
Cytogenetic and Genetic Abnormalities with Diagnostic Value in Myelodysplastic Syndromes (MDS): Focus on the Pre-Messenger RNA Splicing Process

 , and
, and
Abstract
:1. Introduction
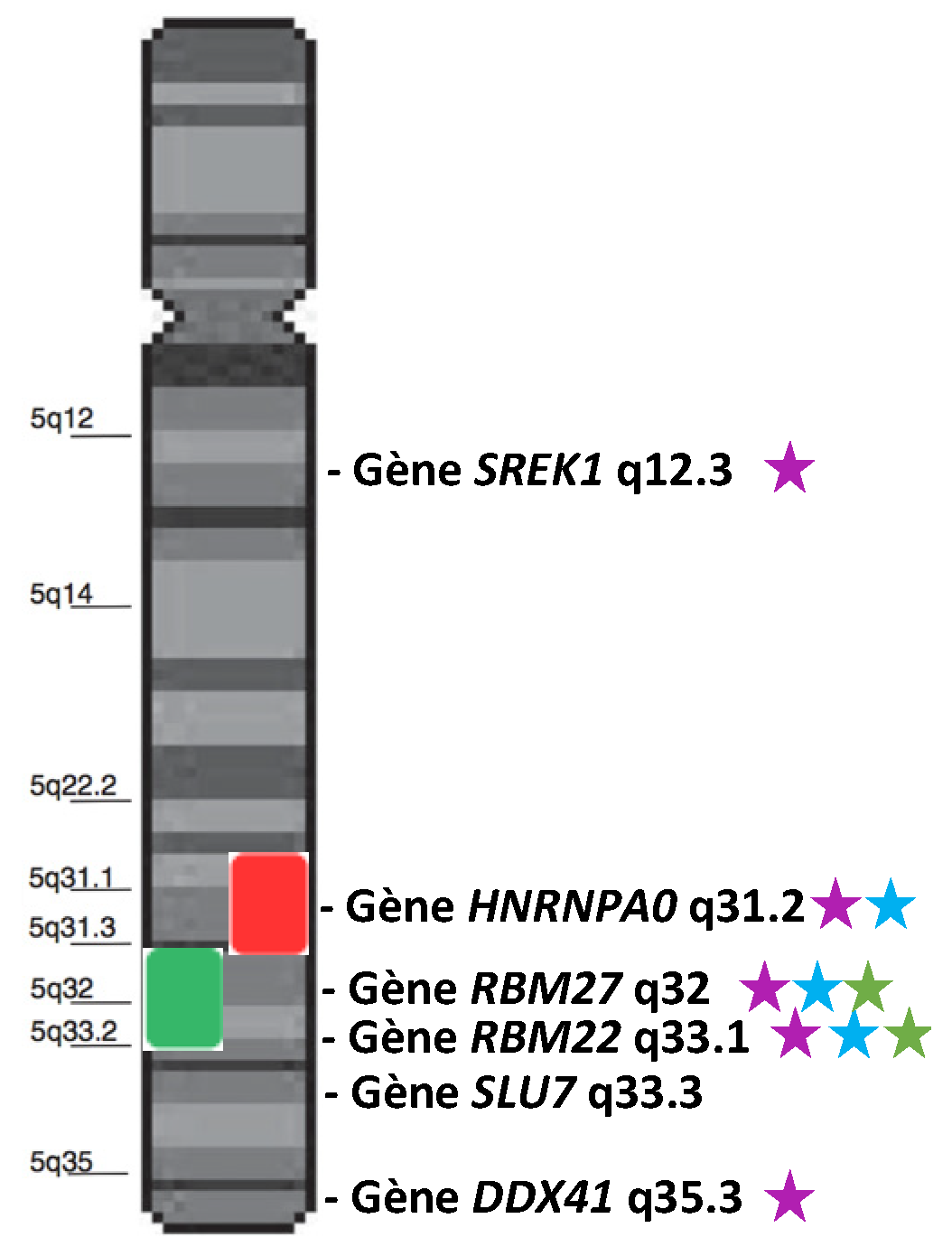
2. Chromosome 5 and Its Genes Implicated in Splicing
2.1. The Diagnostic Value of Chromosome 5 Deletion (del(5q) MDS)
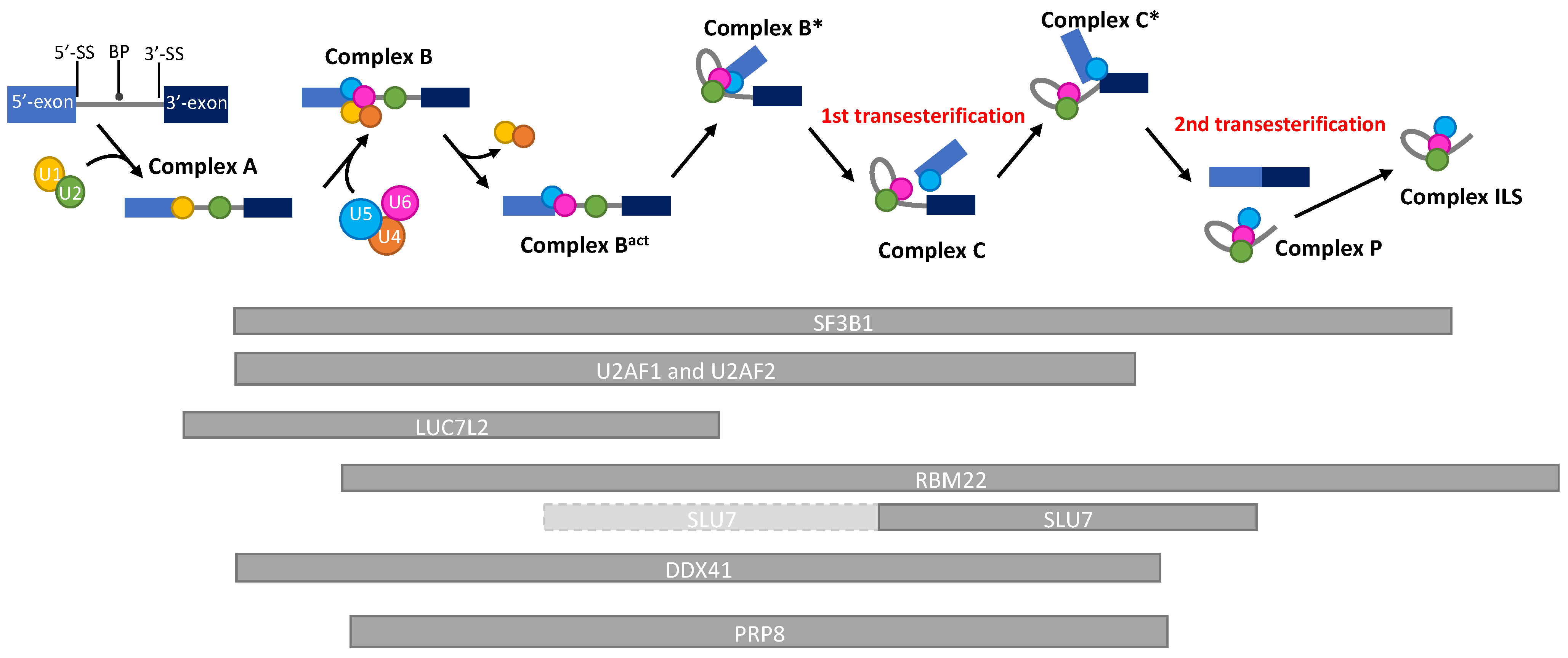
2.2. Splicing Genes Located in the Commonly Deleted Region of the Deletion of 5q
2.2.1. HNRNPA0 (Heterogeneous Nuclear Ribonucleoprotein A0)
2.2.2. RBM27 (RNA-Binding Motif 27)
2.2.3. RBM22 (RNA-Binding Motif 22)
2.3. Splicing Genes in the Variably Deleted Region of the 5q
2.3.1. SLU7 (Synergistic Lethal with U5 snRNA 7)
2.3.2. DDX41 (DEAD-Box Helicase 41)
3. Chromosome 7 and Splicing Genes
3.1. Diagnostic and Prognostic Values
3.2. LUC7L2 Gene
LUC7L2 (Lethal unless CBC 7-Like 2)
4. The Only Gene with, So Far, a Diagnostic Value Is a Splicing Gene: SF3B1
SF3B1 (Splicing Factor 3B Subunit 1)
5. Other Mutated Splicing Genes with a Prognostic Value: SRSF2, U2AF1, ZRSR2, U2AF2 and PRPF8
5.1. The Most Frequently Mutated Splicing Genes with a Prognostic Value: SRSF2, U2AF1, ZRSR2
5.1.1. SRSF2 (Serine and Arginine Rich Splicing Factor 2)
5.1.2. U2AF1 (U2 Small Nuclear RNA Auxiliary Factor 1)
5.1.3. ZRSR2 (Zinc Finger CCCH-Type, RNA Binding Motif and Serine/Arginine Rich 2)
5.2. Other Mutated Splicing Genes with a Prognostic Value: U2AF2, PRPF8
5.2.1. U2AF2 Gene (U2 Small Nuclear RNA Auxiliary Factor 2)
5.2.2. PRPF8 (Pre-MRNA Processing Factor 8)
6. Perspectives and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellström-Lindberg, E.; Tefferi, A.; et al. The 2008 Revision of the World Health Organization (WHO) Classification of Myeloid Neoplasms and Acute Leukemia: Rationale and Important Changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 Revision to the World Health Organization Classification of Myeloid Neoplasms and Acute Leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Nguyen-Khac, F.; Bidet, A.; Daudignon, A.; Lafage-Pochitaloff, M.; Ameye, G.; Bilhou-Nabéra, C.; Chapiro, E.; Collonge-Rame, M.A.; Cuccuini, W.; Douet-Guilbert, N.; et al. The Complex Karyotype in Hematological Malignancies: A Comprehensive Overview by the Francophone Group of Hematological Cytogenetics (GFCH). Leukemia 2022, 36, 1451–1466. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, P.L.; Tuechler, H.; Schanz, J.; Sanz, G.; Garcia-Manero, G.; Solé, F.; Bennett, J.M.; Bowen, D.; Fenaux, P.; Dreyfus, F.; et al. Revised International Prognostic Scoring System for Myelodysplastic Syndromes. Blood 2012, 120, 2454–2465. [Google Scholar] [CrossRef]
- Schanz, J.; Tüchler, H.; Solé, F.; Mallo, M.; Luño, E.; Cervera, J.; Granada, I.; Hildebrandt, B.; Slovak, M.L.; Ohyashiki, K.; et al. New Comprehensive Cytogenetic Scoring System for Primary Myelodysplastic Syndromes (MDS) and Oligoblastic Acute Myeloid Leukemia after MDS Derived from an International Database Merge. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 820–829. [Google Scholar] [CrossRef]
- Yoshida, K.; Sanada, M.; Shiraishi, Y.; Nowak, D.; Nagata, Y.; Yamamoto, R.; Sato, Y.; Sato-Otsubo, A.; Kon, A.; Nagasaki, M.; et al. Frequent Pathway Mutations of Splicing Machinery in Myelodysplasia. Nature 2011, 478, 64–69. [Google Scholar] [CrossRef]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of Genetic Lesions in 944 Patients with Myelodysplastic Syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Hosono, N.; Makishima, H.; Mahfouz, R.; Przychodzen, B.; Yoshida, K.; Jerez, A.; LaFramboise, T.; Polprasert, C.; Clemente, M.J.; Shiraishi, Y.; et al. Recurrent Genetic Defects on Chromosome 5q in Myeloid Neoplasms. Oncotarget 2017, 8, 6483–6495. [Google Scholar] [CrossRef] [Green Version]
- Van Den Berghe, H.; Cassiman, J.-J.; David, G.; Fryns, J.-P.; Michaux, J.-L.; Sokal, G. Distinct Haematological Disorder with Deletion of Long Arm of No. 5 Chromosome. Nature 1974, 251, 437–438. [Google Scholar] [CrossRef]
- Bernard, E.; Nannya, Y.; Hasserjian, R.P.; Devlin, S.M.; Tuechler, H.; Medina-Martinez, J.S.; Yoshizato, T.; Shiozawa, Y.; Saiki, R.; Malcovati, L.; et al. Implications of TP53 Allelic State for Genome Stability, Clinical Presentation and Outcomes in Myelodysplastic Syndromes. Nat. Med. 2020, 26, 1549–1556. [Google Scholar] [CrossRef]
- Bersanelli, M.; Travaglino, E.; Meggendorfer, M.; Matteuzzi, T.; Sala, C.; Mosca, E.; Chiereghin, C.; Di Nanni, N.; Gnocchi, M.; Zampini, M.; et al. Classification and Personalized Prognostic Assessment on the Basis of Clinical and Genomic Features in Myelodysplastic Syndromes. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 1223–1233. [Google Scholar] [CrossRef]
- Giagounidis, A.A.N.; Aul, C. The 5q- Syndrome. Cancer Treat. Res. 2008, 142, 133–148. [Google Scholar]
- Holtan, S.G.; Santana-Davila, R.; Dewald, G.W.; Khetterling, R.P.; Knudson, R.A.; Hoyer, J.D.; Chen, D.; Hanson, C.A.; Porrata, L.; Tefferi, A.; et al. Myelodysplastic Syndromes Associated with Interstitial Deletion of Chromosome 5q: Clinicopathologic Correlations and New Insights from the Pre-Lenalidomide Era. Am. J. Hematol. 2008, 83, 708–713. [Google Scholar] [CrossRef]
- Boultwood, J.; Pellagatti, A.; Cattan, H.; Lawrie, C.H.; Giagounidis, A.; Malcovati, L.; Porta, M.G.D.; Jädersten, M.; Killick, S.; Fidler, C.; et al. Gene Expression Profiling of CD34+ Cells in Patients with the 5q− Syndrome. Br. J. Haematol. 2007, 139, 578–589. [Google Scholar] [CrossRef]
- Eisenmann, K.M.; Dykema, K.J.; Matheson, S.F.; Kent, N.F.; DeWard, A.D.; West, R.A.; Tibes, R.; Furge, K.A.; Alberts, A.S. 5q- Myelodysplastic Syndromes: Chromosome 5q Genes Direct a Tumor-Suppression Network Sensing Actin Dynamics. Oncogene 2009, 28, 3429–3441. [Google Scholar] [CrossRef] [Green Version]
- Douet-Guilbert, N.; De Braekeleer, E.; Basinko, A.; Herry, A.; Gueganic, N.; Bovo, C.; Trillet, K.; Dos Santos, A.; Le Bris, M.J.; Morel, F.; et al. Molecular Characterization of Deletions of the Long Arm of Chromosome 5 (Del(5q)) in 94 MDS/AML Patients. Leukemia 2012, 26, 1695–1697. [Google Scholar] [CrossRef]
- Adema, V.; Palomo, L.; Walter, W.; Mallo, M.; Hutter, S.; Framboise, T.L.; Arenillas, L.; Meggendorfer, M.; Radivoyevitch, T.; Xicoy, B.; et al. Pathophysiologic and Clinical Implications of Molecular Profiles Resultant from Deletion 5q. eBioMedicine 2022, 80, 104059. [Google Scholar] [CrossRef]
- Pellagatti, A.; Cazzola, M.; Giagounidis, A.A.N.; Malcovati, L.; Porta, M.G.D.; Killick, S.; Campbell, L.J.; Wang, L.; Langford, C.F.; Fidler, C.; et al. Gene Expression Profiles of CD34+ Cells in Myelodysplastic Syndromes: Involvement of Interferon-Stimulated Genes and Correlation to FAB Subtype and Karyotype. Blood 2006, 108, 337–345. [Google Scholar] [CrossRef] [Green Version]
- Myer, V.E.; Steitz, J.A. Isolation and Characterization of a Novel, Low Abundance HnRNP Protein: A0. RNA 1995, 1, 171–182. [Google Scholar]
- Cannell, I.G.; Merrick, K.A.; Morandell, S.; Zhu, C.-Q.; Braun, C.J.; Grant, R.A.; Cameron, E.R.; Tsao, M.-S.; Hemann, M.T.; Yaffe, M.B. A Pleiotropic RNA-Binding Protein Controls Distinct Cell Cycle Checkpoints to Drive Resistance of P53-Defective Tumors to Chemotherapy. Cancer Cell 2015, 28, 623–637. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Kong, L.; Guo, S.; Bu, M.; Guo, Q.; Xiong, Y.; Zhu, N.; Qiu, C.; Yan, X.; Chen, Q.; et al. HnRNPs and ELAVL1 Cooperate with UORFs to Inhibit Protein Translation. Nucleic Acids Res. 2017, 45, 2849–2864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, D.J.; Stoddart, A.; Nakitandwe, J.; Chen, S.-C.; Qian, Z.; Downing, J.R.; Beau, M.M.L. Knockdown of Hnrnpa0, a Del(5q) Gene, Alters Myeloid Cell Fate in Murine Cells through Regulation of AU-Rich Transcripts. Haematologica 2014, 99, 1032–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Z.; Joslin, J.M.; Tennant, T.R.; Reshmi, S.C.; Young, D.J.; Stoddart, A.; Larson, R.A.; Le Beau, M.M. Cytogenetic and Genetic Pathways in Therapy-Related Acute Myeloid Leukemia. Chem. Biol. Interact. 2010, 184, 50–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Nostrand, E.L.; Freese, P.; Pratt, G.A.; Wang, X.; Wei, X.; Xiao, R.; Blue, S.M.; Chen, J.-Y.; Cody, N.A.L.; Dominguez, D.; et al. A Large-Scale Binding and Functional Map of Human RNA-Binding Proteins. Nature 2020, 583, 711–719. [Google Scholar] [CrossRef]
- Silla, T.; Schmid, M.; Dou, Y.; Garland, W.; Milek, M.; Imami, K.; Johnsen, D.; Polak, P.; Andersen, J.S.; Selbach, M.; et al. The Human ZC3H3 and RBM26/27 Proteins Are Critical for PAXT-Mediated Nuclear RNA Decay. Nucleic Acids Res. 2020, 48, 2518–2530. [Google Scholar] [CrossRef] [Green Version]
- Rasche, N.; Dybkov, O.; Schmitzová, J.; Akyildiz, B.; Fabrizio, P.; Lührmann, R. Cwc2 and Its Human Homologue RBM22 Promote an Active Conformation of the Spliceosome Catalytic Centre. EMBO J. 2012, 31, 1591–1604. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Yan, C.; Hang, J.; Finci, L.I.; Lei, J.; Shi, Y. An Atomic Structure of the Human Spliceosome. Cell 2017, 169, 918–929.e14. [Google Scholar] [CrossRef] [Green Version]
- Hogg, R.; McGrail, J.C.; O’Keefe, R.T. The Function of the NineTeen Complex (NTC) in Regulating Spliceosome Conformations and Fidelity During Pre-MRNA Splicing. Biochem. Soc. Trans. 2010, 38, 1110–1115. [Google Scholar] [CrossRef] [Green Version]
- Kastner, B.; Will, C.L.; Stark, H.; Lührmann, R. Structural Insights into Nuclear Pre-MRNA Splicing in Higher Eukaryotes. Cold Spring Harb. Perspect. Biol. 2019, 11, a032417. [Google Scholar] [CrossRef]
- van der Feltz, C.; Nikolai, B.; Schneider, C.; Paulson, J.C.; Fu, X.; Hoskins, A.A. Saccharomyces Cerevisiae Ecm2 Modulates the Catalytic Steps of Pre-MRNA Splicing. RNA 2021, 27, 591–603. [Google Scholar] [CrossRef]
- McGrail, J.C.; Krause, A.; O’Keefe, R.T. The RNA Binding Protein Cwc2 Interacts Directly with the U6 SnRNA to Link the Nineteen Complex to the Spliceosome during Pre-MRNA Splicing. Nucleic Acids Res. 2009, 37, 4205–4217. [Google Scholar] [CrossRef] [Green Version]
- Bertram, K.; Agafonov, D.E.; Liu, W.-T.; Dybkov, O.; Will, C.L.; Hartmuth, K.; Urlaub, H.; Kastner, B.; Stark, H.; Lührmann, R. Cryo-EM Structure of a Human Spliceosome Activated for Step 2 of Splicing. Nature 2017, 542, 318–323. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Li, X.; Zhang, L.; Jiang, J.; Hill, R.C.; Cui, Y.; Hansen, K.C.; Zhou, Z.H.; Zhao, R. Structure of the Yeast Spliceosomal Postcatalytic P Complex. Science 2017, 358, 1278–1283. [Google Scholar] [CrossRef] [Green Version]
- Wan, R.; Yan, C.; Bai, R.; Lei, J.; Shi, Y. Structure of an Intron Lariat Spliceosome from Saccharomyces Cerevisiae. Cell 2017, 171, 120–132.e12. [Google Scholar] [CrossRef] [Green Version]
- Park, J.W.; Parisky, K.; Celotto, A.M.; Reenan, R.A.; Graveley, B.R. Identification of Alternative Splicing Regulators by RNA Interference in Drosophila. Proc. Natl. Acad. Sci. USA 2004, 101, 15974–15979. [Google Scholar] [CrossRef] [Green Version]
- Montaville, P.; Dai, Y.; Cheung, C.Y.; Giller, K.; Becker, S.; Michalak, M.; Webb, S.E.; Miller, A.L.; Krebs, J. Nuclear Translocation of the Calcium-Binding Protein ALG-2 Induced by the RNA-Binding Protein RBM22. Biochim. Biophys. Acta 2006, 1763, 1335–1343. [Google Scholar] [CrossRef] [Green Version]
- Janowicz, A.; Michalak, M.; Krebs, J. Stress Induced Subcellular Distribution of ALG-2, RBM22 and HSlu7. Biochim. Biophys. Acta BBA Mol. Cell Res. 2011, 1813, 1045–1049. [Google Scholar] [CrossRef] [Green Version]
- Xiao, R.; Chen, J.-Y.; Liang, Z.; Luo, D.; Chen, G.; Lu, Z.J.; Chen, Y.; Zhou, B.; Li, H.; Du, X.; et al. Pervasive Chromatin-RNA Binding Protein Interactions Enable RNA-Based Regulation of Transcription. Cell 2019, 178, 107–121.e18. [Google Scholar] [CrossRef]
- Fagerberg, L.; Hallström, B.M.; Oksvold, P.; Kampf, C.; Djureinovic, D.; Odeberg, J.; Habuka, M.; Tahmasebpoor, S.; Danielsson, A.; Edlund, K.; et al. Analysis of the Human Tissue-Specific Expression by Genome-Wide Integration of Transcriptomics and Antibody-Based Proteomics. Mol. Cell. Proteom. MCP 2014, 13, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Ebert, B.L.; Pretz, J.; Bosco, J.; Chang, C.Y.; Tamayo, P.; Galili, N.; Raza, A.; Root, D.E.; Attar, E.; Ellis, S.R.; et al. Identification of RPS14 as a 5q- Syndrome Gene by RNA Interference Screen. Nature 2008, 451, 335–339. [Google Scholar] [CrossRef]
- Yamauchi, T.; Masuda, T.; Canver, M.C.; Seiler, M.; Semba, Y.; Shboul, M.; Al-Raqad, M.; Maeda, M.; Schoonenberg, V.A.C.; Cole, M.A.; et al. Genome-Wide CRISPR-Cas9 Screen Identifies Leukemia-Specific Dependence on a Pre-MRNA Metabolic Pathway Regulated by DCPS. Cancer Cell 2018, 33, 386–400.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Ahmed, T.; Krysiak, K.; Shirai, C.L.; Shao, J.; Nunley, R.; Bucala, R.; McKenzie, A.; Ndonwi, M.; Walter, M.J. Haploinsufficiency of Multiple Del(5q) Genes Induce B Cell Abnormalities in Mice. Leuk. Res. 2020, 96, 106428. [Google Scholar] [CrossRef] [PubMed]
- Soubise, B.; Jiang, Y.; Douet-Guilbert, N.; Troadec, M.-B. RBM22, a Key Player of Pre-MRNA Splicing and Gene Expression Regulation, Is Altered in Cancer. Cancers 2022, 14, 643. [Google Scholar] [CrossRef] [PubMed]
- Frank, D.; Patterson, B.; Guthrie, C. Synthetic Lethal Mutations Suggest Interactions between U5 Small Nuclear RNA and Four Proteins Required for the Second Step of Splicing. Mol. Cell. Biol. 1992, 12, 5197–5205. [Google Scholar] [CrossRef]
- Ohrt, T.; Odenwälder, P.; Dannenberg, J.; Prior, M.; Warkocki, Z.; Schmitzová, J.; Karaduman, R.; Gregor, I.; Enderlein, J.; Fabrizio, P.; et al. Molecular Dissection of Step 2 Catalysis of Yeast Pre-MRNA Splicing Investigated in a Purified System. RNA 2013, 19, 902–915. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.-S.; Tseng, C.-K.; Lai, Y.-H.; Wang, H.-F.; Newman, A.J.; Cheng, S.-C. Dynamic Protein-RNA Interactions in Mediating Splicing Catalysis. Nucleic Acids Res. 2019, 47, 899–910. [Google Scholar] [CrossRef] [Green Version]
- James, S.-A.; Turner, W.; Schwer, B. How Slu7 and Prp18 Cooperate in the Second Step of Yeast Pre-MRNA Splicing. RNA 2002, 8, 1068–1077. [Google Scholar] [CrossRef] [Green Version]
- Chua, K.; Reed, R. Human Step II Splicing Factor HSlu7 Functions in Restructuring the Spliceosome between the Catalytic Steps of Splicing. Genes Dev. 1999, 13, 841–850. [Google Scholar] [CrossRef] [Green Version]
- Brys, A.; Schwer, B. Requirement for SLU7 in Yeast Pre-MRNA Splicing Is Dictated by the Distance between the Branchpoint and the 3′ Splice Site. RNA 1996, 2, 707–717. [Google Scholar]
- Shomron, N.; Alberstein, M.; Reznik, M.; Ast, G. Stress Alters the Subcellular Distribution of HSlu7 and Thus Modulates Alternative Splicing. J. Cell Sci. 2005, 118, 1151–1159. [Google Scholar] [CrossRef] [Green Version]
- Jiménez, M.; Urtasun, R.; Elizalde, M.; Azkona, M.; Latasa, M.U.; Uriarte, I.; Arechederra, M.; Alignani, D.; Bárcena-Varela, M.; Álvarez-Sola, G.; et al. Splicing Events in the Control of Genome Integrity: Role of SLU7 and Truncated SRSF3 Proteins. Nucleic Acids Res. 2019, 47, 3450–3466. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Friesen, J.D. Splicing Factor Slt11p and Its Involvement in Formation of U2/U6 Helix II in Activation of the Yeast Spliceosome. Mol. Cell. Biol. 2001, 21, 1011–1023. [Google Scholar] [CrossRef] [Green Version]
- Quesada, A.E.; Routbort, M.J.; DiNardo, C.D.; Bueso-Ramos, C.E.; Kanagal-Shamanna, R.; Khoury, J.D.; Thakral, B.; Zuo, Z.; Yin, C.C.; Loghavi, S.; et al. DDX41 Mutations in Myeloid Neoplasms Are Associated with Male Gender, TP53 Mutations and High-Risk Disease. Am. J. Hematol. 2019, 94, 757–766. [Google Scholar] [CrossRef]
- Polprasert, C.; Schulze, I.; Sekeres, M.A.; Makishima, H.; Przychodzen, B.; Hosono, N.; Singh, J.; Padgett, R.A.; Gu, X.; Phillips, J.G.; et al. Inherited and Somatic Defects in DDX41 in Myeloid Neoplasms. Cancer Cell 2015, 27, 658–670. [Google Scholar] [CrossRef] [Green Version]
- Makishima, H.; Visconte, V.; Sakaguchi, H.; Jankowska, A.M.; Abu Kar, S.; Jerez, A.; Przychodzen, B.; Bupathi, M.; Guinta, K.; Afable, M.G.; et al. Mutations in the Spliceosome Machinery, a Novel and Ubiquitous Pathway in Leukemogenesis. Blood 2012, 119, 3203–3210. [Google Scholar] [CrossRef] [Green Version]
- Singh, H.; Lane, A.A.; Correll, M.; Przychodzen, B.; Sykes, D.B.; Stone, R.M.; Ballen, K.K.; Amrein, P.C.; Maciejewski, J.; Attar, E.C. Putative RNA-Splicing Gene LUC7L2 on 7q34 Represents a Candidate Gene in Pathogenesis of Myeloid Malignancies. Blood Cancer J. 2013, 3, e117. [Google Scholar] [CrossRef]
- Hershberger, C.E.; Moyer, D.C.; Adema, V.; Kerr, C.M.; Walter, W.; Hutter, S.; Meggendorfer, M.; Baer, C.; Kern, W.; Nadarajah, N.; et al. Complex Landscape of Alternative Splicing in Myeloid Neoplasms. Leukemia 2021, 35, 1108–1120. [Google Scholar] [CrossRef]
- Kurtovic-Kozaric, A.; Przychodzen, B.; Singh, J.; Konarska, M.M.; Clemente, M.J.; Otrock, Z.K.; Nakashima, M.; Hsi, E.D.; Yoshida, K.; Shiraishi, Y.; et al. PRPF8 Defects Cause Missplicing in Myeloid Malignancies. Leukemia 2015, 29, 126–136. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Cazzola, M.; Boultwood, J.; Malcovati, L.; Vyas, P.; Bowen, D.; Pellagatti, A.; Wainscoat, J.S.; Hellstrom-Lindberg, E.; Gambacorti-Passerini, C.; et al. Somatic SF3B1 Mutation in Myelodysplasia with Ring Sideroblasts. N. Engl. J. Med. 2011, 365, 1384–1395. [Google Scholar] [CrossRef] [Green Version]
- Thol, F.; Kade, S.; Schlarmann, C.; Löffeld, P.; Morgan, M.; Krauter, J.; Wlodarski, M.W.; Kölking, B.; Wichmann, M.; Görlich, K.; et al. Frequency and Prognostic Impact of Mutations in SRSF2, U2AF1, and ZRSR2 in Patients with Myelodysplastic Syndromes. Blood 2012, 119, 3578–3584. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.-W.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.-B.; Murphy, M.E.; et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015, 27, 617–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Mudireddy, M.; Finke, C.M.; Nicolosi, M.; Lasho, T.L.; Hanson, C.A.; Patnaik, M.M.; Pardanani, A.; Gangat, N. U2AF1 Mutation Variants in Myelodysplastic Syndromes and Their Clinical Correlates. Am. J. Hematol. 2018, 93, E146–E148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Guo, Y.; Dong, Z.; Li, T.; Xie, X.; Wan, D.; Jiang, Z.; Yu, J.; Guo, R. Differential U2AF1 Mutation Sites, Burden and Co-Mutation Genes Can Predict Prognosis in Patients with Myelodysplastic Syndrome. Sci. Rep. 2020, 10, 18622. [Google Scholar] [CrossRef] [PubMed]
- Maji, D.; Glasser, E.; Henderson, S.; Galardi, J.; Pulvino, M.J.; Jenkins, J.L.; Kielkopf, C.L. Representative Cancer-Associated U2AF2 Mutations Alter RNA Interactions and Splicing. J. Biol. Chem. 2020, 295, 17148–17157. [Google Scholar] [CrossRef]
- Antony-Debré, I.; Steidl, U. Functionally Relevant RNA Helicase Mutations in Familial and Sporadic Myeloid Malignancies. Cancer Cell 2015, 27, 609–611. [Google Scholar] [CrossRef] [Green Version]
- Kadono, M.; Kanai, A.; Nagamachi, A.; Shinriki, S.; Kawata, J.; Iwato, K.; Kyo, T.; Oshima, K.; Yokoyama, A.; Kawamura, T.; et al. Biological Implications of Somatic DDX41 p.R525H Mutation in Acute Myeloid Leukemia. Exp. Hematol. 2016, 44, 745–754.e4. [Google Scholar] [CrossRef] [Green Version]
- Sébert, M.; Passet, M.; Raimbault, A.; Rahmé, R.; Raffoux, E.; Sicre de Fontbrune, F.; Cerrano, M.; Quentin, S.; Vasquez, N.; Da Costa, M.; et al. Germline DDX41 Mutations Define a Significant Entity within Adult MDS/AML Patients. Blood 2019, 134, 1441–1444. [Google Scholar] [CrossRef]
- Alkhateeb, H.B.; Nanaa, A.; Viswanatha, D.; Foran, J.M.; Badar, T.; Sproat, L.; He, R.; Nguyen, P.; Jevremovic, D.; Salama, M.E.; et al. Genetic Features and Clinical Outcomes of Patients with Isolated and Comutated DDX41-Mutated Myeloid Neoplasms. Blood Adv. 2022, 6, 528–532. [Google Scholar] [CrossRef]
- Negoro, E.; Radivoyevitch, T.; Polprasert, C.; Adema, V.; Hosono, N.; Makishima, H.; Przychodzen, B.; Hirsch, C.; Clemente, M.J.; Nazha, A.; et al. Molecular Predictors of Response in Patients with Myeloid Neoplasms Treated with Lenalidomide. Leukemia 2016, 30, 2405–2409. [Google Scholar] [CrossRef] [Green Version]
- Abou Dalle, I.; Kantarjian, H.; Bannon, S.A.; Kanagal-Shamanna, R.; Routbort, M.; Patel, K.P.; Hu, S.; Bhalla, K.; Garcia-Manero, G.; DiNardo, C.D. Successful Lenalidomide Treatment in High Risk Myelodysplastic Syndrome with Germline DDX41 Mutation. Am. J. Hematol. 2020, 95, 227–229. [Google Scholar] [CrossRef]
- Fortes, P.; Bilbao-Cortés, D.; Fornerod, M.; Rigaut, G.; Raymond, W.; Séraphin, B.; Mattaj, I.W. Luc7p, a Novel Yeast U1 SnRNP Protein with a Role in 5′ Splice Site Recognition. Genes Dev. 1999, 13, 2425–2438. [Google Scholar] [CrossRef] [Green Version]
- Howell, V.M.; Jones, J.M.; Bergren, S.K.; Li, L.; Billi, A.C.; Avenarius, M.R.; Meisler, M.H. Evidence for a Direct Role of the Disease Modifier SCNM1 in Splicing. Hum. Mol. Genet. 2007, 16, 2506–2516. [Google Scholar] [CrossRef]
- Puig, O.; Bragado-Nilsson, E.; Koski, T.; Séraphin, B. The U1 SnRNP-Associated Factor Luc7p Affects 5′ Splice Site Selection in Yeast and Human. Nucleic Acids Res. 2007, 35, 5874–5885. [Google Scholar] [CrossRef] [Green Version]
- Bai, R.; Wan, R.; Yan, C.; Lei, J.; Shi, Y. Structures of the Fully Assembled Saccharomyces Cerevisiae Spliceosome before Activation. Science 2018, 360, 1423–1429. [Google Scholar] [CrossRef] [Green Version]
- Plaschka, C.; Lin, P.-C.; Charenton, C.; Nagai, K. Prespliceosome Structure Provides Insights into Spliceosome Assembly and Regulation. Nature 2018, 559, 419–422. [Google Scholar] [CrossRef]
- Daniels, N.J.; Hershberger, C.E.; Gu, X.; Schueger, C.; DiPasquale, W.M.; Brick, J.; Saunthararajah, Y.; Maciejewski, J.P.; Padgett, R.A. Functional Analyses of Human LUC7-like Proteins Involved in Splicing Regulation and Myeloid Neoplasms. Cell Rep. 2021, 35, 108989. [Google Scholar] [CrossRef]
- Jourdain, A.A.; Begg, B.E.; Mick, E.; Shah, H.; Calvo, S.E.; Skinner, O.S.; Sharma, R.; Blue, S.M.; Yeo, G.W.; Burge, C.B.; et al. Loss of LUC7L2 and U1 SnRNP Subunits Shifts Energy Metabolism from Glycolysis to OXPHOS. Mol. Cell 2021, 81, 1905–1919.e12. [Google Scholar] [CrossRef]
- Li, C.; Feng, L.; Luo, W.-W.; Lei, C.-Q.; Li, M.; Shu, H.-B. The RNA-Binding Protein LUC7L2 Mediates MITA/STING Intron Retention to Negatively Regulate Innate Antiviral Response. Cell Discov. 2021, 7, 46. [Google Scholar] [CrossRef]
- Kotini, A.G.; Chang, C.-J.; Boussaad, I.; Delrow, J.J.; Dolezal, E.K.; Nagulapally, A.B.; Perna, F.; Fishbein, G.A.; Klimek, V.M.; Hawkins, R.D.; et al. Functional Analysis of a Chromosomal Deletion Associated with Myelodysplastic Syndromes Using Isogenic Human Induced Pluripotent Stem Cells. Nat. Biotechnol. 2015, 33, 646–655. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.; Szankasi, P.; Sederberg, M.; Schumacher, J.; Frizzell, K.A.; Gee, E.P.; Patel, J.L.; South, S.T.; Xu, X.; Kelley, T.W. Concurrent Detection of Targeted Copy Number Variants and Mutations Using a Myeloid Malignancy next Generation Sequencing Panel Allows Comprehensive Genetic Analysis Using a Single Testing Strategy. Br. J. Haematol. 2016, 173, 49–58. [Google Scholar] [CrossRef]
- Hershberger, C.E.; Hosono, N.; Singh, J.; Dietrich, R.C.; Gu, X.; Makishima, H.; Saunthararajah, Y.; Maciejewski, J.P.; Padgett, R.A. The Role of LUC7L2 in Splicing and MDS. Blood 2016, 128, 5504. [Google Scholar] [CrossRef]
- Visconte, V.; O Nakashima, M.; J Rogers, H. Mutations in Splicing Factor Genes in Myeloid Malignancies: Significance and Impact on Clinical Features. Cancers 2019, 11, 1844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jerez, A.; Sugimoto, Y.; Makishima, H.; Verma, A.; Jankowska, A.M.; Przychodzen, B.; Visconte, V.; Tiu, R.V.; O’Keefe, C.L.; Mohamedali, A.M.; et al. Loss of Heterozygosity in 7q Myeloid Disorders: Clinical Associations and Genomic Pathogenesis. Blood 2012, 119, 6109–6117. [Google Scholar] [CrossRef]
- Madan, V.; Li, J.; Zhou, S.; Teoh, W.W.; Han, L.; Meggendorfer, M.; Malcovati, L.; Cazzola, M.; Ogawa, S.; Haferlach, T.; et al. Distinct and Convergent Consequences of Splice Factor Mutations in Myelodysplastic Syndromes. Am. J. Hematol. 2020, 95, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Kfir, N.; Lev-Maor, G.; Glaich, O.; Alajem, A.; Datta, A.; Sze, S.K.; Meshorer, E.; Ast, G. SF3B1 Association with Chromatin Determines Splicing Outcomes. Cell Rep. 2015, 11, 618–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriel-Carretero, M.; Ovejero, S.; Gérus-Durand, M.; Vryzas, D.; Constantinou, A. Fanconi Anemia FANCD2 and FANCI Proteins Regulate the Nuclear Dynamics of Splicing Factors. J. Cell Biol. 2017, 216, 4007–4026. [Google Scholar] [CrossRef] [PubMed]
- De La Garza, A.; Cameron, R.C.; Nik, S.; Payne, S.G.; Bowman, T.V. Spliceosomal Component Sf3b1 Is Essential for Hematopoietic Differentiation in Zebrafish. Exp. Hematol. 2016, 44, 826–837.e4. [Google Scholar] [CrossRef] [Green Version]
- Malcovati, L.; Papaemmanuil, E.; Bowen, D.T.; Boultwood, J.; Della Porta, M.G.; Pascutto, C.; Travaglino, E.; Groves, M.J.; Godfrey, A.L.; Ambaglio, I.; et al. Clinical Significance of SF3B1 Mutations in Myelodysplastic Syndromes and Myelodysplastic/Myeloproliferative Neoplasms. Blood 2011, 118, 6239–6246. [Google Scholar] [CrossRef] [Green Version]
- Malcovati, L.; Karimi, M.; Papaemmanuil, E.; Ambaglio, I.; Jädersten, M.; Jansson, M.; Elena, C.; Gallì, A.; Walldin, G.; Della Porta, M.G.; et al. SF3B1 Mutation Identifies a Distinct Subset of Myelodysplastic Syndrome with Ring Sideroblasts. Blood 2015, 126, 233–241. [Google Scholar] [CrossRef] [Green Version]
- Cretu, C.; Schmitzová, J.; Ponce-Salvatierra, A.; Dybkov, O.; De Laurentiis, E.I.; Sharma, K.; Will, C.L.; Urlaub, H.; Lührmann, R.; Pena, V. Molecular Architecture of SF3b and Structural Consequences of Its Cancer-Related Mutations. Mol. Cell 2016, 64, 307–319. [Google Scholar] [CrossRef] [Green Version]
- Seiler, M.; Peng, S.; Agrawal, A.A.; Palacino, J.; Teng, T.; Zhu, P.; Smith, P.G.; Buonamici, S.; Yu, L.; Caesar-Johnson, S.J.; et al. Somatic Mutational Landscape of Splicing Factor Genes and Their Functional Consequences across 33 Cancer Types. Cell Rep. 2018, 23, 282–296.e4. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.C.-W.; North, K.; Kim, E.; Jang, E.; Obeng, E.; Lu, S.X.; Liu, B.; Inoue, D.; Yoshimi, A.; Ki, M.; et al. Synthetic Lethal and Convergent Biological Effects of Cancer-Associated Spliceosomal Gene Mutations. Cancer Cell 2018, 34, 225–241.e8. [Google Scholar] [CrossRef] [Green Version]
- Darman, R.B.; Seiler, M.; Agrawal, A.A.; Lim, K.H.; Peng, S.; Aird, D.; Bailey, S.L.; Bhavsar, E.B.; Chan, B.; Colla, S.; et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3′ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 2015, 13, 1033–1045. [Google Scholar] [CrossRef] [Green Version]
- Bondu, S.; Alary, A.-S.; Lefèvre, C.; Houy, A.; Jung, G.; Lefebvre, T.; Rombaut, D.; Boussaid, I.; Bousta, A.; Guillonneau, F.; et al. A Variant Erythroferrone Disrupts Iron Homeostasis in SF3B1-Mutated Myelodysplastic Syndrome. Sci. Transl. Med. 2019, 11, eaav5467. [Google Scholar] [CrossRef]
- Bergot, T.; Lippert, E.; Douet-Guilbert, N.; Commet, S.; Corcos, L.; Bernard, D.G. Human Cancer-Associated Mutations of SF3B1 Lead to a Splicing Modification of Its Own RNA. Cancers 2020, 12, 652. [Google Scholar] [CrossRef] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and Biological Implications of Driver Mutations in Myelodysplastic Syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef]
- Mian, S.A.; Rouault-Pierre, K.; Smith, A.E.; Seidl, T.; Pizzitola, I.; Kizilors, A.; Kulasekararaj, A.G.; Bonnet, D.; Mufti, G.J. SF3B1 Mutant MDS-Initiating Cells May Arise from the Haematopoietic Stem Cell Compartment. Nat. Commun. 2015, 6, 10004. [Google Scholar] [CrossRef]
- Malcovati, L.; Stevenson, K.; Papaemmanuil, E.; Neuberg, D.; Bejar, R.; Boultwood, J.; Bowen, D.T.; Campbell, P.J.; Ebert, B.L.; Fenaux, P.; et al. SF3B1 -Mutant MDS as a Distinct Disease Subtype: A Proposal from the International Working Group for the Prognosis of MDS. Blood 2020, 136, 157–170. [Google Scholar] [CrossRef]
- Dalton, W.B.; Helmenstine, E.; Pieterse, L.; Li, B.; Gocke, C.D.; Donaldson, J.; Xiao, Z.; Gondek, L.P.; Ghiaur, G.; Gojo, I.; et al. The K666N Mutation in SF3B1 Is Associated with Increased Progression of MDS and Distinct RNA Splicing. Blood Adv. 2020, 4, 1192–1196. [Google Scholar] [CrossRef]
- Kanagal-Shamanna, R.; Montalban-Bravo, G.; Sasaki, K.; Darbaniyan, F.; Jabbour, E.; Bueso-Ramos, C.; Wei, Y.; Chien, K.; Kadia, T.; Ravandi, F.; et al. Only SF3B1 Mutation Involving K700E Independently Predicts Overall Survival in Myelodysplastic Syndromes. Cancer 2021, 127, 3552–3565. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, J.; Sun, Y.; Perea-Chamblee, T.E.; Manley, J.L.; Rabadan, R. Pan-Cancer Analysis Identifies Mutations in SUGP1 That Recapitulate Mutant SF3B1 Splicing Dysregulation. Proc. Natl. Acad. Sci. USA 2020, 117, 10305–10312. [Google Scholar] [CrossRef]
- Venkataramany, A.S.; Schieffer, K.M.; Lee, K.; Cottrell, C.E.; Wang, P.Y.; Mardis, E.R.; Cripe, T.P.; Chandler, D.S. Alternative RNA Splicing Defects in Pediatric Cancers: New Insights in Tumorigenesis and Potential Therapeutic Vulnerabilities. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2022, 33, 578–592. [Google Scholar] [CrossRef]
- Effenberger, K.A.; Urabe, V.K.; Jurica, M.S. Modulating Splicing with Small Molecular Inhibitors of the Spliceosome. Wiley Interdiscip. Rev. RNA 2017, 8, e1381. [Google Scholar] [CrossRef] [Green Version]
- Bapat, A.; Keita, N.; Martelly, W.; Kang, P.; Seet, C.; Jacobsen, J.R.; Stoilov, P.; Hu, C.; Crooks, G.M.; Sharma, S. Myeloid Disease Mutations of Splicing Factor SRSF2 Cause G2-M Arrest and Skewed Differentiation of Human Hematopoietic Stem and Progenitor Cells. Stem Cells 2018, 36, 1663–1675. [Google Scholar] [CrossRef]
- Slišković, I.; Eich, H.; Müller-McNicoll, M. Exploring the Multifunctionality of SR Proteins. Biochem. Soc. Trans. 2022, 50, 187–198. [Google Scholar] [CrossRef]
- Rahman, M.A.; Lin, K.-T.; Bradley, R.K.; Abdel-Wahab, O.; Krainer, A.R. Recurrent SRSF2 Mutations in MDS Affect Both Splicing and NMD. Genes Dev. 2020, 34, 413–427. [Google Scholar] [CrossRef]
- Chen, L.; Chen, J.-Y.; Huang, Y.-J.; Gu, Y.; Qiu, J.; Qian, H.; Shao, C.; Zhang, X.; Hu, J.; Li, H.; et al. The Augmented R-Loop Is a Unifying Mechanism for Myelodysplastic Syndromes Induced by High-Risk Splicing Factor Mutations. Mol. Cell 2018, 69, 412–425.e6. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Zhan, Z.; Naren, D.; Li, J.; Yan, T.; Gong, Y. Prognostic Value of SRSF2 Mutations in Patients with de Novo Myelodysplastic Syndromes: A Meta-Analysis. PLoS ONE 2017, 12, e0185053. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Luo, Y.; Zhu, S.; Wang, L.; Ma, L.; Zhang, H.; Shen, C.; Yang, W.; Ren, Y.; Zhou, X.; et al. Mutation Status and Burden Can Improve Prognostic Prediction of Patients with Lower-risk Myelodysplastic Syndromes. Cancer Sci. 2020, 111, 580–591. [Google Scholar] [CrossRef] [Green Version]
- Goll, J.B.; Jensen, T.L.; Lindsley, R.C.; Bejar, R.; Walker, J.; Fulton, R.; Abel, G.A.; Al Baghdadi, T.; Deeg, H.J.; DeZern, A.E.; et al. Targeted Sequencing of 7 Genes Can Help Reduce Pathologic Misclassification of MDS. Blood 2020, 136, 32–33. [Google Scholar] [CrossRef]
- Dutta, A.; Yang, Y.; Le, B.T.; Zhang, Y.; Abdel-Wahab, O.; Zang, C.; Mohi, G. U2af1 Is Required for Survival and Function of Hematopoietic Stem/Progenitor Cells. Leukemia 2021, 35, 2382–2398. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Lasho, T.L.; Patnaik, M.M.; Saeed, L.; Mudireddy, M.; Idossa, D.; Finke, C.; Ketterling, R.P.; Pardanani, A.; Gangat, N. Targeted Next-Generation Sequencing in Myelodysplastic Syndromes and Prognostic Interaction between Mutations and IPSS-R. Am. J. Hematol. 2017, 92, 1311–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okeyo-Owuor, T.; White, B.S.; Chatrikhi, R.; Mohan, D.R.; Kim, S.; Griffith, M.; Ding, L.; Ketkar-Kulkarni, S.; Hundal, J.; Laird, K.M.; et al. U2AF1 Mutations Alter Sequence Specificity of Pre-MRNA Binding and Splicing. Leukemia 2015, 29, 909–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellagatti, A.; Armstrong, R.N.; Steeples, V.; Sharma, E.; Repapi, E.; Singh, S.; Sanchi, A.; Radujkovic, A.; Horn, P.; Dolatshad, H.; et al. Impact of Spliceosome Mutations on RNA Splicing in Myelodysplasia: Dysregulated Genes/Pathways and Clinical Associations. Blood 2018, 132, 1225–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.-J.; Tang, J.-L.; Lin, C.-T.; Kuo, Y.-Y.; Li, L.-Y.; Tseng, M.-H.; Huang, C.-F.; Lai, Y.-J.; Lee, F.-Y.; Liu, M.-C.; et al. Clinical Implications of U2AF1 Mutation in Patients with Myelodysplastic Syndrome and Its Stability during Disease Progression. Am. J. Hematol. 2013, 88, E277–E282. [Google Scholar] [CrossRef] [PubMed]
- Adema, V.; Hershberger, C.E.; Walter, W.; Kerr, C.M.; Hutter, S.; Nagata, Y.; Awada, H.; Kongkiatkamon, S.; Snider, C.; Co, M.; et al. Hotspot U2AF1 Mutations Determine Missplicing Selectivity: Novel Mechanisms Altering Splicing Factors. Blood 2019, 134, 2985. [Google Scholar] [CrossRef]
- Madan, V.; Kanojia, D.; Jia, L.; Okamoto, R.; Sato-Otsubo, A.; Kohlmann, A.; Sanada, M.; Grossmann, V.; Sundaresan, J.; Shiraishi, Y.; et al. ZRSR2 Mutations Cause Dysregulated RNA Splicing in MDS. Blood 2014, 124, 4609. [Google Scholar] [CrossRef]
- Madan, V.; Kanojia, D.; Li, J.; Okamoto, R.; Sato-Otsubo, A.; Kohlmann, A.; Sanada, M.; Grossmann, V.; Sundaresan, J.; Shiraishi, Y.; et al. Aberrant Splicing of U12-Type Introns Is the Hallmark of ZRSR2 Mutant Myelodysplastic Syndrome. Nat. Commun. 2015, 6, 6042. [Google Scholar] [CrossRef]
- Tronchre, H.; Wang, J.; Fu, X.-D. A Protein Related to Splicing Factor U2AF35 That Interacts with U2AF65 and SR Proteins in Splicing of Pre-MRNA. Nature 1997, 388, 397–400. [Google Scholar] [CrossRef]
- Inoue, D.; Polaski, J.T.; Taylor, J.; Castel, P.; Chen, S.; Kobayashi, S.; Hogg, S.J.; Hayashi, Y.; Bello Pineda, J.M.; Penson, A.V.; et al. ZRSR2 Mutation Induced Minor Intron Retention Drives MDS and Diverse Cancer Predisposition Via Aberrant Splicing of LZTR1. Blood 2020, 136, 10–11. [Google Scholar] [CrossRef]
- Warnasooriya, C.; Feeney, C.F.; Laird, K.M.; Ermolenko, D.N.; Kielkopf, C.L. A Splice Site-Sensing Conformational Switch in U2AF2 Is Modulated by U2AF1 and Its Recurrent Myelodysplasia-Associated Mutation. Nucleic Acids Res. 2020, 48, 5695–5709. [Google Scholar] [CrossRef]
- Corsini, L.; Bonnal, S.; Bonna, S.; Basquin, J.; Hothorn, M.; Scheffzek, K.; Valcárcel, J.; Sattler, M. U2AF-Homology Motif Interactions Are Required for Alternative Splicing Regulation by SPF45. Nat. Struct. Mol. Biol. 2007, 14, 620–629. [Google Scholar] [CrossRef]
- Selenko, P.; Gregorovic, G.; Sprangers, R.; Stier, G.; Rhani, Z.; Krämer, A.; Sattler, M. Structural Basis for the Molecular Recognition between Human Splicing Factors U2AF65 and SF1/MBBP. Mol. Cell 2003, 11, 965–976. [Google Scholar] [CrossRef]
- Yano, K.; Takahashi, R.-U.; Shiotani, B.; Abe, J.; Shidooka, T.; Sudo, Y.; Yamamoto, Y.; Kan, S.; Sakagami, H.; Tahara, H. PRPF19 Regulates P53-Dependent Cellular Senescence by Modulating Alternative Splicing of MDM4 MRNA. J. Biol. Chem. 2021, 297, 100882. [Google Scholar] [CrossRef]
- Larsson, C.A.; Cote, G.; Quintás-Cardama, A. The Changing Mutational Landscape of Acute Myeloid Leukemia and Myelodysplastic Syndrome. Mol. Cancer Res. MCR 2013, 11, 815–827. [Google Scholar] [CrossRef] [Green Version]
- Bernard, E.; Tuechler, H.; Greenberg, P.L.; Hasserjian, R.P.; Arango, O.J.E.; Nannya, Y.; Devlin, S.M.; Creignou, M.; Pinel, P.; Monnier, L.; et al. Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Evid. 2022, 1, EVIDoa2200008. [Google Scholar] [CrossRef]
- Stanley, R.F.; Abdel-Wahab, O. Dysregulation and Therapeutic Targeting of RNA Splicing in Cancer. Nat. Cancer 2022, 3, 536–546. [Google Scholar] [CrossRef]
- Reyes, J.L.; Kois, P.; Konforti, B.B.; Konarska, M.M. The Canonical GU Dinucleotide at the 5′ Splice Site Is Recognized by P220 of the U5 SnRNP within the Spliceosome. RNA 1996, 2, 213–225. [Google Scholar]
- Reyes, J.L.; Gustafson, E.H.; Luo, H.R.; Moore, M.J.; Konarska, M.M. The C-Terminal Region of HPrp8 Interacts with the Conserved GU Dinucleotide at the 5′ Splice Site. RNA 1999, 5, 167–179. [Google Scholar] [CrossRef] [Green Version]
- MacRae, A.J.; Mayerle, M.; Hrabeta-Robinson, E.; Chalkley, R.J.; Guthrie, C.; Burlingame, A.L.; Jurica, M.S. Prp8 Positioning of U5 SnRNA Is Linked to 5′ Splice Site Recognition. RNA 2018, 24, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Townsend, C.; Leelaram, M.N.; Agafonov, D.E.; Dybkov, O.; Will, C.L.; Bertram, K.; Urlaub, H.; Kastner, B.; Stark, H.; Lührmann, R. Mechanism of Protein-Guided Folding of the Active Site U2/U6 RNA during Spliceosome Activation. Science 2020, 370, eabc3753. [Google Scholar] [CrossRef]
- Grainger, R.J.; Beggs, J.D. Prp8 Protein: At the Heart of the Spliceosome. RNA 2005, 11, 533–557. [Google Scholar] [CrossRef] [Green Version]
- Keightley, M.-C.; Crowhurst, M.O.; Layton, J.E.; Beilharz, T.; Markmiller, S.; Varma, S.; Hogan, B.M.; de Jong-Curtain, T.A.; Heath, J.K.; Lieschke, G.J. In Vivo Mutation of Pre-MRNA Processing Factor 8 (Prpf8) Affects Transcript Splicing, Cell Survival and Myeloid Differentiation. FEBS Lett. 2013, 587, 2150–2157. [Google Scholar] [CrossRef] [Green Version]
- Dolatshad, H.; Pellagatti, A.; Fernandez-Mercado, M.; Yip, B.H.; Malcovati, L.; Attwood, M.; Przychodzen, B.; Sahgal, N.; Kanapin, A.A.; Lockstone, H.; et al. Disruption of SF3B1 Results in Deregulated Expression and Splicing of Key Genes and Pathways in Myelodysplastic Syndrome Hematopoietic Stem and Progenitor Cells. Leukemia 2015, 29, 1092–1103. [Google Scholar] [CrossRef]
- Hosono, N. Genetic Abnormalities and Pathophysiology of MDS. Int. J. Clin. Oncol. 2019, 24, 885–892. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Malcovati, L.; Gallì, A.; Sato-Otsubo, A.; Kataoka, K.; Sato, Y.; Watatani, Y.; Suzuki, H.; Yoshizato, T.; Yoshida, K.; et al. Aberrant Splicing and Defective MRNA Production Induced by Somatic Spliceosome Mutations in Myelodysplasia. Nat. Commun. 2018, 9, 3649. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Gene | Most Important Pathogenic Mutations | References |
|---|---|---|
| DDX41 | Germline mutations: p.M1I Somatic mutations: p.R525H Possible hemizygous deletion: del(5q) | Quesada et al., 2019 [53] Polprasert et al., 2015 [54] |
| HNRNPA0 | Hemizygous deletion: del(5q) Haploinsufficiency reported | Pellagatti et al., 2006 [18] |
| LUC7L2 | Hemizygous deletion: del(7q) or -7 No hot spot somatic mutations: mutations along the gene ex: p.R27X, p.R71H, p.R271, p.R279X | Makishima et al., 2012 [55] Singh et al., 2013 [56] Hershberger et al., 2021 [57] |
| PRPF8 | Somatic mutation: p.D1598N | Haferlach et al., 2014 [7] Kurtovic-Kozaric et al., 2015 [58] |
| RBM22 | Hemizygous deletion: del(5q) No mutation described so far (not specifically searched) Haploinsufficiency reported | Pellagatti et al., 2006 [18] Boultwood et al., 2007 [14] Soubise et al., 2022 [43] |
| RBM27 | Hemizygous deletion: del(5q) No mutation described so far (not specifically searched) Haploinsufficiency reported | Boultwood et al., 2007 [14] |
| SF3B1 | Somatic mutations: p.K700E, p.K666N, p.R625H/C | Papaemmanuil et al., 2011 [59] Yoshida et al., 2011 [6] |
| SLU7 | Possible hemizygous deletion: del(5q) No mutation described so far (not specifically searched) Possible haploinsufficiency when deleted | |
| SRSF2 | Somatic mutations: p.P95H, p.P95L, p.P95R | Yoshida et al., 2011 [6] Thol et al., 2012 [60] Kim et al., 2015 [61] |
| U2AF1 | Somatic mutations: p.S34F, p.S34Y, p.Q157R, p.Q157P | Yoshida et al., 2011 [6] Tefferi et al., 2017 [62] Wang et al., 2020 [63] |
| U2AF2 | Somatic mutations: p.N196K, p.G301D | Maji et al., 2021 [64] |
| ZRSR2 | No hot spot somatic mutations: mutations along the gene (loss-of-function) | Yoshida et al., 2011 [6] Thol et al., 2012 [60] Haferlach et al., 2014 [7] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Douet-Guilbert, N.; Soubise, B.; Bernard, D.G.; Troadec, M.-B. Cytogenetic and Genetic Abnormalities with Diagnostic Value in Myelodysplastic Syndromes (MDS): Focus on the Pre-Messenger RNA Splicing Process. Diagnostics 2022, 12, 1658. https://doi.org/10.3390/diagnostics12071658
Douet-Guilbert N, Soubise B, Bernard DG, Troadec M-B. Cytogenetic and Genetic Abnormalities with Diagnostic Value in Myelodysplastic Syndromes (MDS): Focus on the Pre-Messenger RNA Splicing Process. Diagnostics. 2022; 12(7):1658. https://doi.org/10.3390/diagnostics12071658
Chicago/Turabian StyleDouet-Guilbert, Nathalie, Benoît Soubise, Delphine G. Bernard, and Marie-Bérengère Troadec. 2022. "Cytogenetic and Genetic Abnormalities with Diagnostic Value in Myelodysplastic Syndromes (MDS): Focus on the Pre-Messenger RNA Splicing Process" Diagnostics 12, no. 7: 1658. https://doi.org/10.3390/diagnostics12071658