
Immunoinformatics Approach for Epitope-Based Vaccine Design: Key Steps for Breast Cancer Vaccine
{kind=link}
Abstract
:1. Introduction
2. Design Strategy for Breast Cancer Vaccine
3. Immune Response to Epitope-Based Peptide Vaccine
4. Cancer Vaccine Candidate Criteria
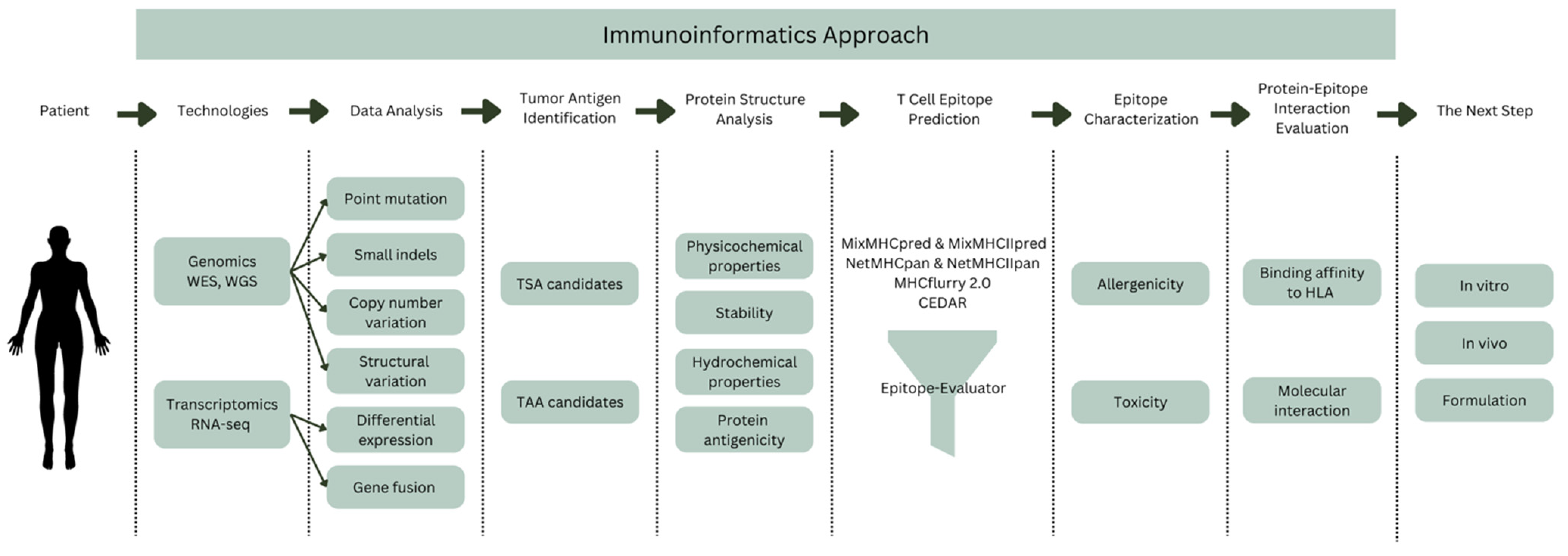
5. Immunoinformatics Approach
5.1. Tumor Antigen Identification
5.2. Protein Structure Analysis
5.3. T Cell Epitope Prediction
5.4. Epitope Characterization
5.5. Protein–Epitope Interaction Evaluation
6. The Next Step
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Observatory, G. Breast Fact Sheet; 2020; Volume 419. [Google Scholar]
- Gautama, W. Breast Cancer in Indonesia in 2022: 30 Years of Marching in Place. Indones. J. Cancer 2022, 16, 1–2. [Google Scholar] [CrossRef]
- Arnold, M.; Morgan, E.; Rumgay, H.; Mafra, A.; Singh, D.; Laversanne, M.; Vignat, J.; Gralow, J.R.; Cardoso, F.; Siesling, S.; et al. Current and Future Burden of Breast Cancer: Global Statistics for 2020 and 2040. Breast 2022, 66, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Behravan, J.; Razazan, A.; Behravan, G. Towards Breast Cancer Vaccines, Progress and Challenges. Curr. Drug Discov. Technol. 2019, 16, 251–258. [Google Scholar] [CrossRef] [PubMed]
- McKittrick, G.; Shepherd, P.; Gilleece, T. Management of Breast Cancer: An Overview for Therapeutic Radiographers. J. Radiother. Pract. 2021, 20, 99–107. [Google Scholar] [CrossRef]
- Fadilah, F.; Erlina, L.; Paramita, R.I.; Istiadi, K.A. Immunoinformatics Studies and Design of Breast Cancer Multiepitope Peptide Vaccines: Diversity Analysis Approach. J. Appl. Pharm. Sci. 2021, 11, 35–45. [Google Scholar] [CrossRef]
- Abdelmoneim, A.H.; Mustafa, M.I.; Abdelmageed, M.I.; Murshed, N.S.; Dawoud, E.D.; Ahmed, E.M.; Kamal Eldein, R.M.; Elfadol, N.M.; Sati, A.O.M.; Makhawi, A.M. Immunoinformatics Design of Multiepitopes Peptide-Based Universal Cancer Vaccine Using Matrix Metalloproteinase-9 Protein as a Target. Immunol. Med. 2020, 44, 35–52. [Google Scholar] [CrossRef]
- Fleri, W.; Paul, S.; Dhanda, S.K.; Mahajan, S.; Xu, X.; Peters, B.; Sette, A. The Immune Epitope Database and Analysis Resource in Epitope Discovery and Synthetic Vaccine Design. Front. Immunol. 2017, 8, 278. [Google Scholar] [CrossRef] [Green Version]
- Kanampalliwar, A.M.; Soni, R.; Girdhar, A.; Tiwari, A. Reverse Vaccinology: Basics and Applications. J. Vaccines Vaccin 2013, 4, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Fu, M.; Wang, M.; Wan, D.; Wei, Y.; Wei, X. Cancer Vaccines as Promising Immuno-Therapeutics: Platforms and Current Progress. J. Hematol. Oncol. 2022, 15, 28. [Google Scholar] [CrossRef]
- Graham, D.B.; Luo, C.; O’Connell, D.J.; Lefkovith, A.; Brown, E.M.; Yassour, M.; Varma, M.; Abelin, J.G.; Conway, K.L.; Jasso, G.J.; et al. Antigen Discovery and Specification of Immunodominance Hierarchies for MHCII-Restricted Epitopes. Nat. Med. 2018, 24, 1762–1772. [Google Scholar] [CrossRef]
- Tay, R.E.; Richardson, E.K.; Toh, H.C. Revisiting the Role of CD4+ T Cells in Cancer Immunotherapy—New Insights into Old Paradigms. Cancer Gene Ther. 2020, 28, 5–17. [Google Scholar] [CrossRef] [PubMed]
- van der Burg Franken, H.; Cornelis, J.M.; Melief, R.; Offringa, S.; Martijn, S.; Bijker, S.J.F.; van den Eeden, K.L. CD8+ CTL Priming by Exact Peptide Epitopes in Incomplete Freund’s Adjuvant Induces a Vanishing CTL Response, Whereas Long Peptides Induce Sustained CTL Reactivity. J. Immunol. 2007, 179, 5033–5040. [Google Scholar] [CrossRef] [Green Version]
- Bijker, M.S.; van den Eeden, S.J.F.; Franken, K.L.; Melief, C.J.M.; van der Burg, S.H.; Offringa, R. Superior Induction of Anti-Tumor CTL Immunity by Extended Peptide Vaccines Involves Prolonged, DC-Focused Antigen Presentation. Eur. J. Immunol. 2008, 38, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Rosenberg, S.A. Adoptive T-Cell Therapy for Cancer. Adv. Immunol. 2016, 130, 279–294. [Google Scholar] [CrossRef]
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen Presentation in Cancer: Insights into Tumour Immunogenicity and Immune Evasion. Nat. Rev. Cancer 2021, 21, 298–312. [Google Scholar] [CrossRef]
- Radford, K.J.; Higgins, D.E.; Pasquini, S.; Cheadle, E.J.; Carta, L.; Jackson, A.M.; Lemoine, N.R.; Vassaux, G. A Recombinant E. coli Vaccine to Promote MHC Class I-Dependent Antigen Presentation: Application to Cancer Immunotherapy. Gene 2002, 9, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Wong-Arce, A.; González-Ortega, O.; Rosales-Mendoza, S. Plant-Made Vaccines in the Fight Against Cancer. Trends Biotechnol. 2017, 35, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Heery, C.R.; Singh, B.H.; Rauckhorst, M.; Marté, J.L.; Donahue, R.N.; Grenga, I.; Rodell, T.C.; Dahut, W.; Arlen, P.M.; Madan, R.A.; et al. Phase I Trial of a Yeast-Based Therapeutic Cancer Vaccine (GI-6301) Targeting the Transcription Factor Brachyury. Cancer Immunol. Res. 2015, 3, 1248–1256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betting, D.J.; Mu, X.Y.; Kafi, K.; McDonnel, D.; Rosas, F.; Gold, D.P.; Timmerman, J.M. Enhanced Immune Stimulation by a Therapeutic Lymphoma Tumor Antigen Vaccine Produced in Insect Cells Involves Mannose Receptor Targeting to Antigen Presenting Cells. Vaccine 2009, 27, 250. [Google Scholar] [CrossRef]
- Song, C.; Zheng, X.J.; Liu, C.C.; Zhou, Y.; Ye, X.S. A Cancer Vaccine Based on Fluorine-Modified Sialyl-Tn Induces Robust Immune Responses in a Murine Model. Oncotarget 2017, 8, 47330. [Google Scholar] [CrossRef]
- Buonaguro, L.; Tagliamonte, M. Selecting Target Antigens for Cancer Vaccine Development. Vaccines 2020, 8, 615. [Google Scholar] [CrossRef] [PubMed]
- Benavides, L.C.; Gates, J.D.; Carmichael, M.G.; Patel, R.; Holmes, J.P.; Hueman, M.T.; Mittendorf, E.A.; Craig, D.; Stojadinovic, A.; Ponniah, S.; et al. The Impact of HER2/Neu Expression Level on Response to the E75 Vaccine: From U.S. Military Cancer Institute Clinical Trials Group Study I-01 and I-02. Clin. Cancer Res. 2009, 15, 2895–2904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittendorf, E.A.; Storrer, C.E.; Foley, R.J.; Harris, K.; Jama, Y.; Shriver, C.D.; Ponniah, S.; Peoples, G.E. Evaluation of the HER2/Neu-Derived Peptide GP2 for Use in a Peptide-Based Breast Cancer Vaccine Trial. Cancer 2006, 106, 2309–2317. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.A.; Mittendorf, E.A.; Hale, D.F.; Myers, J.W.; Peace, K.M.; Jackson, D.O.; Greene, J.M.; Vreeland, T.J.; Clifton, G.T.; Ardavanis, A.; et al. Prospective, Randomized, Single-Blinded, Multi-Center Phase II Trial of Two HER2 Peptide Vaccines, GP2 and AE37, in Breast Cancer Patients to Prevent Recurrence. Breast Cancer Res. Treat. 2020, 181, 391. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2018, 37, 173–200. [Google Scholar] [CrossRef]
- Yarchoan, M.; Johnson, B.A.; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting Neoantigens to Augment Antitumour Immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in Cancer Immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Reddehase, M.J.; Rothbard, J.B.; Koszinowski, U.H. A Pentapeptide as Minimal Antigenic Determinant for MHC Class I-Restricted T Lymphocytes. Nature 1989, 337, 651–653. [Google Scholar] [CrossRef] [Green Version]
- Hemmer, B.; Kondo, T.; Gran, B.; Pinilla, C.; Cortese, I.; Pascal, J.; Tzou, A.; McFarland, H.F.; Houghten, R.; Martin, R. Minimal Peptide Length Requirements for CD4+ T Cell Clones—Implications for Molecular Mimicry and T Cell Survival. Int. Immunol. 2000, 12, 375–383. [Google Scholar] [CrossRef]
- Fishman, J.M.; Wiles, K.; Wood, K.J. The Acquired Immune System Response to Biomaterials, Including Both Naturally Occurring and Synthetic Biomaterials. In Host Response to Biomaterials: The Impact of Host Response on Biomaterial Selection; Academic Press: Cambridge, MA, USA, 2015; pp. 151–187. [Google Scholar] [CrossRef]
- Malonis, R.J.; Lai, J.R.; Vergnolle, O. Peptide-Based Vaccines: Current Progress and Future Challenges. Chem. Rev. 2020, 120, 3210–3229. [Google Scholar] [CrossRef] [Green Version]
- Chowell, D.; Morris, L.G.T.; Grigg, C.M.; Weber, J.K.; Samstein, R.M.; Makarov, V.; Kuo, F.; Kendall, S.M.; Requena, D.; Riaz, N.; et al. Patient HLA Class I Genotype Influences Cancer Response to Checkpoint Blockade Immunotherapy. Science 2018, 359, 582–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bui, H.-H.; Sidney, J.; Dinh, K.; Southwood, S.; Newman, M.J.; Sette, A. Predicting Population Coverage of T-Cell Epitope-Based Diagnostics and Vaccines. BMC Bioinform. 2006, 7, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Galarza, F.F.; McCabe, A.; Melo dos Santos, E.J.; Jones, A.R.; Middleton, D. A Snapshot of Human Leukocyte Antigen (HLA) Diversity Using Data from the Allele Frequency Net Database. Hum. Immunol. 2021, 82, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Galarza, F.F.; McCabe, A.; dos Santos, E.J.M.; Jones, J.; Takeshita, L.; Ortega-Rivera, N.D.; Cid-Pavon, G.M.D.; Ramsbottom, K.; Ghattaoraya, G.; Alfirevic, A.; et al. Allele Frequency Net Database (AFND) 2020 Update: Gold-Standard Data Classification, Open Access Genotype Data and New Query Tools. Nucleic Acids Res. 2020, 48, D783. [Google Scholar] [CrossRef]
- Martini, S.; Nielsen, M.; Peters, B.; Sette, A. The Immune Epitope Database and Analysis Resource Program 2003–2018: Reflections and Outlook. Immunogenetics 2020, 72, 57. [Google Scholar] [CrossRef] [Green Version]
- Requena, D.; Médico, A.; Chacón, R.D.; Ramírez, M.; Marín-Sánchez, O. Identification of Novel Candidate Epitopes on SARS-CoV-2 Proteins for South America: A Review of HLA Frequencies by Country. Front. Immunol. 2020, 11, 2008. [Google Scholar] [CrossRef] [PubMed]
- Janse van Rensburg, W.J.; de Kock, A.; Bester, C.; Kloppers, J.F. HLA Major Allele Group Frequencies in a Diverse Population of the Free State Province, South Africa. Heliyon 2021, 7, e06850. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.J.; Svensson-Arvelund, J.; Lubitz, G.S.; Marabelle, A.; Melero, I.; Brown, B.D.; Brody, J.D. Cancer Vaccines: The next Immunotherapy Frontier. Nat. Cancer 2022, 3, 911–926. [Google Scholar] [CrossRef]
- Hatziioannou, A.; Alissafi, T.; Verginis, P. Myeloid-Derived Suppressor Cells and T Regulatory Cells in Tumors: Unraveling the Dark Side of the Force. J. Leukoc. Biol. 2017, 102, 407–421. [Google Scholar] [CrossRef] [Green Version]
- Minor, P. Considerations for Setting the Specifications of Vaccines. Expert Rev. Vaccines 2012, 11, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yin, B.; Wang, H.Y.; Wang, R.F. Current Advances in T-Cell-Based Cancer Immunotherapy. Immunotherapy 2014, 6, 1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braden, B.C.; Dall’Acqua, W.; Eisenstein, E.; Fields, B.A.; Goldbaum, F.A.; Malchiodi, E.L.; Mariuzza, R.A.; Schwarz, F.P.; Ysern, X.; Poljak, R.J. Protein Motion and Lock and Key Complementarity in Antigen-Antibody Reactions. Pharm. Acta Helv. 1995, 69, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Garboczi, D.N.; Ghosh, P.; Utz, U.; Fan, Q.R.; Biddison, W.E.; Wiley, D.C. Structure of the Complex between Human T-Cell Receptor, Viral Peptide and HLA-A2. Nature 1996, 384, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Sidney, J.; del Guercio, M.F.; Southwood, S.; Engelhard, V.H.; Appella, E.; Rammensee, H.G.; Falk, K.; Rötzschke, O.; Takiguchi, M.; Kubo, R.T. Several HLA Alleles Share Overlapping Peptide Specificities. J. Immunol. 1995, 154, 247–259. [Google Scholar] [PubMed]
- Chesnut, R.W.; Grey, H.M.; Guercio, A.S.; Appella, E.; Hoffman, S.; Kubo, R.T.; Southwood, S.; Sidney, J.; Kondo, A.; Del, M.-F. Overlapping Peptide Binding Repertoires Several Common HLA-DR Types Share Largely. J. Immunol. Ref. 2022, 163, 3363–3373. [Google Scholar]
- Diao, L.; Meibohm, B. Pharmacokinetics and Pharmacokinetic–Pharmacodynamic Correlations of Therapeutic Peptides. Clin. Pharmacokinet. 2013, 52, 855–868. [Google Scholar] [CrossRef]
- Di, L. Strategic Approaches to Optimizing Peptide ADME Properties. AAPS J. 2015, 17, 134–143. [Google Scholar] [CrossRef] [Green Version]
- Robbins, P.F.; Lu, Y.C.; El-Gamil, M.; Li, Y.F.; Gross, C.; Gartner, J.; Lin, J.C.; Teer, J.K.; Cliften, P.; Tycksen, E.; et al. Mining Exomic Sequencing Data to Identify Mutated Antigens Recognized by Adoptively Transferred Tumor-Reactive T Cells. Nat. Med. 2013, 19, 747–752. [Google Scholar] [CrossRef]
- Cohen, C.J.; Gartner, J.J.; Horovitz-Fried, M.; Shamalov, K.; Trebska-McGowan, K.; Bliskovsky, V.V.; Parkhurst, M.R.; Ankri, C.; Prickett, T.D.; Crystal, J.S.; et al. Isolation of Neoantigen-Specific T Cells from Tumor and Peripheral Lymphocytes. J. Clin. Investig. 2015, 125, 3981. [Google Scholar] [CrossRef] [Green Version]
- Vollers, S.S.; Stern, L.J. Class II Major Histocompatibility Complex Tetramer Staining: Progress, Problems, and Prospects. Immunology 2008, 123, 305. [Google Scholar] [CrossRef]
- Garcia-Garijo, A.; Fajardo, C.A.; Gros, A. Determinants for Neoantigen Identification. Front. Immunol. 2019, 10, 1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koşaloğlu-Yalçın, Z.; Lanka, M.; Frentzen, A.; Logandha Ramamoorthy Premlal, A.; Sidney, J.; Vaughan, K.; Greenbaum, J.; Robbins, P.; Gartner, J.; Sette, A.; et al. Predicting T Cell Recognition of MHC Class I Restricted Neoepitopes. Oncoimmunology 2018, 7, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, E.; Robbins, P.F.; Rosenberg, S.A. ‘Final Common Pathway’ of Human Cancer Immunotherapy: Targeting Random Somatic Mutations. Nat. Immunol. 2017, 18, 255. [Google Scholar] [CrossRef]
- Varshavsky, A. The N-End Rule Pathway of Protein Degradation. Genes Cells 1997, 2, 13–28. [Google Scholar] [CrossRef] [PubMed]
- Guruprasad, K.; Reddy, B.V.B.; Pandit, M.W. Correlation between Stability of a Protein and Its Dipeptide Composition: A Novel Approach for Predicting in Vivo Stability of a Protein from Its Primary Sequence. Protein Eng. Des. Sel. 1990, 4, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J.; Doolittle, R.F. A Simple Method for Displaying the Hydropathic Character of a Protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Doytchinova, I.A.; Flower, D.R. VaxiJen: A Server for Prediction of Protective Antigens, Tumour Antigens and Subunit Vaccines. BMC Bioinform. 2007, 8, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, B.; Reynisson, B.; Barra, C.; Buus, S.; Ternette, N.; Connelley, T.; Andreatta, M.; Nielsen, M. NNAlign_MA; MHC Peptidome Deconvolution for Accurate MHC Binding Motif Characterization and Improved T-Cell Epitope Predictions. Mol. Cell. Proteom. 2019, 18, 2459–2477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynisson, B.; Alvarez, B.; Paul, S.; Peters, B.; Nielsen, M. NetMHCpan-4.1 and NetMHCIIpan-4.0: Improved Predictions of MHC Antigen Presentation by Concurrent Motif Deconvolution and Integration of MS MHC Eluted Ligand Data. Nucleic Acids Res. 2020, 48, W449. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, T.J.; Rubinsteyn, A.; Laserson, U. MHCflurry 2.0: Improved Pan-Allele Prediction of MHC Class I-Presented Peptides by Incorporating Antigen Processing. Cell Syst. 2020, 11, 42–48. [Google Scholar] [CrossRef]
- Wolf-Levy, H.; Javitt, A.; Eisenberg-Lerner, A.; Kacen, A.; Ulman, A.; Sheban, D.; Dassa, B.; Fishbain-Yoskovitz, V.; Carmona-Rivera, C.; Kramer, M.P.; et al. Revealing the Cellular Degradome by Mass Spectrometry Analysis of Proteasome-Cleaved Peptides. Nat. Biotechnol. 2018, 36, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Bassani-Sternberg, M.; Chong, C.; Guillaume, P.; Solleder, M.; Pak, H.S.; Gannon, P.O.; Kandalaft, L.E.; Coukos, G.; Gfeller, D. Deciphering HLA-I Motifs across HLA Peptidomes Improves Neo-Antigen Predictions and Identifies Allostery Regulating HLA Specificity. PLoS Comput. Biol. 2017, 13, e1005725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Racle, J.; Michaux, J.; Rockinger, G.A.; Arnaud, M.; Bobisse, S.; Chong, C.; Guillaume, P.; Coukos, G.; Harari, A.; Jandus, C.; et al. HLA-II Motif Deconvolution for Robust Epitope Predictions Deep Motif Deconvolution of HLA-II Peptidomes for Robust Class II Epitope Predictions Running Title: HLA-II Motif Deconvolution for Robust Epitope Predictions. bioRxiv 2019. [Google Scholar] [CrossRef]
- Soto, L.F.; Requena, D.; Fuxman Bass, J.I. Epitope-Evaluator: An Interactive Web Application to Study Predicted T-Cell Epitopes. PLoS ONE 2022, 17, e0273577. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Sher, X. Systematically Benchmarking Peptide-MHC Binding Predictors: From Synthetic to Naturally Processed Epitopes. PLoS Comput. Biol. 2018, 14, e1006457. [Google Scholar] [CrossRef] [PubMed]
- Mei, S.; Li, F.; Leier, A.; Marquez-Lago, T.T.; Giam, K.; Croft, N.P.; Akutsu, T.; Ian Smith, A.; Li, J.; Rossjohn, J.; et al. A Comprehensive Review and Performance Evaluation of Bioinformatics Tools for HLA Class I Peptide-Binding Prediction. Brief. Bioinform. 2020, 21, 1119–1135. [Google Scholar] [CrossRef] [PubMed]
- Koşaloğlu-Yalçın, Z.; Blazeska, N.; Carter, H.; Nielsen, M.; Cohen, E.; Kufe, D.; Conejo-Garcia, J.; Robbins, P.; Schoenberger, S.P.; Peters, B.; et al. The Cancer Epitope Database and Analysis Resource: A Blueprint for the Establishment of a New Bioinformatics Resource for Use by the Cancer Immunology Community. Front. Immunol. 2021, 12, 735609. [Google Scholar] [CrossRef]
- Dimitrov, I.; Naneva, L.; Doytchinova, I.; Bangov, I. AllergenFP: Allergenicity Prediction by Descriptor Fingerprints. Bioinformatics 2014, 30, 846–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Kapoor, P.; Chaudhary, K.; Gautam, A.; Kumar, R.; Raghava, G.P.S. In Silico Approach for Predicting Toxicity of Peptides and Proteins. PLoS ONE 2013, 8, e73957. [Google Scholar] [CrossRef] [Green Version]
- Meng, X.-Y.; Zhang, H.-X.; Mezei, M.; Cui, M. Molecular Docking: A Powerful Approach for Structure-Based Drug Discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef]
- Lee, H.; Heo, L.; Lee, M.S.; Seok, C. GalaxyPepDock: A Protein–Peptide Docking Tool Based on Interaction Similarity and Energy Optimization. Nucleic Acids Res. 2015, 43, W431–W435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, N.; Schueler-Furman, O. Modeling Peptide-Protein Structure and Binding Using Monte Carlo Sampling Approaches: Rosetta Flexpepdock and Flexpepbind. Methods Mol. Biol. 2017, 1561, 139–169. [Google Scholar] [CrossRef] [PubMed]
- London, N.; Raveh, B.; Cohen, E.; Fathi, G.; Schueler-Furman, O. Rosetta FlexPepDock Web Server—High Resolution Modeling of Peptide–Protein Interactions. Nucleic Acids Res. 2011, 39, W249–W253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antes, I. DynaDock: A New Molecular Dynamics-Based Algorithm for Protein–Peptide Docking Including Receptor Flexibility. Proteins Struct. Funct. Bioinform. 2010, 78, 1084–1104. [Google Scholar] [CrossRef]
- Raveh, B.; London, N.; Zimmerman, L.; Schueler-Furman, O. Rosetta FlexPepDock Ab-Initio: Simultaneous Folding, Docking and Refinement of Peptides onto Their Receptors. PLoS ONE 2011, 6, e18934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, R.; Renard, B.Y. Docking Small Peptides Remains a Great Challenge: An Assessment Using AutoDock Vina. Brief. Bioinform. 2015, 16, 1045–1056. [Google Scholar] [CrossRef] [Green Version]
- Yan, C.; Xu, X.; Zou, X. Fully Blind Docking at the Atomic Level for Protein-Peptide Complex Structure Prediction. Structure 2016, 24, 1842–1853. [Google Scholar] [CrossRef] [Green Version]
- de Vries, S.J.; Rey, J.; Schindler, C.E.M.; Zacharias, M.; Tuffery, P. The PepATTRACT Web Server for Blind, Large-Scale Peptide–Protein Docking. Nucleic Acids Res. 2017, 45, W361–W364. [Google Scholar] [CrossRef]
- Ben-Shimon, A.; Niv, M.Y. AnchorDock: Blind and Flexible Anchor-Driven Peptide Docking. Structure 2015, 23, 929–940. [Google Scholar] [CrossRef]
- Kurcinski, M.; Jamroz, M.; Blaszczyk, M.; Kolinski, A.; Kmiecik, S. CABS-Dock Web Server for the Flexible Docking of Peptides to Proteins without Prior Knowledge of the Binding Site. Nucleic Acids Res. 2015, 43, W419–W424. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, D.I.; le Nours, J.; Andrews, D.M.; Uldrich, A.P.; Rossjohn, J. Unconventional T Cell Targets for Cancer Immunotherapy. Immunity 2018, 48, 453–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeiffer, C.; Stein, J.; Southwood, S.; Ketelaar, H.; Sette, A.; Bottomly, K. Altered Peptide Ligands Can Control CD4 T Lymphocyte Differentiation in Vivo. J. Exp. Med. 1995, 181, 1569–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholz, C.; Hlllsberg, P.; Sette, A.; Hager, D.A. Modulation of Cytokine Patterns of Human Autoreactive T Cell Clones by a Single Amino Acid Substitution of Their Peptide Ligand. Immunity 1995, 2, 373–380. [Google Scholar]
- Robinson, N.E.; Robinson, A.B. Use of Merrifield Solid Phase Peptide Synthesis in Investigations of Biological Deamidation of Peptides and Proteins. Biopolymers 2008, 90, 297–306. [Google Scholar] [CrossRef]
- Fabian, H.; Schultz, C.P. Fourier Transform Infrared Spectroscopy in Peptide and Protein Analysis. In Encyclopedia of Analytical Chemistry: Applications, Theory and Instrumentation; Wiley Online Library: Hoboken, NJ, USA, 2000. [Google Scholar] [CrossRef]
- Siligardi, G.; Hussain, R. CD Spectroscopy: An Essential Tool for Quality Control of Protein Folding. Methods Mol. Biol. 2015, 1261, 255–276. [Google Scholar] [CrossRef]
- Acikara, O.B.; Acikara, O.B. Ion-Exchange Chromatography and Its Applications. Column. Chromatogr. 2013, 10, 55744. [Google Scholar] [CrossRef] [Green Version]
- Camperi, S.A.; Marani, M.; Camila, M.; Ceron, M.; Albericio, F. Protein Purification by Affinity Chromatography with Peptide Ligands Selected from the Screening of Combinatorial Libraries. Trends Chromatogr. 2010, 6, 11–22. [Google Scholar]
- Calvo Tardón, M.; Allard, M.; Dutoit, V.; Dietrich, P.Y.; Walker, P.R. Peptides as Cancer Vaccines. Curr. Opin. Pharm. 2019, 47, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.Y.; Yu, K. da Breast Cancer Vaccines: Disappointing or Promising? Front. Immunol. 2022, 13, 828386. [Google Scholar] [CrossRef] [PubMed]
- Fisk, B.; Blevins, T.L.; Wharton, J.T.; Ioannides, C.G. Identification of an Immunodominant Peptide of HER-2/Neu Protooncogene Recognized by Ovarian Tumor-Specific Cytotoxic T Lymphocyte Lines. J. Exp. Med. 1995, 181, 2109. [Google Scholar] [CrossRef]
- Peoples, G.E.; Gurney, J.M.; Hueman, M.T.; Woll, M.M.; Ryan, G.B.; Storrer, C.E.; Fisher, C.; Shriver, C.D.; Ioannides, C.G.; Ponniah, S. Clinical Trial Results of a HER2/Neu (E75) Vaccine to Prevent Recurrence in High-Risk Breast Cancer Patients. J. Clin. Oncol. 2005, 23, 7536–7545. [Google Scholar] [CrossRef]
- Peoples, G.E.; Holmes, J.P.; Hueman, M.T.; Mittendorf, E.A.; Amin, A.; Khoo, S.; Dehqanzada, Z.A.; Gurney, J.M.; Woll, M.M.; Ryan, G.B.; et al. Combined Clinical Trial Results of a HER2/Neu (E75) Vaccine for the Prevention of Recurrence in High-Risk Breast Cancer Patients: U.S. Military Cancer Institute Clinical Trials Group Study I-01 and I-02. Clin. Cancer Res. 2008, 14, 797–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittendorf, E.A.; Clifton, G.T.; Holmes, J.P.; Schneble, E.; van Echo, D.; Ponniah, S.; Peoples, G.E. Final Report of the Phase I/II Clinical Trial of the E75 (Nelipepimut-S) Vaccine with Booster Inoculations to Prevent Disease Recurrence in High-Risk Breast Cancer Patients. Ann. Oncol. 2014, 25, 1735. [Google Scholar] [CrossRef] [PubMed]
- Clifton, G.T.; Hale, D.; Vreeland, T.J.; Hickerson, A.T.; Litton, J.K.; Alatrash, G.; Murthy, R.K.; Qiao, N.; Philips, A.V.; Lukas, J.J.; et al. Results of a Randomized Phase IIb Trial of Nelipepimut-S + Trastuzumab vs Trastuzumab to Prevent Recurrences in High-Risk HER2 Low-Expressing Breast Cancer Patients. Clin. Cancer Res. 2020, 26, 2515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmichael, M.G.; Benavides, L.C.; Holmes, J.P.; Gates, J.D.; Mittendorf, E.A.; Ponniah, S.; Peoples, G.E. Results of the First Phase 1 Clinical Trial of the HER-2/Neu Peptide (GP2) Vaccine in Disease-Free Breast Cancer Patients: United States Military Cancer Institute Clinical Trials Group Study I-04. Cancer 2010, 116, 292–301. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Ardavanis, A.; Litton, J.K.; Shumway, N.M.; Hale, D.F.; Murray, J.L.; Perez, S.A.; Ponniah, S.; Baxevanis, C.N.; Papamichail, M.; et al. Primary Analysis of a Prospective, Randomized, Single-Blinded Phase II Trial Evaluating the HER2 Peptide GP2 Vaccine in Breast Cancer Patients to Prevent Recurrence. Oncotarget 2016, 7, 66192. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.S.; McWilliams, D.B.; Patel, M.S.; Fischette, C.T.; Thompson, J.; Daugherty, F.J. Five Year Median Follow-up Data from a Prospective, Randomized, Placebo-Controlled, Single-Blinded, Multicenter, Phase IIb Study Evaluating the Reduction of Recurrences Using HER2/Neu Peptide GP2 + GM-CSF vs. GM-CSF Alone after Adjuvant Trastuzumab in HER2 Positive Women with Operable Breast Cancer. Cancer Res. 2021, 81, PS10-23. [Google Scholar] [CrossRef]
- Humphreys, R.E.; Adams, S.; Koldzic, G.; Nedelescu, B.; von Hofe, E.; Xu, M. Increasing the Potency of MHC Class II-Presented Epitopes by Linkage to Ii-Key Peptide. Vaccine 2000, 18, 2693–2697. [Google Scholar] [CrossRef]
- Holmes, J.P.; Benavides, L.C.; Gates, J.D.; Carmichael, M.G.; Hueman, M.T.; Mittendorf, E.A.; Murray, J.L.; Amin, A.; Craig, D.; von Hofe, E.; et al. Results of the First Phase I Clinical Trial of the Novel Ii-Key Hybrid Preventive HER-2/Neu Peptide (AE37) Vaccine. J. Clin. Oncol. 2008, 26, 3426–3433. [Google Scholar] [CrossRef]
- Gates, J.D.; Clifton, G.T.; Benavides, L.C.; Sears, A.K.; Carmichael, M.G.; Hueman, M.T.; Holmes, J.P.; Jama, Y.H.; Mursal, M.; Zacharia, A.; et al. Circulating Regulatory T Cells (CD4+CD25+FOXP3+) Decrease in Breast Cancer Patients after Vaccination with a Modified MHC Class II HER2/Neu (AE37) Peptide. Vaccine 2010, 28, 7476–7482. [Google Scholar] [CrossRef]
- Luo, C.; Wang, P.; He, S.; Zhu, J.; Shi, Y.; Wang, J. Progress and Prospect of Immunotherapy for Triple-Negative Breast Cancer. Front. Oncol. 2022, 12, 919072. [Google Scholar] [CrossRef] [PubMed]
- Hijikata, Y.; Okazaki, T.; Tanaka, Y.; Murahashi, M.; Yamada, Y.; Yamada, K.; Takahashi, A.; Inoue, H.; Kishimoto, J.; Nakanishi, Y.; et al. A Phase I Clinical Trial of RNF43 Peptide-Related Immune Cell Therapy Combined with Low-Dose Cyclophosphamide in Patients with Advanced Solid Tumors. PLoS ONE 2018, 13, e0187878. [Google Scholar] [CrossRef] [Green Version]
- Weber, J.S.; Kudchadkar, R.R.; Yu, B.; Gallenstein, D.; Horak, C.E.; Inzunza, H.D.; Zhao, X.; Martinez, A.J.; Wang, W.; Gibney, G.; et al. Safety, Efficacy, and Biomarkers of Nivolumab with Vaccine in Ipilimumab-Refractory or -Naive Melanoma. J. Clin. Oncol. 2013, 31, 4311–4318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naito, M.; Itoh, K.; Komatsu, N.; Yamashita, Y.; Shirakusa, T.; Yamada, A.; Moriya, F.; Ayatuka, H.; Mohamed, E.R.; Matsuoka, K.; et al. Dexamethasone Did Not Suppress Immune Boosting by Personalized Peptide Vaccination for Advanced Prostate Cancer Patients. Prostate 2008, 68, 1753–1762. [Google Scholar] [CrossRef]
- Rettig, L.; Seidenberg, S.; Parvanova, I.; Samaras, P.; Knuth, A.; Pascolo, S. Gemcitabine Depletes Regulatory T-Cells in Human and Mice and Enhances Triggering of Vaccine-Specific Cytotoxic T-Cells. Int. J. Cancer 2011, 129, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Shirahama, T.; Muroya, D.; Matsueda, S.; Yamada, A.; Shichijo, S.; Naito, M.; Yamashita, T.; Sakamoto, S.; Okuda, K.; Itoh, K.; et al. A Randomized Phase II Trial of Personalized Peptide Vaccine with Low Dose Cyclophosphamide in Biliary Tract Cancer. Cancer Sci. 2017, 108, 838–845. [Google Scholar] [CrossRef] [Green Version]
- Kono, K.; Sato, E.; Naganuma, H.; Takahashi, A.; Mimura, K.; Nukui, H.; Fujii, H. Trastuzumab (Herceptin) Enhances Class I-Restricted Antigen Presentation Recognized by HER-2/Neu-Specific T Cytotoxic Lymphocytes. Clin. Cancer Res. 2004, 10, 2538–2544. [Google Scholar] [CrossRef] [Green Version]
- Petricevic, B.; Laengle, J.; Singer, J.; Sachet, M.; Fazekas, J.; Steger, G.; Bartsch, R.; Jensen-Jarolim, E.; Bergmann, M. Trastuzumab Mediates Antibody-Dependent Cell-Mediated Cytotoxicity and Phagocytosis to the Same Extent in Both Adjuvant and Metastatic HER2/Neu Breast Cancer Patients. J. Transl. Med. 2013, 11, 307. [Google Scholar] [CrossRef] [Green Version]
- Gall, V.A.; Philips, A.V.; Qiao, N.; Clise-Dwyer, K.; Perakis, A.A.; Zhang, M.; Clifton, G.T.; Sukhumalchandra, P.; Ma, Q.; Reddy, S.M.; et al. Trastuzumab Increases HER2 Uptake and Cross-Presentation by Dendritic Cells. Cancer Res. 2017, 77, 5374–5383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickerson, A.; Clifton, G.T.; Hale, D.F.; Peace, K.M.; Holmes, J.P.; Vreeland, T.J.; Litton, J.K.; Murthy, R.K.; Lukas, J.J.; Mittendorf, E.A.; et al. Final Analysis of Nelipepimut-S plus GM-CSF with Trastuzumab versus Trastuzumab Alone to Prevent Recurrences in High-Risk, HER2 Low-Expressing Breast Cancer: A Prospective, Randomized, Blinded, Multicenter Phase IIb Trial. J. Clin. Oncol. 2019, 37, 1. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prawiningrum, A.F.; Paramita, R.I.; Panigoro, S.S. Immunoinformatics Approach for Epitope-Based Vaccine Design: Key Steps for Breast Cancer Vaccine. Diagnostics 2022, 12, 2981. https://doi.org/10.3390/diagnostics12122981
Prawiningrum AF, Paramita RI, Panigoro SS. Immunoinformatics Approach for Epitope-Based Vaccine Design: Key Steps for Breast Cancer Vaccine. Diagnostics. 2022; 12(12):2981. https://doi.org/10.3390/diagnostics12122981
Chicago/Turabian StylePrawiningrum, Aisyah Fitriannisa, Rafika Indah Paramita, and Sonar Soni Panigoro. 2022. "Immunoinformatics Approach for Epitope-Based Vaccine Design: Key Steps for Breast Cancer Vaccine" Diagnostics 12, no. 12: 2981. https://doi.org/10.3390/diagnostics12122981