
Diagnosis of Myelodysplastic Syndromes: From Immunological Observations to Clinical Applications



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Innate Immune Signaling in Response to Inflamm-Aging in the Context of CHIP
3. Selective Expansion of MDS Clones at the Early Phase of MDS Is Driven by Innate Immune Dysregulation
4. Clinical Immune Manifestation in MDS
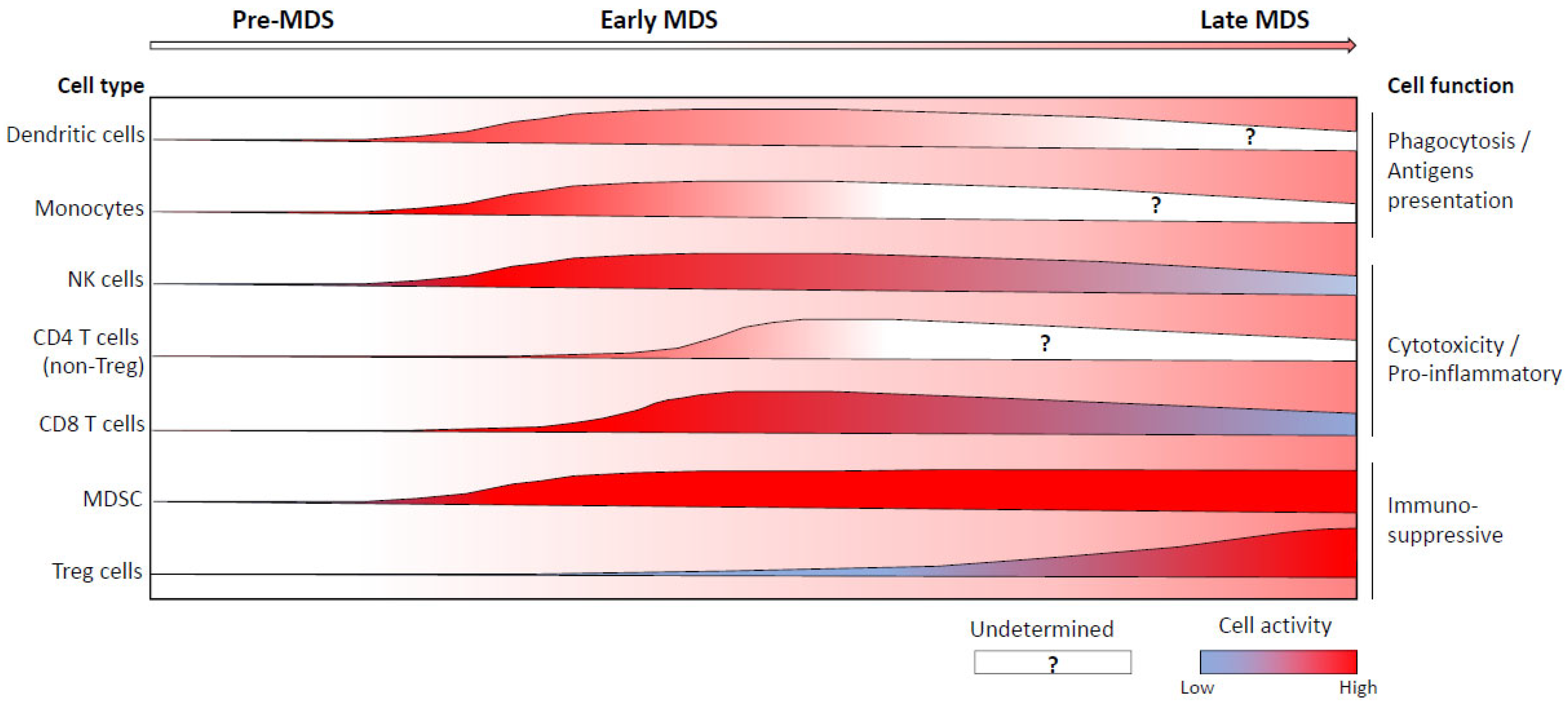
5. Abnormal Immune Cell Repartition and/or Functions during the Course of MDS
5.1. Natural Killer Cells
5.2. T Lymphoid Cells
5.3. CD8+ Cytotoxic T Cells
5.4. CD4+ Helper T Cells
5.5. Regulatory T Cells
5.6. Myeloid-Derived Suppressor Cells
5.7. Mesenchymal Stem Cells
5.8. Monocytes and Macrophages
6. Perspective on Immune Monitoring at MDS Diagnosis
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Cazzola, M. Myelodysplastic Syndromes. N. Engl. J. Med. 2020, 383, 1358–1374. [Google Scholar] [CrossRef] [PubMed]
- Adès, L.; Itzykson, R.; Fenaux, P. Myelodysplastic Syndromes. Lancet Lond. Engl. 2014, 383, 2239–2252. [Google Scholar] [CrossRef]
- Jaiswal, S.; Ebert, B.L. Clonal Hematopoiesis in Human Aging and Disease. Science 2019, 366, eaan4673. [Google Scholar] [CrossRef] [PubMed]
- Sperling, A.S.; Gibson, C.J.; Ebert, B.L. The Genetics of Myelodysplastic Syndrome: From Clonal Haematopoiesis to Secondary Leukaemia. Nat. Rev. Cancer 2017, 17, 5–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeZern, A.E.; Malcovati, L.; Ebert, B.L. CHIP, CCUS, and Other Acronyms: Definition, Implications, and Impact on Practice. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 400–410. [Google Scholar] [CrossRef]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised International Prognostic Scoring System for Myelodysplastic Syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef]
- Bersanelli, M.; Travaglino, E.; Meggendorfer, M.; Matteuzzi, T.; Sala, C.; Mosca, E.; Chiereghin, C.; Di Nanni, N.; Gnocchi, M.; Zampini, M.; et al. Classification and Personalized Prognostic Assessment on the Basis of Clinical and Genomic Features in Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1223–1233. [Google Scholar] [CrossRef]
- Pellagatti, A.; Armstrong, R.N.; Steeples, V.; Sharma, E.; Repapi, E.; Singh, S.; Sanchi, A.; Radujkovic, A.; Horn, P.; Dolatshad, H.; et al. Impact of Spliceosome Mutations on RNA Splicing in Myelodysplasia: Dysregulated Genes/Pathways and Clinical Associations. Blood 2018, 132, 1225–1240. [Google Scholar] [CrossRef] [Green Version]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of Genetic Lesions in 944 Patients with Myelodysplastic Syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Itzykson, R.; Kosmider, O.; Cluzeau, T.; Mansat-De Mas, V.; Dreyfus, F.; Beyne-Rauzy, O.; Quesnel, B.; Vey, N.; Gelsi-Boyer, V.; Raynaud, S.; et al. Impact of TET2 Mutations on Response Rate to Azacitidine in Myelodysplastic Syndromes and Low Blast Count Acute Myeloid Leukemias. Leukemia 2011, 25, 1147–1152. [Google Scholar] [CrossRef] [Green Version]
- Malcovati, L.; Stevenson, K.; Papaemmanuil, E.; Neuberg, D.; Bejar, R.; Boultwood, J.; Bowen, D.T.; Campbell, P.J.; Ebert, B.L.; Fenaux, P.; et al. SF3B1-Mutant MDS as a Distinct Disease Subtype: A Proposal from the International Working Group for the Prognosis of MDS. Blood 2020, 136, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Sallman, D.A.; List, A. The Central Role of Inflammatory Signaling in the Pathogenesis of Myelodysplastic Syndromes. Blood 2019, 133, 1039–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Comont, T.; Treiner, E.; Vergez, F. From Immune Dysregulations to Therapeutic Perspectives in Myelodysplastic Syndromes: A Review. Diagnostics 2021, 11, 1982. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, A.M.; Shallis, R.M.; Wang, R.; Davidoff, A.; Ma, X. Epidemiology of Myelodysplastic Syndromes: Why Characterizing the Beast Is a Prerequisite to Taming It. Available online: https://pubmed-ncbi-nlm-nih-gov.proxy.insermbiblio.inist.fr/30314642/ (accessed on 9 May 2022).
- Chen, Y.J.; Liao, Y.J.; Tram, V.T.N.; Lin, C.H.; Liao, K.C.; Liu, C.L. Alterations of Specific Lymphocytic Subsets with Aging and Age-Related Metabolic and Cardiovascular Diseases. Life 2020, 10, 246. [Google Scholar] [CrossRef] [PubMed]
- Kovtonyuk, L.V.; Fritsch, K.; Feng, X.; Manz, M.G.; Takizawa, H. Inflamm-Aging of Hematopoiesis, Hematopoietic Stem Cells, and the Bone Marrow Microenvironment. Front. Immunol. 2016, 7, 502. [Google Scholar] [CrossRef] [Green Version]
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging as Two Sides of the Same Coin: Friends or Foes? Front. Immunol. 2018, 8, 1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bick, A.G.; Weinstock, J.S.; Nandakumar, S.K.; Fulco, C.P.; Bao, E.L.; Zekavat, S.M.; Szeto, M.D.; Liao, X.; Leventhal, M.J.; Nasser, J.; et al. Inherited Causes of Clonal Hematopoiesis in 97,691 TOPMed Whole Genomes. Nature 2020, 586, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. Available online: https://www-nejm-org.proxy.insermbiblio.inist.fr/doi/10.1056/NEJMoa1701719 (accessed on 5 May 2022).
- Buscarlet, M.; Provost, S.; Zada, Y.F.; Barhdadi, A.; Bourgoin, V.; Lépine, G.; Mollica, L.; Szuber, N.; Dubé, M.-P.; Busque, L. DNMT3A and TET2 Dominate Clonal Hematopoiesis and Demonstrate Benign Phenotypes and Different Genetic Predispositions. Blood 2017, 130, 753–762. [Google Scholar] [CrossRef] [Green Version]
- Miller, P.G.; Qiao, D.; Rojas-Quintero, J.; Honigberg, M.C.; Sperling, A.S.; Gibson, C.J.; Bick, A.G.; Niroula, A.; McConkey, M.E.; Sandoval, B.; et al. Association of Clonal Hematopoiesis with Chronic Obstructive Pulmonary Disease. Blood 2022, 139, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Bonnefond, A.; Skrobek, B.; Lobbens, S.; Eury, E.; Thuillier, D.; Cauchi, S.; Lantieri, O.; Balkau, B.; Riboli, E.; Marre, M.; et al. Association between Large Detectable Clonal Mosaicism and Type 2 Diabetes with Vascular Complications. Nat. Genet. 2013, 45, 1040–1043. [Google Scholar] [CrossRef] [PubMed]
- Fuster, J.J.; Zuriaga, M.A.; Zorita, V.; MacLauchlan, S.; Polackal, M.N.; Viana-Huete, V.; Ferrer-Pérez, A.; Matesanz, N.; Herrero-Cervera, A.; Sano, S.; et al. TET2-Loss-of-Function-Driven Clonal Hematopoiesis Exacerbates Experimental Insulin Resistance in Aging and Obesity. Cell Rep. 2020, 33, 108326. [Google Scholar] [CrossRef]
- Arends, C.M.; Galan-Sousa, J.; Hoyer, K.; Chan, W.; Jäger, M.; Yoshida, K.; Seemann, R.; Noerenberg, D.; Waldhueter, N.; Fleischer-Notter, H.; et al. Hematopoietic Lineage Distribution and Evolutionary Dynamics of Clonal Hematopoiesis. Leukemia 2018, 32, 1908–1919. [Google Scholar] [CrossRef]
- Buscarlet, M.; Provost, S.; Zada, Y.F.; Bourgoin, V.; Mollica, L.; Dubé, M.-P.; Busque, L. Lineage Restriction Analyses in CHIP Indicate Myeloid Bias for TET2 and Multipotent Stem Cell Origin for DNMT3A. Blood 2018, 132, 277–280. [Google Scholar] [CrossRef] [PubMed]
- Couronné, L.; Bastard, C.; Bernard, O.A. TET2 and DNMT3A Mutations in Human T-Cell Lymphoma. N. Engl. J. Med. 2012, 366, 95–96. [Google Scholar] [CrossRef]
- Fukumoto, K.; Nguyen, T.B.; Chiba, S.; Sakata-Yanagimoto, M. Review of the Biologic and Clinical Significance of Genetic Mutations in Angioimmunoblastic T-Cell Lymphoma. Cancer Sci. 2018, 109, 490–496. [Google Scholar] [CrossRef] [Green Version]
- Gibson, C.J.; Kim, H.T.; Zhao, L.; Murdock, H.M.; Hambley, B.; Ogata, A.; Madero-Marroquin, R.; Wang, S.; Green, L.; Fleharty, M.; et al. Donor Clonal Hematopoiesis and Recipient Outcomes After Transplantation. J. Clin. Oncol. 2022, 40, 189–201. [Google Scholar] [CrossRef]
- Frick, M.; Chan, W.; Arends, C.M.; Hablesreiter, R.; Halik, A.; Heuser, M.; Michonneau, D.; Blau, O.; Hoyer, K.; Christen, F.; et al. Role of Donor Clonal Hematopoiesis in Allogeneic Hematopoietic Stem-Cell Transplantation. J. Clin. Oncol. 2019, 37, 375–385. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 Is Required to Resolve Inflammation by Recruiting Hdac2 to Specifically Repress IL-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef] [Green Version]
- Cull, A.H.; Snetsinger, B.; Buckstein, R.; Wells, R.A.; Rauh, M.J. Tet2 Restrains Inflammatory Gene Expression in Macrophages. Exp. Hematol. 2017, 55, 56–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.-L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal Hematopoiesis Associated with TET2 Deficiency Accelerates Atherosclerosis Development in Mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, S.; Oshima, K.; Wang, Y.; Katanasaka, Y.; Sano, M.; Walsh, K. CRISPR-Mediated Gene Editing to Assess the Roles of Tet2 and Dnmt3a in Clonal Hematopoiesis and Cardiovascular Disease. Circ. Res. 2018, 123, 335–341. [Google Scholar] [CrossRef]
- Leoni, C.; Montagner, S.; Rinaldi, A.; Bertoni, F.; Polletti, S.; Balestrieri, C.; Monticelli, S. Dnmt3a Restrains Mast Cell Inflammatory Responses. Proc. Natl. Acad. Sci. USA 2017, 114, E1490–E1499. [Google Scholar] [CrossRef] [Green Version]
- Cook, E.K.; Izukawa, T.; Young, S.; Rosen, G.; Jamali, M.; Zhang, L.; Johnson, D.; Bain, E.; Hilland, J.; Ferrone, C.K.; et al. Comorbid and Inflammatory Characteristics of Genetic Subtypes of Clonal Hematopoiesis. Blood Adv. 2019, 3, 2482–2486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.; Zheng, Y.; Xu, L.; Cao, C.; Dong, B.; Chen, X. The Inflammatory Cytokine Profile of Myelodysplastic Syndromes. Medicine 2019, 98, e15844. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, X.; Guo, J.; Xu, F.; He, Q.; Zhao, Y.; Yang, Y.; Gu, S.; Zhang, Y.; Wu, L.; et al. Interleukin-17 Enhances the Production of Interferon-γ and Tumour Necrosis Factor-α by Bone Marrow T Lymphocytes from Patients with Lower Risk Myelodysplastic Syndromes. Eur. J. Haematol. 2013, 90, 375–384. [Google Scholar] [CrossRef]
- Iwasaki, A.; Medzhitov, R. Control of Adaptive Immunity by the Innate Immune System. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef]
- Nagai, Y.; Garrett, K.P.; Ohta, S.; Bahrun, U.; Kouro, T.; Akira, S.; Takatsu, K.; Kincade, P.W. Toll-Like Receptors on Hematopoietic Progenitor Cells Stimulate Innate Immune System Replenishment. Immunity 2006, 24, 801–812. [Google Scholar] [CrossRef] [Green Version]
- Esplin, B.L.; Shimazu, T.; Welner, R.S.; Garrett, K.P.; Nie, L.; Zhang, Q.; Humphrey, M.B.; Yang, Q.; Borghesi, L.A.; Kincade, P.W. Chronic Exposure to a TLR Ligand Injures Hematopoietic Stem Cells. J. Immunol. 2011, 186, 5367–5375. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Mu, W.-C.; Karki, R.; Chiang, H.-H.; Mohrin, M.; Shin, J.J.; Ohkubo, R.; Ito, K.; Kanneganti, T.-D.; Chen, D. Mitochondrial Stress-Initiated Aberrant Activation of the NLRP3 Inflammasome Regulates the Functional Deterioration of Hematopoietic Stem Cell Aging. Cell Rep. 2019, 26, 945–954.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barreyro, L.; Chlon, T.M.; Starczynowski, D.T. Chronic Immune Response Dysregulation in MDS Pathogenesis. Blood 2018, 132, 1553–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellagatti, A.; Cazzola, M.; Giagounidis, A.; Perry, J.; Malcovati, L.; Della Porta, M.G.; Jädersten, M.; Killick, S.; Verma, A.; Hellström-Lindberg, E.; et al. Deregulated Gene Expression Pathways in Myelodysplastic Syndrome Hematopoietic Stem Cells. Leukemia 2010, 24, 756–764. [Google Scholar] [CrossRef] [Green Version]
- Lc, P.; Da, M.; Zj, G.; Dac, F.; Mj, W.; St, O.; Lg, S. Toll-like Receptor and Cytokine Expression throughout the Bone Marrow Differs between Patients with Low- and High-Risk Myelodysplastic Syndromes. Available online: https://pubmed-ncbi-nlm-nih-gov.proxy.insermbiblio.inist.fr/35367529/ (accessed on 24 May 2022).
- Wei, Y.; Dimicoli, S.; Bueso-Ramos, C.; Chen, R.; Yang, H.; Neuberg, D.; Pierce, S.; Jia, Y.; Zheng, H.; Wang, H.; et al. Toll-like Receptor Alterations in Myelodysplastic Syndrome. Leukemia 2013, 27, 1832–1840. [Google Scholar] [CrossRef] [Green Version]
- Monlish, D.A.; Greenberg, Z.J.; Bhatt, S.T.; Leonard, K.M.; Romine, M.P.; Dong, Q.; Bendesky, L.; Duncavage, E.J.; Magee, J.A.; Schuettpelz, L.G. TLR2/6 Signaling Promotes the Expansion of Premalignant Hematopoietic Stem and Progenitor Cells in the NUP98-HOXD13 Mouse Model of MDS. Exp. Hematol. 2020, 88, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Monlish, D.A.; Bhatt, S.T.; Duncavage, E.J.; Greenberg, Z.J.; Keller, J.L.; Romine, M.P.; Yang, W.; Aplan, P.D.; Walter, M.J.; Schuettpelz, L.G. Loss of Toll-like Receptor 2 Results in Accelerated Leukemogenesis in the NUP98-HOXD13 Mouse Model of MDS. Blood 2018, 131, 1032–1035. [Google Scholar] [CrossRef] [PubMed]
- Maratheftis, C.I.; Andreakos, E.; Moutsopoulos, H.M.; Voulgarelis, M. Toll-like Receptor-4 Is Up-Regulated in Hematopoietic Progenitor Cells and Contributes to Increased Apoptosis in Myelodysplastic Syndromes. Clin. Cancer Res. 2007, 13, 1154–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratajczak, M.Z.; Bujko, K.; Cymer, M.; Thapa, A.; Adamiak, M.; Ratajczak, J.; Abdel-Latif, A.K.; Kucia, M. The Nlrp3 Inflammasome as a “Rising Star” in Studies of Normal and Malignant Hematopoiesis. Leukemia 2020, 34, 1512–1523. [Google Scholar] [CrossRef] [Green Version]
- Basiorka, A.A.; McGraw, K.L.; Eksioglu, E.A.; Chen, X.; Johnson, J.; Zhang, L.; Zhang, Q.; Irvine, B.A.; Cluzeau, T.; Sallman, D.A.; et al. The NLRP3 Inflammasome Functions as a Driver of the Myelodysplastic Syndrome Phenotype. Blood 2016, 128, 2960–2975. [Google Scholar] [CrossRef]
- Sallman, D.A.; Cluzeau, T.; Basiorka, A.A.; List, A. Unraveling the Pathogenesis of MDS: The NLRP3 Inflammasome and Pyroptosis Drive the MDS Phenotype. Front. Oncol. 2016, 6, 151. [Google Scholar] [CrossRef]
- Cluzeau, T.; McGraw, K.L.; Irvine, B.; Masala, E.; Ades, L.; Basiorka, A.A.; Maciejewski, J.; Auberger, P.; Wei, S.; Fenaux, P.; et al. Pro-Inflammatory Proteins S100A9 and Tumor Necrosis Factor-α Suppress Erythropoietin Elaboration in Myelodysplastic Syndromes. Haematologica 2017, 102, 2015–2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giudice, V.; Wu, Z.; Kajigaya, S.; del Pilar Fernandez Ibanez, M.; Rios, O.; Cheung, F.; Ito, S.; Young, N.S. Circulating S100A8 and S100A9 Protein Levels in Plasma of Patients with Acquired Aplastic Anemia and Myelodysplastic Syndromes. Cytokine 2019, 113, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Velegraki, M.; Papakonstanti, E.; Mavroudi, I.; Psyllaki, M.; Tsatsanis, C.; Oulas, A.; Iliopoulos, I.; Katonis, P.; Papadaki, H.A. Impaired Clearance of Apoptotic Cells Leads to HMGB1 Release in the Bone Marrow of Patients with Myelodysplastic Syndromes and Induces TLR4-Mediated Cytokine Production. Haematologica 2013, 98, 1206–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basiorka, A.A.; McGraw, K.L.; Abbas-Aghababazadeh, F.; McLemore, A.F.; Vincelette, N.D.; Ward, G.A.; Eksioglu, E.A.; Sallman, D.A.; Al Ali, N.; Padron, E.; et al. Assessment of ASC Specks as a Putative Biomarker of Pyroptosis in Myelodysplastic Syndromes: An Observational Cohort Study. Lancet Haematol. 2018, 5, e393–e402. [Google Scholar] [CrossRef]
- Chen, X.; Eksioglu, E.A.; Zhou, J.; Zhang, L.; Djeu, J.; Fortenbery, N.; Epling-Burnette, P.; Van Bijnen, S.; Dolstra, H.; Cannon, J.; et al. Induction of Myelodysplasia by Myeloid-Derived Suppressor Cells. J. Clin. Invest. 2013, 123, 4595–4611. [Google Scholar] [CrossRef]
- Muto, T.; Walker, C.S.; Choi, K.; Hueneman, K.; Smith, M.A.; Gul, Z.; Garica-Manero, G.; Ma, A.; Zheng, Y.; Statczynowski, D.T. Adaptive Response to Inflammation Contributes to Sustained Myelopoiesis and Confers a Competitive Advantage in Myelodysplastic Syndrome HSCs. Nat. Immunol. 2020, 21, 535–545. [Google Scholar] [CrossRef]
- Varney, M.E.; Niederkorn, M.; Konno, H.; Matsumura, T.; Gohda, J.; Yoshida, N.; Akiyama, T.; Christie, S.; Fang, J.; Miller, D.; et al. Loss of Tifab, a Del(5q) MDS Gene, Alters Hematopoiesis through Derepression of Toll-like Receptor–TRAF6 Signaling. J. Exp. Med. 2015, 212, 1967–1985. [Google Scholar] [CrossRef]
- Starczynowski, D.T.; Kuchenbauer, F.; Argiropoulos, B.; Sung, S.; Morin, R.; Muranyi, A.; Hirst, M.; Hogge, D.; Marra, M.; Wells, R.A.; et al. Identification of MiR-145 and MiR-146a as Mediators of the 5q– Syndrome Phenotype. Nat. Med. 2010, 16, 49–58. [Google Scholar] [CrossRef]
- Ribezzo, F.; Snoeren, I.A.M.; Ziegler, S.; Stoelben, J.; Olofsen, P.A.; Henic, A.; Ferreira, M.V.; Chen, S.; Stalmann, U.S.A.; Buesche, G.; et al. Rps14, Csnk1a1 and MiRNA145/MiRNA146a Deficiency Cooperate in the Clinical Phenotype and Activation of the Innate Immune System in the 5q- Syndrome. Leukemia 2019, 33, 1759–1772. [Google Scholar] [CrossRef]
- Mei, Y.; Zhao, B.; Basiorka, A.A.; Yang, J.; Cao, L.; Zhang, J.; List, A.; Ji, P. Age-Related Inflammatory Bone Marrow Microenvironment Induces Ineffective Erythropoiesis Mimicking Del(5q) MDS. Leukemia 2017, 32, 1023–1033. [Google Scholar] [CrossRef] [Green Version]
- Keerthivasan, G.; Mei, Y.; Zhao, B.; Zhang, L.; Harris, C.E.; Gao, J.; Basiorka, A.A.; Schipma, M.J.; McElherne, J.; Yang, J.; et al. Aberrant Overexpression of CD14 on Granulocytes Sensitizes the Innate Immune Response in MDia1 Heterozygous Del(5q) MDS. Blood 2014, 124, 780–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, Y.; Basiorka, A.; Zhao, B.; Yang, J.; List, A.F.; Ji, P. Dual Deficiency of MDia1 and Mir-146a in an Age-Related Inflammatory Bone Marrow Microenvironment Induces Ineffective Erythropoiesis in Del(5q) MDS. Blood 2016, 128, 3146. [Google Scholar] [CrossRef]
- Schneider, R.K.; Schenone, M.; Ferreira, M.V.; Kramann, R.; Joyce, C.E.; Hartigan, C.; Beier, F.; Brümmendorf, T.H.; Germing, U.; Platzbecker, U.; et al. Rps14 Haploinsufficiency Causes a Block in Erythroid Differentiation Mediated by S100A8 and S100A9. Nat. Med. 2016, 22, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Stoner, S.A.; Yan, M.; Liu, K.T.H.; Arimoto, K.-I.; Shima, T.; Wang, H.-Y.; Johnson, D.T.; Bejar, R.; Jamieson, C.; Guan, K.-L.; et al. Hippo Kinase Loss Contributes to Del(20q) Hematologic Malignancies through Chronic Innate Immune Activation. Blood 2019, 134, 1730–1744. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Choudhary, G.S.; Pellagatti, A.; Choi, K.; Bolanos, L.C.; Bhagat, T.D.; Gordon-Mitchell, S.; Von Ahrens, D.; Pradhan, K.; Steeples, V.; et al. U2AF1 Mutations Induce Oncogenic IRAK4 Isoforms and Activate Innate Immune Pathways in Myeloid Malignancies. Nat. Cell Biol. 2019, 21, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Pollyea, D.A.; Harris, C.; Rabe, J.L.; Hedin, B.R.; Arras, L.D.; Katz, S.; Wheeler, E.; Bejar, R.; Walter, M.J.; Jordan, C.T.; et al. Myelodysplastic Syndrome-Associated Spliceosome Gene Mutations Enhance Innate Immune Signaling. Haematologica 2019, 104, e388–e392. [Google Scholar] [CrossRef] [Green Version]
- Komrokji, R.S.; Kulasekararaj, A.; Al Ali, N.H.; Kordasti, S.; Bart-Smith, E.; Craig, B.M.; Padron, E.; Zhang, L.; Lancet, J.E.; Pinilla-Ibarz, J.; et al. Autoimmune Diseases and Myelodysplastic Syndromes. Am. J. Hematol. 2016, 91, E280–E283. [Google Scholar] [CrossRef] [Green Version]
- Mekinian, A.; Grignano, E.; Braun, T.; Decaux, O.; Liozon, E.; Costedoat-Chalumeau, N.; Kahn, J.-E.; Hamidou, M.; Park, S.; Puéchal, X.; et al. Systemic Inflammatory and Autoimmune Manifestations Associated with Myelodysplastic Syndromes and Chronic Myelomonocytic Leukaemia: A French Multicentre Retrospective Study. Rheumatology 2016, 55, 291–300. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.-P.; Boy, M.; Azoulay, C.; Clappier, E.; Sébert, M.; Amable, L.; Klibi, J.; Benlagha, K.; Espéli, M.; Balabanian, K.; et al. Genomic Landscape of MDS/CMML Associated with Systemic Inflammatory and Autoimmune Disease. Leukemia 2021, 35, 2720–2724. [Google Scholar] [CrossRef]
- Arinobu, Y.; Kashiwado, Y.; Miyawaki, K.; Ayano, M.; Kimoto, Y.; Mitoma, H.; Akahoshi, M.; Miyamoto, T.; Horiuchi, T.; Akashi, K.; et al. Autoimmune Manifestations Associated with Myelodysplastic Syndrome Predict a Poor Prognosis. Medicine 2021, 100, e25406. [Google Scholar] [CrossRef]
- Giannouli, S.; Voulgarelis, M.; Zintzaras, E.; Tzioufas, A.G.; Moutsopoulos, H.M. Autoimmune Phenomena in Myelodysplastic Syndromes: A 4-Yr Prospective Study. Rheumatology 2004, 43, 626–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lietzen, L.W.; Cronin-Fenton, D.; Christiansen, P.; Sørensen, H.T.; Lash, T.L. Autoimmune Diseases and Breast Cancer Recurrence: A Danish Nationwide Cohort Study. Breast Cancer Res. Treat. 2015, 149, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.A.; Gadalla, S.; Morton, L.M.; Landgren, O.; Pfeiffer, R.; Warren, J.L.; Berndt, S.I.; Ricker, W.; Parsons, R.; Engels, E.A. Population-Based Study of Autoimmune Conditions and the Risk of Specific Lymphoid Malignancies. Int. J. Cancer 2009, 125, 398–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eaton, W.W.; Rose, N.R.; Kalaydjian, A.; Pedersen, M.G.; Mortensen, P.B. Epidemiology of Autoimmune Diseases in Denmark. J. Autoimmun. 2007, 29, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kristinsson, S.Y.; Björkholm, M.; Hultcrantz, M.; Derolf, Å.R.; Landgren, O.; Goldin, L.R. Chronic Immune Stimulation Might Act as a Trigger for the Development of Acute Myeloid Leukemia or Myelodysplastic Syndromes. J. Clin. Oncol. 2011, 29, 2897–2903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, L.A.; Pfeiffer, R.M.; Landgren, O.; Gadalla, S.; Berndt, S.I.; Engels, E.A. Risks of Myeloid Malignancies in Patients with Autoimmune Conditions. Br. J. Cancer 2009, 100, 822–828. [Google Scholar] [CrossRef] [Green Version]
- Stahl, M.; DeVeaux, M.; de Witte, T.; Neukirchen, J.; Sekeres, M.A.; Brunner, A.M.; Roboz, G.J.; Steensma, D.P.; Bhatt, V.R.; Platzbecker, U.; et al. The Use of Immunosuppressive Therapy in MDS: Clinical Outcomes and Their Predictors in a Large International Patient Cohort. Blood Adv. 2018, 2, 1765–1772. [Google Scholar] [CrossRef] [Green Version]
- Passweg, J.R.; Giagounidis, A.A.N.; Simcock, M.; Aul, C.; Dobbelstein, C.; Stadler, M.; Ossenkoppele, G.; Hofmann, W.-K.; Schilling, K.; Tichelli, A.; et al. Immunosuppressive Therapy for Patients with Myelodysplastic Syndrome: A Prospective Randomized Multicenter Phase III Trial Comparing Antithymocyte Globulin Plus Cyclosporine With Best Supportive Care—SAKK 33/99. J. Clin. Oncol. 2010, 29, 303–309. [Google Scholar] [CrossRef] [Green Version]
- Komrokji, R.S.; Mailloux, A.W.; Chen, D.-T.; Sekeres, M.A.; Paquette, R.; Fulp, W.J.; Sugimori, C.; Paleveda-Pena, J.; Maciejewski, J.P.; List, A.F.; et al. A Phase II Multicenter Rabbit Anti-Thymocyte Globulin Trial in Patients with Myelodysplastic Syndromes Identifying a Novel Model for Response Prediction. Haematologica 2014, 99, 1176–1183. [Google Scholar] [CrossRef] [Green Version]
- Lim, Z.Y.; Killick, S.; Germing, U.; Cavenagh, J.; Culligan, D.; Bacigalupo, A.; Marsh, J.; Mufti, G.J. Low IPSS Score and Bone Marrow Hypocellularity in MDS Patients Predict Hematological Responses to Antithymocyte Globulin. Leukemia 2007, 21, 1436–1441. [Google Scholar] [CrossRef]
- Georgin-Lavialle, S.; Terrier, B.; Guedon, A.F.; Heiblig, M.; Comont, T.; Lazaro, E.; Lacombe, V.; Terriou, L.; Ardois, S.; Bouaziz, J.-D.; et al. Further Characterization of Clinical and Laboratory Features in VEXAS Syndrome: Large-Scale Analysis of a Multicentre Case Series of 116 French Patients. Br. J. Dermatol. 2022, 186, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Beck, D.B.; Ferrada, M.A.; Sikora, K.A.; Ombrello, A.K.; Collins, J.C.; Pei, W.; Balanda, N.; Ross, D.L.; Cardona, D.O.; Wu, Z.; et al. Somatic Mutations in UBA1 and Severe Adult-Onset Autoinflammatory Disease. N. Engl. J. Med. 2020, 383, 2628–2638. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.-P.; Schell, B.; Sébert, M.; Kim, R.; Lemaire, P.; Boy, M.; Mathis, S.; Larcher, L.; Chauvel, C.; Dhouaieb, M.B.; et al. Prevalence of UBA1 Mutations in MDS/CMML Patients with Systemic Inflammatory and Auto-Immune Disease. Leukemia 2021, 35, 2731–2733. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Shimizu, K.; Klimek, V.; Geller, M.D.; Nimer, S.D.; Dhodapkar, M.V. Severe and Selective Deficiency of Interferon-γ-Producing Invariant Natural Killer T Cells in Patients with Myelodysplastic Syndromes. Br. J. Haematol. 2003, 122, 617–622. [Google Scholar] [CrossRef]
- Kiladjian, J.-J.; Bourgeois, E.; Lobe, I.; Braun, T.; Visentin, G.; Bourhis, J.-H.; Fenaux, P.; Chouaib, S.; Caignard, A. Cytolytic Function and Survival of Natural Killer Cells Are Severely Altered in Myelodysplastic Syndromes. Leukemia 2006, 20, 463–470. [Google Scholar] [CrossRef] [Green Version]
- Hejazi, M.; Manser, A.R.; Fröbel, J.; Kündgen, A.; Zhao, X.; Schönberg, K.; Germing, U.; Haas, R.; Gattermann, N.; Uhrberg, M. Impaired Cytotoxicity Associated with Defective Natural Killer Cell Differentiation in Myelodysplastic Syndromes. Haematologica 2015, 100, 643–652. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, N.; Swerdlow, S.H.; TenEyck, S.P.; Boyiadzis, M.; Felgar, R.E. Natural Killer Cell (NK) Subsets and NK-like T-Cell Populations in Acute Myeloid Leukemias and Myelodysplastic Syndromes. Cytometry B Clin. Cytom. 2016, 90, 349–357. [Google Scholar] [CrossRef]
- Cianga, V.A.; Campos Catafal, L.; Cianga, P.; Pavel Tanasa, M.; Cherry, M.; Collet, P.; Tavernier, E.; Guyotat, D.; Rusu, C.; Aanei, C.M. Natural Killer Cell Subpopulations and Inhibitory Receptor Dynamics in Myelodysplastic Syndromes and Acute Myeloid Leukemia. Front. Immunol. 2021, 12, 665541. [Google Scholar] [CrossRef]
- Epling-Burnette, P.K.; Bai, F.; Painter, J.S.; Rollison, D.E.; Salih, H.R.; Krusch, M.; Zou, J.; Ku, E.; Zhong, B.; Boulware, D.; et al. Reduced Natural Killer (NK) Function Associated with High-Risk Myelodysplastic Syndrome (MDS) and Reduced Expression of Activating NK Receptors. Blood 2007, 109, 4816–4824. [Google Scholar] [CrossRef]
- Carlsten, M.; Järås, M. Natural Killer Cells in Myeloid Malignancies: Immune Surveillance, NK Cell Dysfunction, and Pharmacological Opportunities to Bolster the Endogenous NK Cells. Front. Immunol. 2019, 10, 2357. [Google Scholar] [CrossRef] [Green Version]
- Sohlberg, E.; Pfefferle, A.; Andersson, S.; Baumann, B.C.; Hellström-Lindberg, E.; Malmberg, K.-J. Imprint of 5-Azacytidine on the Natural Killer Cell Repertoire during Systemic Treatment for High-Risk Myelodysplastic Syndrome. Oncotarget 2015, 6, 34178–34190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsirogianni, M.; Grigoriou, E.; Kapsimalli, V.; Dagla, K.; Stamouli, M.; Gkirkas, K.; Konsta, E.; Karagiannidou, A.; Gkodopoulos, K.; Stavroulaki, G.; et al. Natural Killer Cell Cytotoxicity Is a Predictor of Outcome for Patients with High Risk Myelodysplastic Syndrome and Oligoblastic Acute Myeloid Leukemia Treated with Azacytidine. Leuk. Lymphoma 2019, 60, 2457–2463. [Google Scholar] [CrossRef] [PubMed]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8+ Cytotoxic T Lymphocytes in Cancer Immunotherapy: A Review. J. Cell. Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef] [PubMed]
- Sand, K.; Theorell, J.; Bruserud, Ø.; Bryceson, Y.T.; Kittang, A.O. Reduced Potency of Cytotoxic T Lymphocytes from Patients with High-Risk Myelodysplastic Syndromes. Cancer Immunol. Immunother. 2016, 65, 1135–1147. [Google Scholar] [CrossRef]
- Tao, J.; Li, L.; Wang, Y.; Fu, R.; Wang, H.; Shao, Z. Increased TIM3+CD8+T Cells in Myelodysplastic Syndrome Patients Displayed Less Perforin and Granzyme B Secretion and Higher CD95 Expression. Leuk. Res. 2016, 51, 49–55. [Google Scholar] [CrossRef]
- Cheng, P.; Eksioglu, E.A.; Chen, X.; Kandell, W.; Le Trinh, T.; Cen, L.; Qi, J.; Sallman, D.A.; Zhang, Y.; Tu, N.; et al. S100A9-Induced Overexpression of PD-1/PD-L1 Contributes to Ineffective Hematopoiesis in Myelodysplastic Syndromes. Leukemia 2019, 33, 2034–2046. [Google Scholar] [CrossRef]
- Fu, R.; Li, L.; Hu, J.; Wang, Y.; Tao, J.; Liu, H.; Liu, Z.; Zhang, W. Elevated TIM3 Expression of T Helper Cells Affects Immune System in Patients with Myelodysplastic Syndrome. J. Investig. Med. 2019, 67, 1125–1130. [Google Scholar] [CrossRef]
- Tao, J.; Han, D.; Gao, S.; Zhang, W.; Yu, H.; Liu, P.; Fu, R.; Li, L.; Shao, Z. CD8+ T Cells Exhaustion Induced by Myeloid-derived Suppressor Cells in Myelodysplastic Syndromes Patients Might Be through TIM3/Gal-9 Pathway. J. Cell. Mol. Med. 2020, 24, 1046–1058. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Li, L.; Lu, F.; Yue, J.; Liu, Z.; Zhang, W.; Fu, R. Overexpression of TIGIT in NK and T Cells Contributes to Tumor Immune Escape in Myelodysplastic Syndromes. Front. Oncol. 2020, 10, 1595. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, S.; Zhang, B.; Qiao, L.; Zhang, Y.; Zhang, Y. T Cell Dysfunction and Exhaustion in Cancer. Front. Cell Dev. Biol. 2020, 8, 17. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Ma, L.; Zhang, X.; Huang, L.; Wei, J. Targeting PD-1/PD-L1 Pathway in Myelodysplastic Syndromes and Acute Myeloid Leukemia. Exp. Hematol. Oncol. 2022, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Giovazzino, A.; Leone, S.; Rubino, V.; Palatucci, A.T.; Cerciello, G.; Alfinito, F.; Pane, F.; Ruggiero, G.; Terrazzano, G. Reduced Regulatory T Cells (Treg) in Bone Marrow Preferentially Associate with the Expansion of Cytotoxic T Lymphocytes in Low Risk MDS Patients. Br. J. Haematol. 2018, 185, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Fozza, C.; Contini, S.; Galleu, A.; Pina Simula, M.; Virdis, P.; Bonfigli, S.; Longinotti, M. Patients with Myelodysplastic Syndromes Display Several T-Cell Expansions, Which Are Mostly Polyclonal in the CD4+ Subset and Oligoclonal in the CD8+ Subset. Exp. Hematol. 2009, 37, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Sloand, E.M.; Melenhorst, J.J.; Tucker, Z.C.G.; Pfannes, L.; Brenchley, J.M.; Yong, A.; Visconte, V.; Wu, C.; Gostick, E.; Scheinberg, P.; et al. T-Cell Immune Responses to Wilms Tumor 1 Protein in Myelodysplasia Responsive to Immunosuppressive Therapy. Blood 2011, 117, 2691–2699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suwabe, T.; Shibasaki, Y.; Sato, H.; Tamura, S.; Katagiri, T.; Nemoto, H.; Kasami, T.; Kozakai, T.; Nanba, A.; Kitajima, T.; et al. WT1-Specific CD8 + Cytotoxic T Cells with the Capacity for Antigen-Specific Expansion Accumulate in the Bone Marrow in MDS. Int. J. Hematol. 2021, 113, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.N.; Ferrari, V.; Tarke, A.; Fields, H.; Ferrari, L.; Ferrari, F.; McCarthy, C.L.; Sanchez, A.P.; Vitiello, A.; Lane, T.A.; et al. Adoptive Transfer of Neoantigen-Specific T-Cell Therapy Is Feasible in Older Patients with Higher-Risk Myelodysplastic Syndrome. Cytotherapy 2021, 23, 236–241. [Google Scholar] [CrossRef]
- Keilholz, U.; Letsch, A.; Busse, A.; Asemissen, A.M.; Bauer, S.; Blau, I.W.; Hofmann, W.-K.; Uharek, L.; Thiel, E.; Scheibenbogen, C. A Clinical and Immunologic Phase 2 Trial of Wilms Tumor Gene Product 1 (WT1) Peptide Vaccination in Patients with AML and MDS. Blood 2009, 113, 6541–6548. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, E.A.; Srivastava, P.; Matsuzaki, J.; Brumberger, Z.; Wang, E.S.; Kocent, J.; Miller, A.; Roloff, G.W.; Wong, H.Y.; Paluch, B.E.; et al. NY-ESO-1 Vaccination in Combination with Decitabine Induces Antigen-Specific T-Lymphocyte Responses in Patients with Myelodysplastic Syndrome. Clin. Cancer Res. 2018, 24, 1019–1029. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, V.; Tarke, A.; Fields, H.; Ferrari, L.; Conley, T.; Ferrari, F.; Koşaloğlu-Yalçın, Z.; Sette, A.; Peters, B.; McCarthy, C.L.; et al. In Vitro Induction of Neoantigen-Specific T Cells in Myelodysplastic Syndrome, a Disease with Low Mutational Burden. Cytotherapy 2021, 23, 320–328. [Google Scholar] [CrossRef]
- Li, T.; Wu, B.; Yang, T.; Zhang, L.; Jin, K. The Outstanding Antitumor Capacity of CD4+ T Helper Lymphocytes. Biochim. Biophys. Acta BBA—Rev. Cancer 2020, 1874, 188439. [Google Scholar] [CrossRef]
- Wang, X.; Wu, D.P.; He, G.; Miao, M.; Sun, A. Research of Subset and Function of Th Cells in Bone Marrow of Myelodysplastic Syndrome Patients. Blood 2005, 106, 4913. [Google Scholar] [CrossRef]
- Li, J.; Yue, L.; Wang, H.; Liu, C.; Liu, H.; Tao, J.; Qi, W.; Wang, Y.; Zhang, W.; Fu, R.; et al. Th17 Cells Exhibit Antitumor Effects in MDS Possibly through Augmenting Functions of CD8+ T Cells. J. Immunol. Res. 2016, 2016, 9404705. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Zhang, L.; Hou, Y.; Yu, S.; Liu, X.; Huang, X.; Sun, Y.; Tian, T.; He, N.; Ma, D.; et al. Th22 Cells as Well as Th17 Cells Expand Differentially in Patients with Early-Stage and Late-Stage Myelodysplastic Syndrome. PLoS ONE 2012, 7, e51339. [Google Scholar] [CrossRef] [PubMed]
- Kordasti, S.Y.; Ingram, W.; Hayden, J.; Darling, D.; Barber, L.; Afzali, B.; Lombardi, G.; Wlodarski, M.W.; Maciejewski, J.P.; Farzaneh, F.; et al. CD4+CD25high Foxp3+ Regulatory T Cells in Myelodysplastic Syndrome (MDS). Blood 2007, 110, 847–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotsianidis, I.; Bouchliou, I.; Nakou, E.; Spanoudakis, E.; Margaritis, D.; Christophoridou, A.V.; Anastasiades, A.; Tsigalou, C.; Bourikas, G.; Karadimitris, A.; et al. Kinetics, Function and Bone Marrow Trafficking of CD4+CD25+FOXP3+ Regulatory T Cells in Myelodysplastic Syndromes (MDS). Leukemia 2009, 23, 510–518. [Google Scholar] [CrossRef] [Green Version]
- Bouchliou, I.; Miltiades, P.; Nakou, E.; Spanoudakis, E.; Goutzouvelidis, A.; Vakalopoulou, S.; Garypidou, V.; Kotoula, V.; Bourikas, G.; Tsatalas, C.; et al. Th17 and Foxp3+ T Regulatory Cell Dynamics and Distribution in Myelodysplastic Syndromes. Clin. Immunol. 2011, 139, 350–359. [Google Scholar] [CrossRef]
- Mailloux, A.W.; Sugimori, C.; Komrokji, R.S.; Yang, L.; Maciejewski, J.P.; Sekeres, M.A.; Paquette, R.; Loughran, T.P.; List, A.F.; Epling-Burnette, P.K. Expansion of Effector Memory Regulatory T Cells Represents a Novel Prognostic Factor in Lower Risk Myelodysplastic Syndrome. J. Immunol. 2012, 189, 3198–3208. [Google Scholar] [CrossRef] [Green Version]
- Kahn, J.D.; Chamuleau, M.E.D.; Westers, T.M.; Van de Ven, P.M.; van Dreunen, L.; van Spronsen, M.; Ossenkoppele, G.J.; van de Loosdrecht, A.A. Regulatory T Cells and Progenitor B Cells Are Independent Prognostic Predictors in Lower Risk Myelodysplastic Syndromes. Haematologica 2015, 100, e220–e222. [Google Scholar] [CrossRef] [Green Version]
- Sallman, D.A.; McLemore, A.F.; Aldrich, A.L.; Komrokji, R.S.; McGraw, K.L.; Dhawan, A.; Geyer, S.; Hou, H.-A.; Eksioglu, E.A.; Sullivan, A.; et al. TP53 Mutations in Myelodysplastic Syndromes and Secondary AML Confer an Immunosuppressive Phenotype. Blood 2020, 136, 2812–2823. [Google Scholar] [CrossRef]
- Mailloux, A.W.; Epling-Burnette, P.K. Effector Memory Regulatory T-Cell Expansion Marks a Pivotal Point of Immune Escape in Myelodysplastic Syndromes. OncoImmunology 2013, 2, e22654. [Google Scholar] [CrossRef] [Green Version]
- Costantini, B.; Kordasti, S.Y.; Kulasekararaj, A.G.; Jiang, J.; Seidl, T.; Abellan, P.P.; Mohamedali, A.; Thomas, N.S.B.; Farzaneh, F.; Mufti, G.J. The Effects of 5-Azacytidine on the Function and Number of Regulatory T Cells and T-Effectors in Myelodysplastic Syndrome. Haematologica 2013, 98, 1196–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bontkes, H.J.; Ruben, J.M.; Alhan, C.; Westers, T.M.; Ossenkoppele, G.J.; van de Loosdrecht, A.A. Azacitidine Differentially Affects CD4pos T-Cell Polarization in Vitro and in Vivo in High Risk Myelodysplastic Syndromes. Leuk. Res. 2012, 36, 921–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-Derived Suppressor Cells in the Era of Increasing Myeloid Cell Diversity. Nat. Rev. Immunol. 2021, 21, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Barreau, S.; Green, A.S.; Dussiau, C.; Alary, A.-S.; Raimbault, A.; Mathis, S.; Willems, L.; Bouscary, D.; Kosmider, O.; Bardet, V.; et al. Phenotypic Landscape of Granulocytes and Monocytes by Multiparametric Flow Cytometry: A Prospective Study of a 1-Tube Panel Strategy for Diagnosis and Prognosis of Patients with MDS. Cytometry B Clin. Cytom. 2020, 98, 226–237. [Google Scholar] [CrossRef]
- Kittang, A.O.; Kordasti, S.; Sand, K.E.; Costantini, B.; Kramer, A.M.; Perezabellan, P.; Seidl, T.; Rye, K.P.; Hagen, K.M.; Kulasekararaj, A.; et al. Expansion of Myeloid Derived Suppressor Cells Correlates with Number of T Regulatory Cells and Disease Progression in Myelodysplastic Syndrome. Oncoimmunology 2015, 5, e1062208. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Jiang, H.; Liu, P.; Xie, N.; Fu, R.; Wang, H.; Liu, C.; Zhang, T.; Wang, H.; Shao, Z. Increased Myeloid-Derived Suppressor Cells in Patients with Myelodysplastic Syndromes Suppress CD8+ T Lymphocyte Function through the STAT3-ARG1 Pathway. Leuk. Lymphoma 2020, 62, 218–223. [Google Scholar] [CrossRef]
- Kapor, S.; Santibanez, J.F. Myeloid-Derived Suppressor Cells and Mesenchymal Stem/Stromal Cells in Myeloid Malignancies. J. Clin. Med. 2021, 10, 2788. [Google Scholar] [CrossRef]
- Han, D.; Tao, J.; Fu, R.; Shao, Z. Myeloid-Derived Suppressor Cell Cytokine Secretion as Prognostic Factor in Myelodysplastic Syndromes. Innate Immun. 2020, 26, 703–715. [Google Scholar] [CrossRef]
- Gleason, M.K.; Ross, J.A.; Warlick, E.D.; Lund, T.C.; Verneris, M.R.; Wiernik, A.; Spellman, S.; Haagenson, M.D.; Lenvik, A.J.; Litzow, M.R.; et al. CD16xCD33 Bispecific Killer Cell Engager (BiKE) Activates NK Cells against Primary MDS and MDSC CD33+ Targets. Blood 2014, 123, 3016–3026. [Google Scholar] [CrossRef]
- Eksioglu, E.A.; Chen, X.; Heider, K.-H.; Rueter, B.; McGraw, K.L.; Basiorka, A.A.; Wei, M.; Burnette, A.; Cheng, P.; Lancet, J.; et al. Novel Therapeutic Approach to Improve Hematopoiesis in Low Risk MDS by Targeting MDSCs with the Fc-Engineered CD33 Antibody BI 836858. Leukemia 2017, 31, 2172–2180. [Google Scholar] [CrossRef] [Green Version]
- Sarhan, D.; Brandt, L.; Felices, M.; Guldevall, K.; Lenvik, T.; Hinderlie, P.; Curtsinger, J.; Warlick, E.; Spellman, S.R.; Blazar, B.R.; et al. 161533 TriKE Stimulates NK-Cell Function to Overcome Myeloid-Derived Suppressor Cells in MDS. Blood Adv. 2018, 2, 1459–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, P.; Chen, X.; Dalton, R.; Calescibetta, A.; So, T.; Gilvary, D.; Ward, G.; Smith, V.; Eckard, S.; Foxa, J.A.; et al. Immunodepletion of MDSC by AMV564, a Novel Bivalent Bispecific CD33/CD3 T-Cell Engager Ex Vivo in MDS and Melanoma. Mol. Ther. J. Am. Soc. Gene Ther. 2022, 30, 2315–2326. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Sun, Z.; Liu, L.; Chen, B.; Cao, Y.; Li, K.; Zhao, R.C. Impairment in Immuno-Modulatory Function of Flk1(+)CD31(−)CD34(−) MSCs from MDS-RA Patients. Available online: https://pubmed-ncbi-nlm-nih-gov.proxy.insermbiblio.inist.fr/17360037/ (accessed on 2 June 2022).
- Zambetti, N.A.; Ping, Z.; Chen, S.; Kenswil, K.J.G.; Mylona, M.A.; Sanders, M.A.; Hoogenboezem, R.M.; Bindels, E.M.; Adisty, M.N.; Van Strien, P.M.H.; et al. Mesenchymal Inflammation Drives Genotoxic Stress in Hematopoietic Stem Cells and Predicts Disease Evolution in Human Pre-Leukemia. Cell Stem Cell 2016, 19, 613–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allampallam, K.; Shetty, V.; Mundle, S.; Dutt, D.; Kravitz, H.; Reddy, P.L.; Alvi, S.; Galili, N.; Saberwal, G.S.; Anthwal, S.; et al. Biological Significance of Proliferation, Apoptosis, Cytokines, and Monocyte/Macrophage Cells in Bone Marrow Biopsies of 145 Patients with Myelodysplastic Syndrome. Available online: https://pubmed-ncbi-nlm-nih-gov.proxy.insermbiblio.inist.fr/11999358/ (accessed on 2 June 2022).
- Han, Y.; Wang, H.; Shao, Z. Monocyte-Derived Macrophages Are Impaired in Myelodysplastic Syndrome. J. Immunol. Res. 2016, 2016, 5479013. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Yang, L.; Han, Y.; Niu, H.; Yan, L.; Shao, Z.; Xing, L.; Wang, H. Abnormal Macrophage Polarization in Patients with Myelodysplastic Syndrome. Available online: https://www.hindawi.com/journals/mi/2021/9913382/ (accessed on 2 June 2022).
- Bento, L.C.; Bacal, N.S.; Rocha, F.A.; Severino, P.; Marti, L.C. Bone Marrow Monocytes and Derived Dendritic Cells from Myelodysplastic Patients Have Functional Abnormalities Associated with Defective Response to Bacterial Infection. J. Immunol. 2020, 204, 2098–2109. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Ceuppens, J.; Kasran, A.; Delforge, M.; Boogaerts, M.; Vandenberghe, P. Immature and Mature Monocyte-Derived Dendritic Cells in Myelodysplastic Syndromes of Subtypes Refractory Anemia or Refractory Anemia with Ringed Sideroblasts Display an Altered Cytokine Profile. Available online: https://pubmed-ncbi-nlm-nih-gov.proxy.insermbiblio.inist.fr/17188353/ (accessed on 2 June 2022).
- Selimoglu-Buet, D.; Wagner-Ballon, O.; Saada, V.; Bardet, V.; Itzykson, R.; Bencheikh, L.; Morabito, M.; Met, E.; Debord, C.; Benayoun, E.; et al. Characteristic Repartition of Monocyte Subsets as a Diagnostic Signature of Chronic Myelomonocytic Leukemia. Blood 2015, 125, 3618–3626. [Google Scholar] [CrossRef] [PubMed]
- Wagner-Ballon, O.; Bettelheim, P.; Lauf, J.; Bellos, F.; Della Porta, M.; Travaglino, E.; Subira, D.; Lopez, I.N.; Tarfi, S.; Westers, T.M.; et al. ELN IMDS Flow Working Group Validation of the Monocyte Assay for Chronic Myelomonocytic Leukemia Diagnosis by Flow Cytometry. Cytometry B Clin. Cytom. 2021, 138, 2602. [Google Scholar] [CrossRef]
- Selimoglu-Buet, D.; Badaoui, B.; Benayoun, E.; Toma, A.; Fenaux, P.; Quesnel, B.; Etienne, G.; Braun, T.; Abermil, N.; Morabito, M.; et al. Accumulation of Classical Monocytes Defines a Subgroup of MDS That Frequently Evolves into CMML. Blood 2017, 130, 832–835. [Google Scholar] [CrossRef]
- Velegraki, M.; Papakonstantinou, N.; Kalaitzaki, L.; Ntoufa, S.; Laidou, S.; Tsagiopoulou, M.; Bizymi, N.; Damianaki, A.; Mavroudi, I.; Pontikoglou, C.; et al. Increased Proportion and Altered Properties of Intermediate Monocytes in the Peripheral Blood of Patients with Lower Risk Myelodysplastic Syndrome. Available online: https://pubmed-ncbi-nlm-nih-gov.proxy.insermbiblio.inist.fr/33032166/ (accessed on 2 June 2022).
- Galon, J.; Costes, A.; Sanchez-Cabo, F.; Kirilovsky, A.; Mlecnik, B.; Lagorce-Pagès, C.; Tosolini, M.; Camus, M.; Berger, A.; Wind, P.; et al. Type, Density, and Location of Immune Cells Within Human Colorectal Tumors Predict Clinical Outcome. Science 2006, 313, 1960–1964. [Google Scholar] [CrossRef] [Green Version]
- Lanzi, A.; Pagès, F.; Lagorce-Pagès, C.; Galon, J. The Consensus Immunoscore: Toward a New Classification of Colorectal Cancer. OncoImmunology 2020, 9, 1789032. [Google Scholar] [CrossRef]
- Mlecnik, B.; Bifulco, C.; Bindea, G.; Marliot, F.; Lugli, A.; Lee, J.J.; Zlobec, I.; Rau, T.T.; Berger, M.D.; Nagtegaal, I.D.; et al. Multicenter International Society for Immunotherapy of Cancer Study of the Consensus Immunoscore for the Prediction of Survival and Response to Chemotherapy in Stage III Colon Cancer. J. Clin. Oncol. 2020, 38, 3638–3651. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, S.; Philip, P.A.; Athanasiadis, I.; Bazarbashi, S.; Shamseddine, A. Classification of Early-Stage Colon Cancer with Immunoscore®: Clinical Evidence and Case Studies. Future Oncol. 2021, 18, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Van de Loosdrecht, A.A.; Kern, W.; Porwit, A.; Valent, P.; Kordasti, S.; Cremers, E.; Alhan, C.; Duetz, C.; Dunlop, A.; Hobo, W.; et al. Clinical Application of Flow Cytometry in Patients with Unexplained Cytopenia and Suspected Myelodysplastic Syndrome: A Report of the European LeukemiaNet International MDS-Flow Cytometry Working Group. Cytometry B Clin. Cytom. 2021. [Google Scholar] [CrossRef] [PubMed]
- Winter, S.; Shoaie, S.; Kordasti, S.; Platzbecker, U. Integrating the “Immunome” in the Stratification of Myelodysplastic Syndromes and Future Clinical Trial Design. J. Clin. Oncol. 2020, 38, 1723–1735. [Google Scholar] [CrossRef]
- Colmenares, R.; Álvarez, N.; Barrio, S.; Martínez-López, J.; Ayala, R. The Minimal Residual Disease Using Liquid Biopsies in Hematological Malignancies. Cancers 2022, 14, 1310. [Google Scholar] [CrossRef]
- Westers, T.M.; Cremers, E.M.P.; Oelschlaegel, U.; Johansson, U.; Bettelheim, P.; Matarraz, S.; Orfao, A.; Moshaver, B.; Brodersen, L.E.; Loken, M.R.; et al. Immunophenotypic Analysis of Erythroid Dysplasia in Myelodysplastic Syndromes. A Report from the IMDSFlow Working Group. Haematologica 2017, 102, 308–319. [Google Scholar] [CrossRef] [Green Version]
- Bardet, V.; Wagner-Ballon, O.; Guy, J.; Morvan, C.; Debord, C.; Trimoreau, F.; Benayoun, E.; Chapuis, N.; Freynet, N.; Rossi, C.; et al. Multicentric Study Underlining the Interest of Adding CD5, CD7 and CD56 Expression Assessment to the Flow Cytometric Ogata Score in Myelodysplastic Syndromes and Myelodysplastic/Myeloproliferative Neoplasms. Haematologica 2015, 100, 472–478. [Google Scholar] [CrossRef] [Green Version]
- Mathis, S.; Chapuis, N.; Debord, C.; Rouquette, A.; Radford-Weiss, I.; Park, S.; Dreyfus, F.; Lacombe, C.; Béné, M.C.; Kosmider, O.; et al. Flow Cytometric Detection of Dyserythropoiesis: A Sensitive and Powerful Diagnostic Tool for Myelodysplastic Syndromes. Leukemia 2013, 27, 1981–1987. [Google Scholar] [CrossRef] [Green Version]
- Shameli, A.; Dharmani-Khan, P.; Luider, J.; Auer, I.; Shabani-Rad, M.-T. Exploring Blast Composition in Myelodysplastic Syndromes and Myelodysplastic/Myeloproliferative Neoplasms: CD45RA and CD371 Improve Diagnostic Value of Flow Cytometry through Assessment of Myeloblast Heterogeneity and Stem Cell Aberrancy. Cytometry B Clin. Cytom. 2021, 100, 574–589. [Google Scholar] [CrossRef]
- Bachas, C.; Duetz, C.; van Spronsen, M.F.; Verhoeff, J.; Garcia Vallejo, J.J.; Jansen, J.H.; Cloos, J.; Westers, T.M.; van de Loosdrecht, A.A. Characterization of Myelodysplastic Syndromes Hematopoietic Stem and Progenitor Cells Using Mass Cytometry. Cytometry B Clin. Cytom. 2022. [Google Scholar] [CrossRef]
- Behbehani, G.K.; Finck, R.; Samusik, N.; Sridhar, K.; Fantl, W.J.; Greenberg, P.L.; Nolan, G.P. Profiling Myelodysplastic Syndromes by Mass Cytometry Demonstrates Abnormal Progenitor Cell Phenotype and Differentiation. Cytometry B Clin. Cytom. 2020, 98, 131–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simoni, Y.; Chapuis, N. Diagnosis of Myelodysplastic Syndromes: From Immunological Observations to Clinical Applications. Diagnostics 2022, 12, 1659. https://doi.org/10.3390/diagnostics12071659
Simoni Y, Chapuis N. Diagnosis of Myelodysplastic Syndromes: From Immunological Observations to Clinical Applications. Diagnostics. 2022; 12(7):1659. https://doi.org/10.3390/diagnostics12071659
Chicago/Turabian StyleSimoni, Yannick, and Nicolas Chapuis. 2022. "Diagnosis of Myelodysplastic Syndromes: From Immunological Observations to Clinical Applications" Diagnostics 12, no. 7: 1659. https://doi.org/10.3390/diagnostics12071659