [CrossRef]
- Ferreira, E.C.; Brito, C.C.; Domingues, R.C.; Bernardes, M.; Marchiori, E.; Gasparetto, E.L. Whole-body MR imaging for the evaluation of mcCune-albright syndrome. J. Magn. Reson. Imaging Off. J. Int. Soc. Magn. Reson. Med. 2010, 31, 706–710. [Google Scholar] [CrossRef]
- Defilippi, C.; Chiappetta, D.; Marzari, D.; Mussa, A.; Lala, R. Image diagnosis in McCune-Albright syndrome. J. Pediatric Endocrinol. Metab. 2006, 19, 561–570. [Google Scholar] [CrossRef]
- Bongers, E.M.; Gubler, M.-C.; Knoers, N.V. Nail-patella syndrome. Overview on clinical and molecular findings. Pediatric Nephrol. 2002, 17, 703–712. [Google Scholar] [CrossRef] [PubMed]
- KZuberi, H.Z.; Angirekula, A.; Akram, M.R.; Kooner, K.S. Nail-Patella Syndrome: Optical Coherence Tomography Angiography Findings. Case Rep. Ophthalmol. 2022, 13, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Al-Dawsari, N.; Al-Mokhadam, A.; Al-Abdulwahed, H.; Al-Sannaa, N. Nail-Patella Syndrome: A Report of a Saudi Arab Family with an Autosomal Recessive Inheritance. J. Cutan. Med. Surg. 2015, 19, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Witzgall, R. Nail-patella syndrome. Pflügers Arch. Eur. J. Physiol. 2017, 469, 927–936. [Google Scholar] [CrossRef]
- Tigchelaar, S.; Rooy Jd Hannink, G.; Koëter, S.; van Kampen, A.; Bongers, E. Radiological characteristics of the knee joint in nail patella syndrome. Bone Jt. J. 2016, 98, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Ann-Louise McDermott, F.; Sunil, N.D.; Chavda, S.V.; Morgan, D. Maffucci’s syndrome: Clinical and radiological features of a rare condition. J. Laryngol. Otol. 2001, 115, 845–847. [Google Scholar]
- Dasgeb, B.; Morris, M.A.; Ring, C.M.; Mehregan, D.; Mulligan, M.E. Musculoskeletal and overgrowth syndromes associated with cutaneous abnormalities. Br. J. Radiol. 2016, 89, 20160521. [Google Scholar] [CrossRef]
- Silve, C.; Jüppner, H. Ollier disease. Orphanet J. Rare Dis. 2006, 1, 37. [Google Scholar] [CrossRef] [PubMed]
- Pope, V.; Dupuis, L.; Kannu, P.; Mendoza-Londono, R.; Sajic, D.; So, J.; Yoon, G.; Lara-Corrales, I. Buschke–Ollendorff syndrome: A novel case series and systematic review. Br. J. Dermatol. 2016, 174, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, M.F.; Elli, M.; Özkan, M.B.; Bilgici, M.C.; Dağdemir, A.; Korkmaz, M.; Tosun, F.C.; Daǧdemir, A. Osteopoikilosis: Report of a familial case and review of the literature. Rheumatol. Int. 2015, 35, 921–924. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, A.; Ochiai, T.; Tan-Kinoshita, M.; Suzuki, H. Buschke–Ollendorff syndrome: Three generations in a Japanese family. Pediatric Dermatol. 2005, 22, 133–137. [Google Scholar] [CrossRef]
- Nevin, N.C.; Thomas, P.S.; Davis, R.I.; Cowie, G.H. Melorheostosis in a family with autosomal dominant osteopoikilosis. Am. J. Med. Genet. 1999, 82, 409–414. [Google Scholar] [CrossRef]
- Ruaro, B.; Sulli, A.; Alessandri, E.; Ravera, F.; Cutolo, M. Coexistence of osteopoikilosis with seronegative spondyloarthritis and Raynaud’s phenomenon: First case report with evaluation of the nailfold capillary bed and literature review. Reumatismo 2012, 64, 335–339. [Google Scholar] [CrossRef]
- Tomas, C.; Soyer, P.; Dohan, A.; Dray, X.; Boudiaf, M.; Hoeffel, C. Update on imaging of Peutz-Jeghers syndrome. World J. Gastroenterol. WJG 2014, 20, 10864. [Google Scholar] [CrossRef]
- Katabathina, V.S.; Menias, C.O.; Khanna, L.; Murphy, L.; Dasyam, A.K.; Lubner, M.G.; Prasad, S.R. Hereditary gastrointestinal cancer syndromes: Role of imaging in screening, diagnosis, and management. RadioGraphics 2019, 39, 1280–1301. [Google Scholar] [CrossRef]
- Tabriz, H.M.; Obohat, M.; Vahedifard, F.; Eftekharjavadi, A. Survey of Mast Cell Density in Transitional Cell Carcinoma. Iran. J. Pathol. 2021, 16, 119. [Google Scholar] [CrossRef]
- Thrash, B.; Patel, M.; Shah, K.R.; Boland, C.R.; Menter, A. Cutaneous manifestations of gastrointestinal disease: Part II. J. Am. Acad. Dermatol. 2013, 68, 211.e1–211.e33. [Google Scholar] [CrossRef]
- Beggs, A.; Latchford, A.; Vasen, H.F.; Moslein, G.; Alonso, A.; Aretz, S.; Bertario, L.; Blanco, I.; Bulow, S.; Burn, J.; et al. Peutz–Jeghers syndrome: A systematic review and recommendations for management. Gut 2010, 59, 975–986. [Google Scholar] [CrossRef]
- Giardiello, F.M.; Trimbath, J.D. Peutz-Jeghers syndrome and management recommendations. Clin. Gastroenterol. Hepatol. 2006, 4, 408–415. [Google Scholar] [CrossRef]
- Patnana, M.; Bronstein, Y.; Szklaruk, J.; Bedi, D.G.; Hwu, W.J.; Gershenwald, J.E.; Prieto, V.G.; Ng, C.S. Multimethod imaging, staging, and spectrum of manifestations of metastatic melanoma. Clin. Radiol. 2011, 66, 224–236. [Google Scholar] [CrossRef]
- Schmid, R.; Schmidt, S.K.; Detsch, R.; Horder, H.; Blunk, T.; Schrüfer, S.; Schubert, D.W.; Fischer, L.; Thievessen, I.; Heltmann-Meyer, S.; et al. A New Printable Alginate/Hyaluronic Acid/Gelatin Hydrogel Suitable for Biofabrication of In Vitro and In Vivo Metastatic Melanoma Models. Adv. Funct. Mater. 2022, 32, 2107993. [Google Scholar] [CrossRef]
- Vyas, R.; Selph, J.; Gerstenblith, M.R. (Eds.) Cutaneous manifestations associated with melanoma. In Seminars in Oncology; Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Flaus, A.; Habouzit, V.; de Leiris, N.; Vuillez, J.P.; Leccia, M.T.; Simonson, M.; Perrot, J.L.; Cachin, F.; Prevot, N. Outcome Prediction at Patient Level Derived from Pre-Treatment 18F-FDG PET Due to Machine Learning in Metastatic Melanoma Treated with Anti-PD1 Treatment. Diagnostics 2022, 12, 388. [Google Scholar] [CrossRef]
- Vahedifard, F.; Hassani, S.; Afrasiabi, A.; Esfe, A.M. Artificial intelligence for radiomics; diagnostic biomarkers for neuro-oncology. World J. Adv. Res. Rev. 2022, 14, 304–310. [Google Scholar] [CrossRef]
- Karabajakian, A.; Ray-Coquard, I.; Blay, J.Y. Molecular Mechanisms of Kaposi Sarcoma Development. Cancers 2022, 14, 1869. [Google Scholar] [CrossRef]
- Restrepo, C.S.; Martínez, S.; Lemos, J.A.; Carrillo, J.A.; Lemos, D.F.; Ojeda, P.; Koshy, P. Imaging manifestations of Kaposi sarcoma. Radiographics 2006, 26, 1169–1185. [Google Scholar] [CrossRef]
- Schwartz, R.A.; Micali, G.; Nasca, M.R.; Scuderi, L. Kaposi sarcoma: A continuing conundrum. J. Am. Acad. Dermatol. 2008, 59, 179–206. [Google Scholar] [CrossRef]
- Fatahzadeh, M. Kaposi sarcoma: Review and medical management update. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2012, 113, 2–16. [Google Scholar] [CrossRef]
- Pesqué, L.; Delyon, J.; Lheure, C.; Baroudjian, B.; Battistella, M.; Merlet, P.; Lebbé, C.; Vercellino, L. Yield of FDG PET/CT for Defining the Extent of Disease in Patients with Kaposi Sarcoma. Cancers 2022, 14, 2189. [Google Scholar] [CrossRef] [PubMed]



























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disorder | Clinical and Dermatologic Findings | Imaging Findings |
|---|---|---|
| Autoimmune/Inflammatory Disorders and Vasculitides | ||
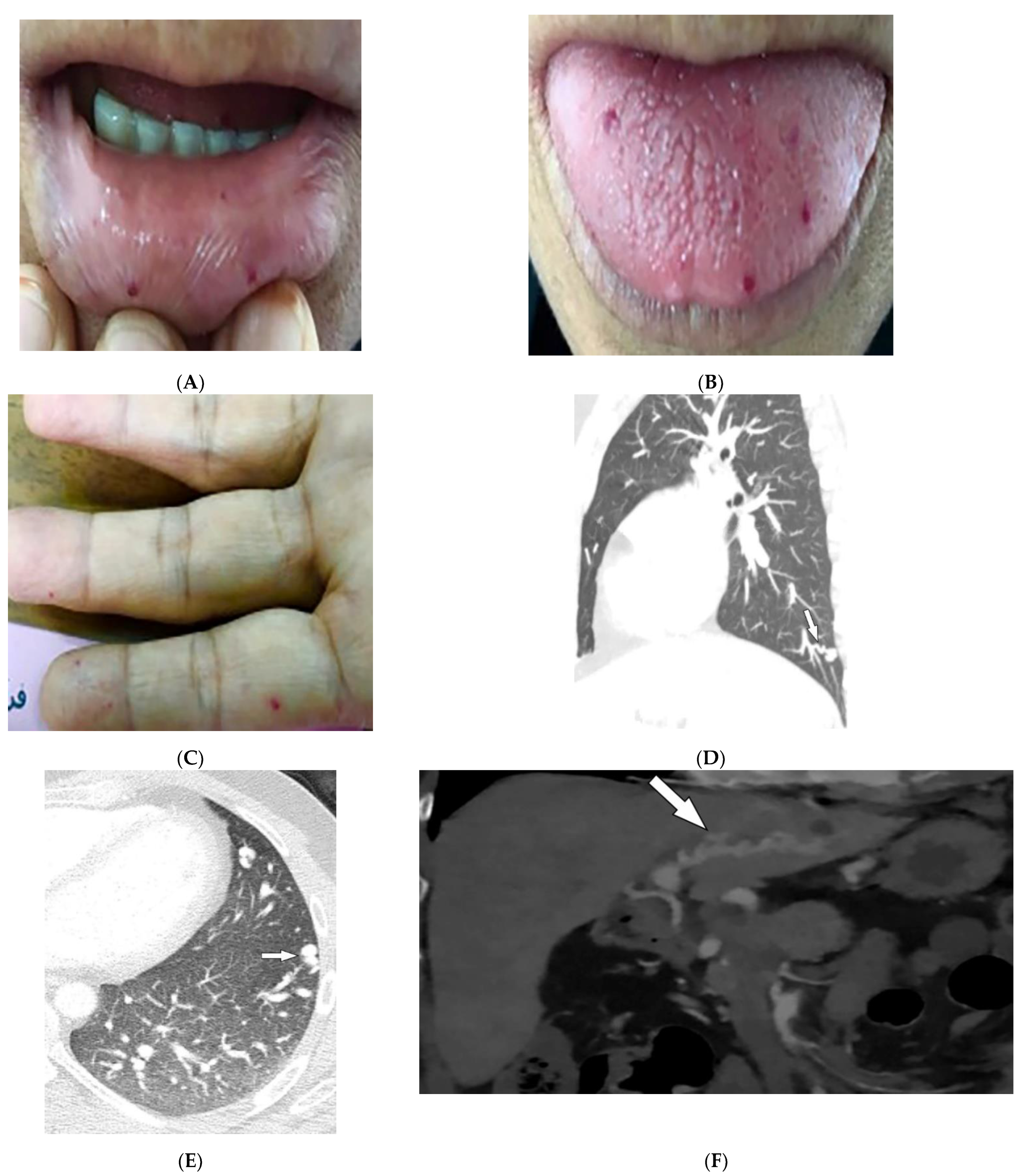
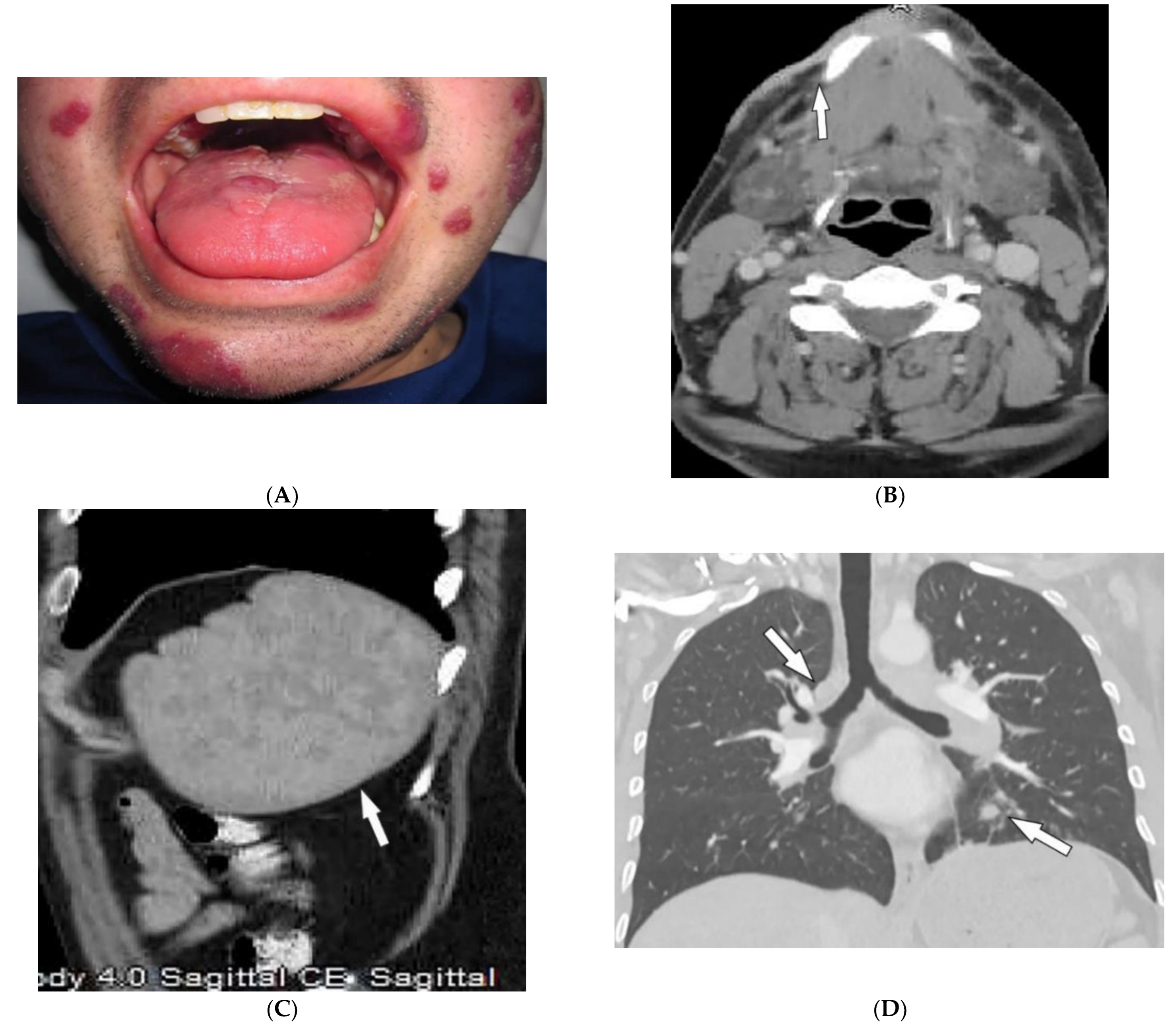
| Dermatomyositis | Atrophic dermal papules of dermatomyositis (Gottron papules), Gottron sign, heliotrope rash, V sign, shawl sign, calcinosis cutis Proximal nailfold erythema, capillary loop dilation and dropout, ragged cuticles Esophageal dysmotility Myositis ILD Malignancy | Calcinosis cutis Feathery edema-like SI of the muscles NSIP, OP, UIP |
| Sarcoidosis | Lupus pernio Erythema nodosum Lung nodules and adenopathy neurosarcoidosis Bone lesions | Reticulonodular lung opacities with upper lobe and peri-lymphatic distribution Leptomeningeal enhancement Lacy lytic bone lesions |
| Scleroderma (diffuse systemic sclerosis) | Raynaud’s phenomenon Skin tightening Sclerodactyly Calcinosis cutis Dilated bowel/esophagus Pulmonary hypertension ILD | Soft-tissue calcifications and acro-osteolysis Lack of peristalsis and esophageal dilation NSIP and UIP |
| Celiac disease | Dermatitis herpetiformis Psoriasis Intestinal manifestations | Small-bowel dilation Reversal of jejunal and ileal folds |
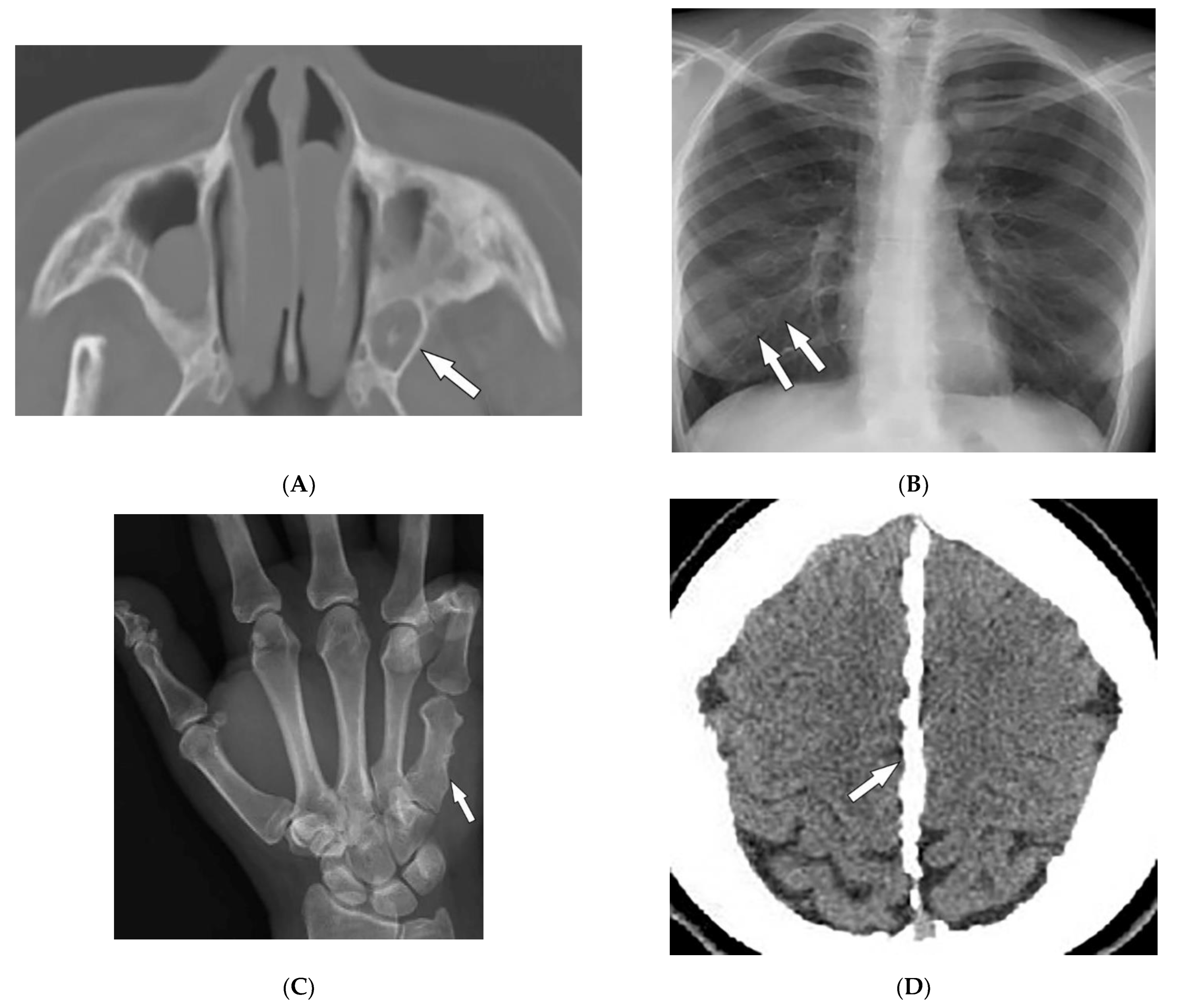
| Granulomatosis with polyangiitis (Wegner’s) | Palpable purpura Subcutaneous nodules Pyoderma-gangrenosum-like ulcerations Lung lesions and hemoptysis Glomerulonephritis Peripheral neuropathy, mononeuritis multiplex Chronic sinusitis and saddle nose deformity | Bilateral cavitary lung lesions with a ground-glass halo sign Mucosal thickening Nasal septal perforation Hyperostosis |
| Polyarteritis nodosa | Palpable purpura Painful nodules on lower legs Livedo reticularis Medium-sized artery vasculitis | Microaneurysms and constrictions of medium-sized arteritis (beaded appearance) |
| Behcet’s disease | Oral and genital ulcers Ocular findings Vasculitis CNS lesions | Thickening of the aorta and SVC Bilateral pulmonary artery aneurysms Basal ganglia and brainstem lesions |
| Genetic/Congenital Disorders | ||
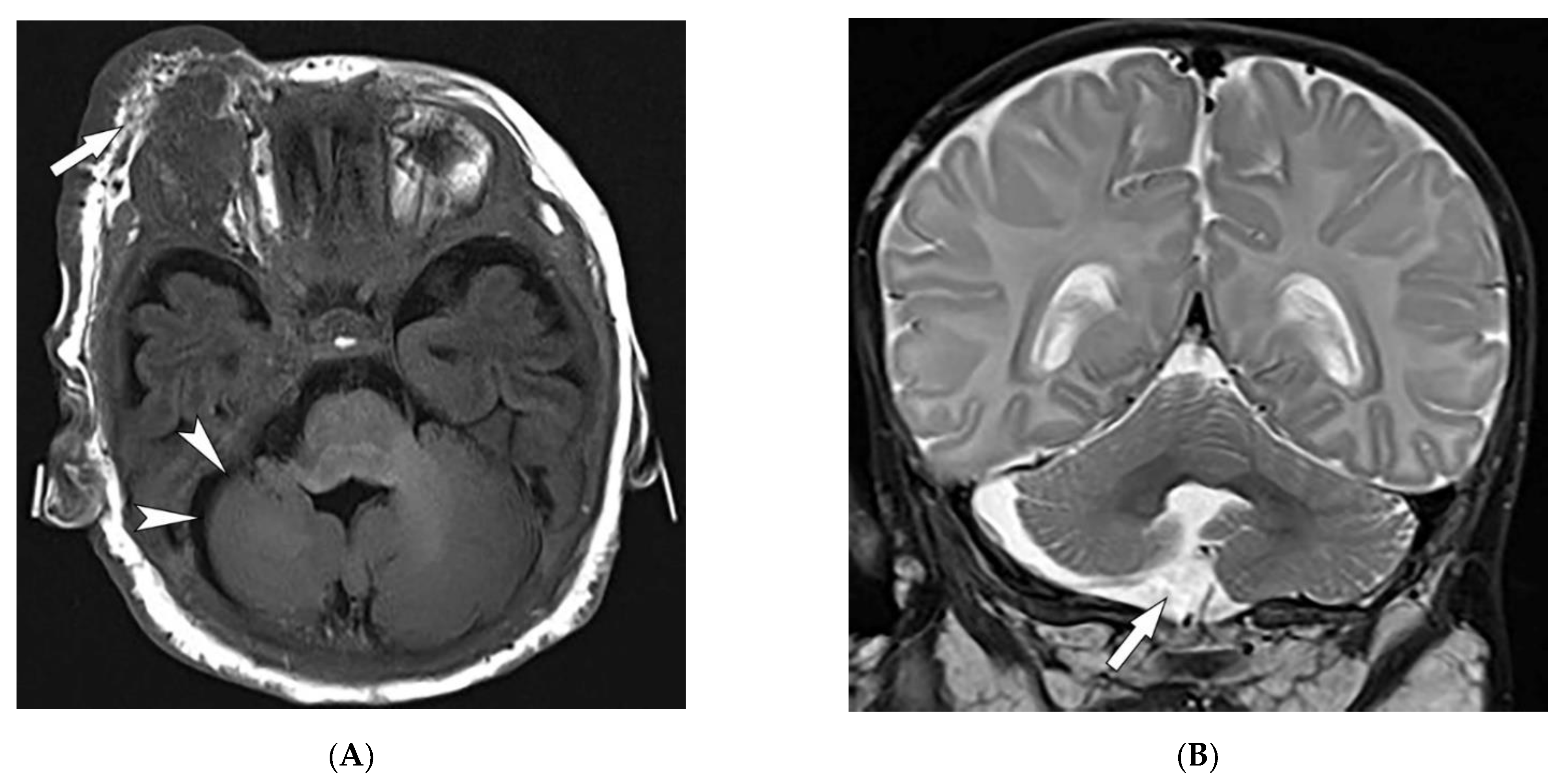
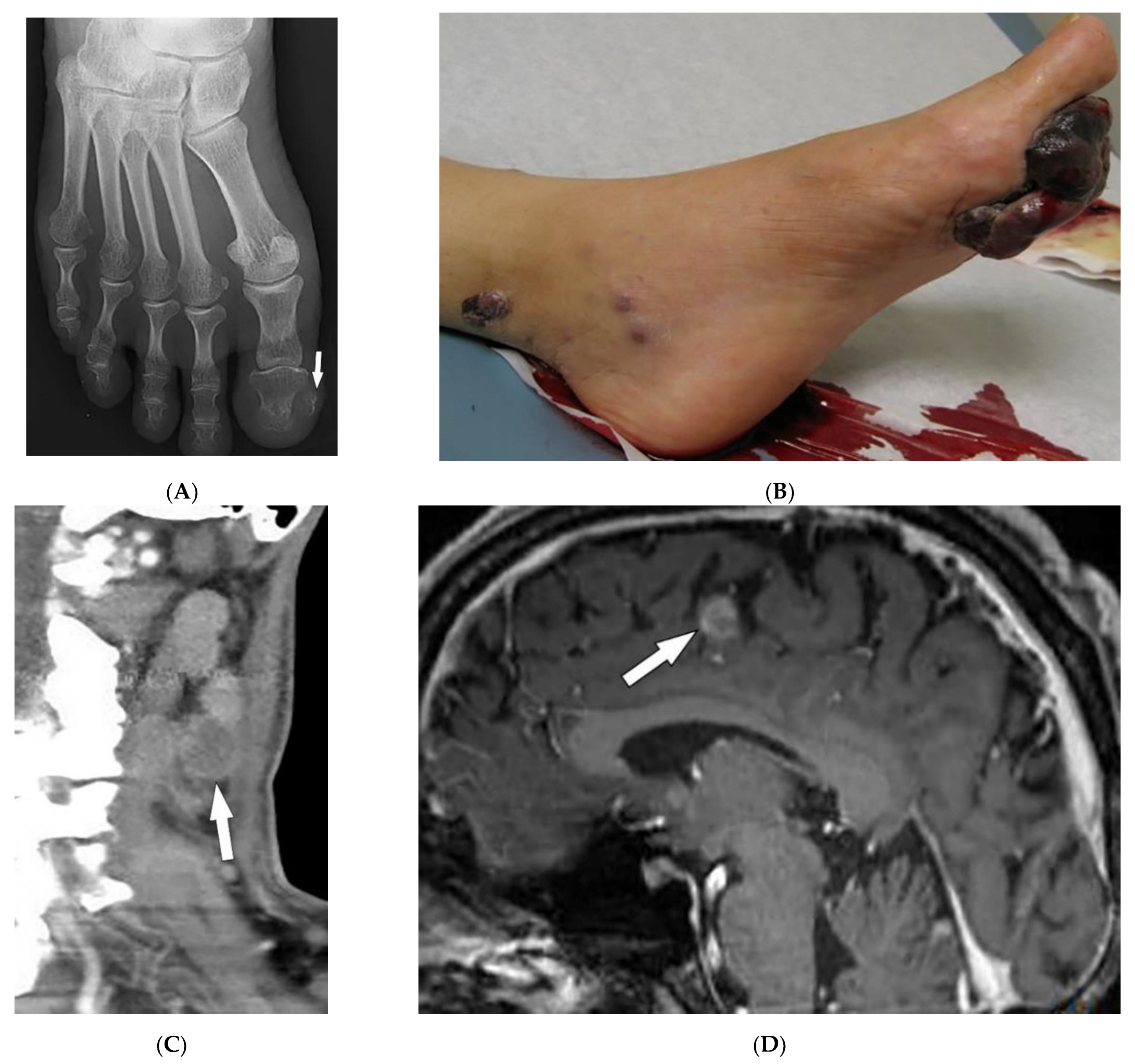
| Tuberous sclerosis complex | Facial angiofibroma Hypopigmented macules Shagreen patches Periungual fibromas Osseous abnormalities CNS hamartomas Renal AML Pulmonary LAM | Tubers, RMLs, SENs, SEGAs of brain Focal sclerotic bone lesions Hypertrophic osteoarthropathy Fat-containing renal mass Thin-walled lung cysts |
| Neurofibromatosis type 1 (NF-1) | Café-au-lait spots Freckling (axillary or inguinal) Lisch nodules Neurofibromas Optic nerve and other gliomas Skeletal abnormalities | Peripheral nerve sheath tumors including cutaneous, spinal, plexiform neuroma Diffuse thickening of the nerve |
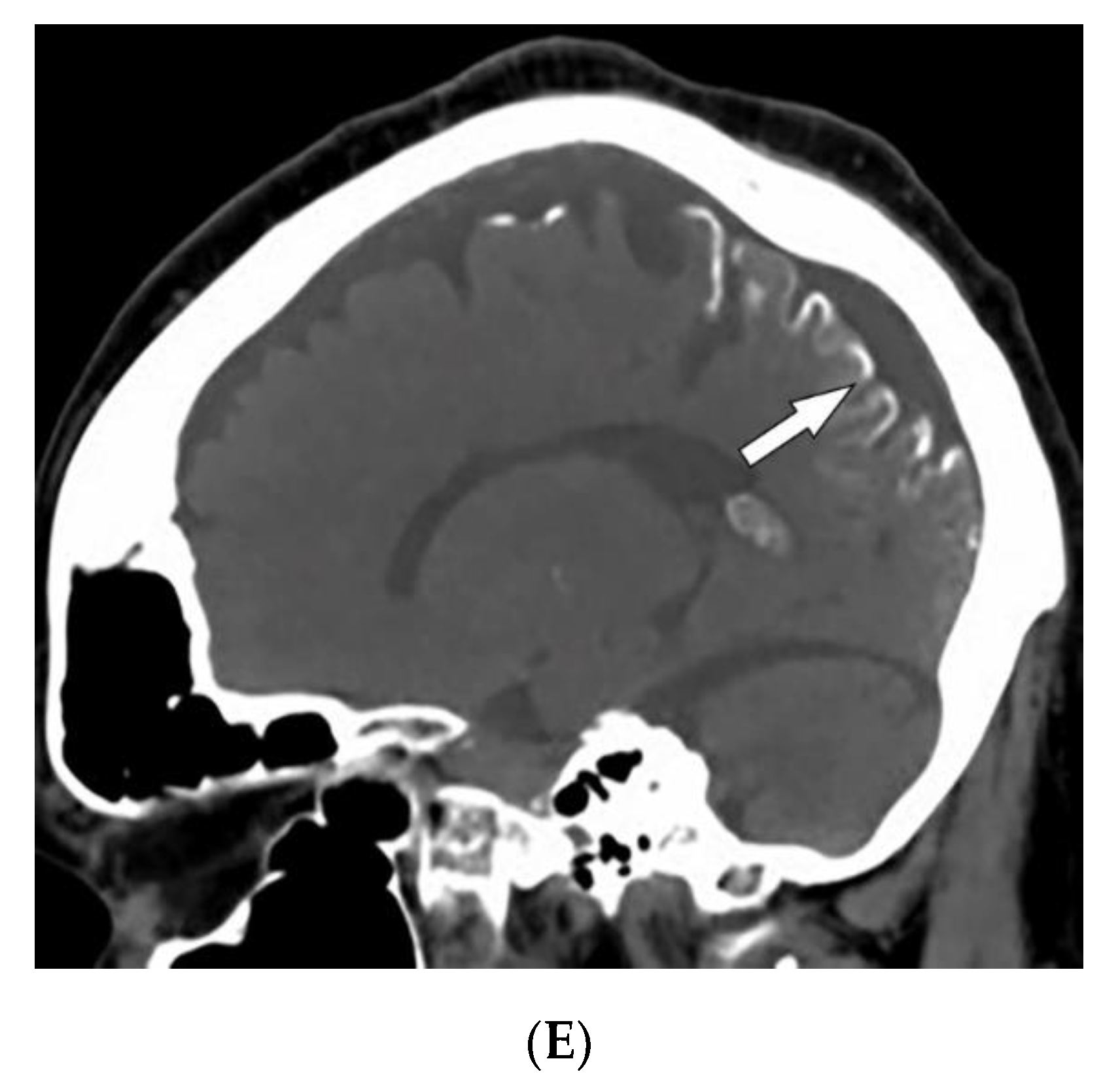
| Sturge–Weber syndrome | Port-wine stains Leptomeningeal capillary malformation Glaucoma | Parieto-occipital cortical hemiatrophy Tram-track calcification Calvarial thickening |
| PHACES syndrome | Craniofacial hemangiomas Posterior fossa malformations Cerebrovascular anomalies Eye anomalies | Ipsilateral cerebellar hemisphere dysplasia Major cerebral vessels dysplasia |
| Basal cell nevus syndrome | Basal cell carcinomas Palmoplantar pits Skeletal abnormalities Brain abnormalities | Keratocystic odontogenic tumors Ribs and metacarpals abnormalities Medulloblastoma, falx cerebri calcification |
| Hereditary hemorrhagic telangiectasia | Recurrent epistaxis Multiple telangiectasias Arteriovenous malformations | Bilateral well-defined lung opacities with lobulated shapes Ground-glass nodule |
| Birt–Hogg–Dube syndrome | Fibrofolliculomas, trichodiscomas, acrochordons Lung cysts (pneumothorax) Renal cysts | Bilateral basilar predominant, thin-walled cysts abutting pleura and pulmonary vessels |
| McCune–Albright syndrome | Cafe’-au-lait macules Fibrous dysplasia Endocrine dysfunction | Medullary ground-glass lytic lesions with thin cortices Various sclerotic to cystic pattern |
| Fong (Nail–patella) syndrome | Hypoplastic nails, triangular lunulae Hypoplastic patellae Focal segmental glomerulosclerosisLester iris | Bilateral absence of patellae Posterior iliac horns (Fong’s prongs) Subluxation of radial heads |
| Maffucci syndrome | Multiple enchondromatosis (Ollier disease) Venous malformations | Multiple osteochondromas |
| Buschke–Ollendorff syndrome | Dermatofibrosis lenticularis disseminata Osteopoikilosis Melorheostosis | Bony islands and multiple sclerotic lesions cause mottled appearance Cortical thickening with undulating bone |
| Peutz–Jeghers syndrome | Mucocutaneous pigmented macules Hamartomatous polyps | Multiple intraluminal filling defects on barium study |
| Neoplasm | ||
| Melanotic melanoma | ABCDE features | Enhancing lesions if they contain a sufficient amount of melanin Multiple well-defined lung nodules |
| Kaposi sarcoma | Erythematous or violaceous macules, plaques, nodules Pulmonary involvement Gastrointestinal involvement | Nodular enhancing masses Peribroncovascular nodules and halo sign |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shafiei, M.; Shomal Zadeh, F.; Mansoori, B.; Pyle, H.; Agim, N.; Hinojosa, J.; Dominguez, A.; Thomas, C.; Chalian, M. Imaging More than Skin-Deep: Radiologic and Dermatologic Presentations of Systemic Disorders. Diagnostics 2022, 12, 2011. https://doi.org/10.3390/diagnostics12082011
Shafiei M, Shomal Zadeh F, Mansoori B, Pyle H, Agim N, Hinojosa J, Dominguez A, Thomas C, Chalian M. Imaging More than Skin-Deep: Radiologic and Dermatologic Presentations of Systemic Disorders. Diagnostics. 2022; 12(8):2011. https://doi.org/10.3390/diagnostics12082011
Chicago/Turabian StyleShafiei, Mehrzad, Firoozeh Shomal Zadeh, Bahar Mansoori, Hunter Pyle, Nnenna Agim, Jorge Hinojosa, Arturo Dominguez, Cristina Thomas, and Majid Chalian. 2022. "Imaging More than Skin-Deep: Radiologic and Dermatologic Presentations of Systemic Disorders" Diagnostics 12, no. 8: 2011. https://doi.org/10.3390/diagnostics12082011