
The Analysis of Relevant Gene Networks Based on Driver Genes in Breast Cancer

 ,
,  , ,
, ,
Abstract
:1. Introduction
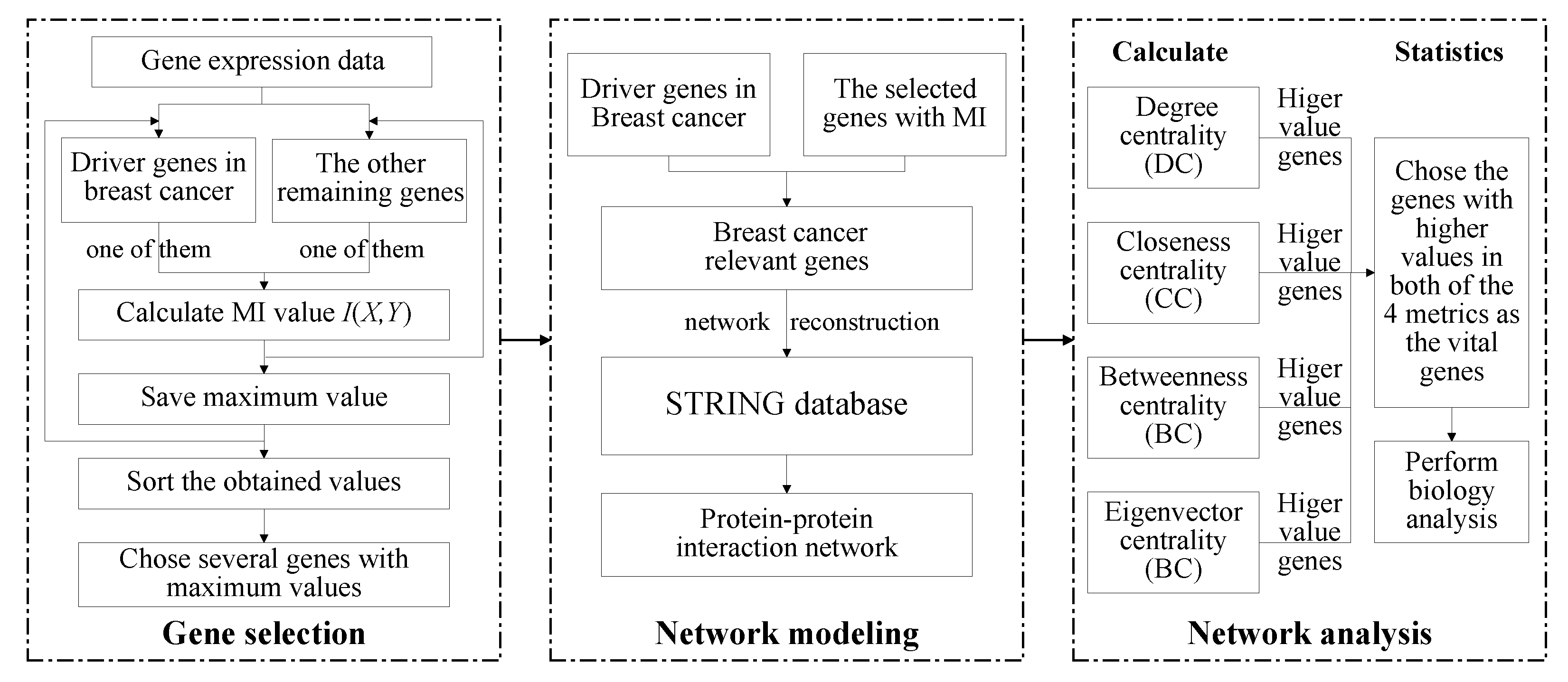
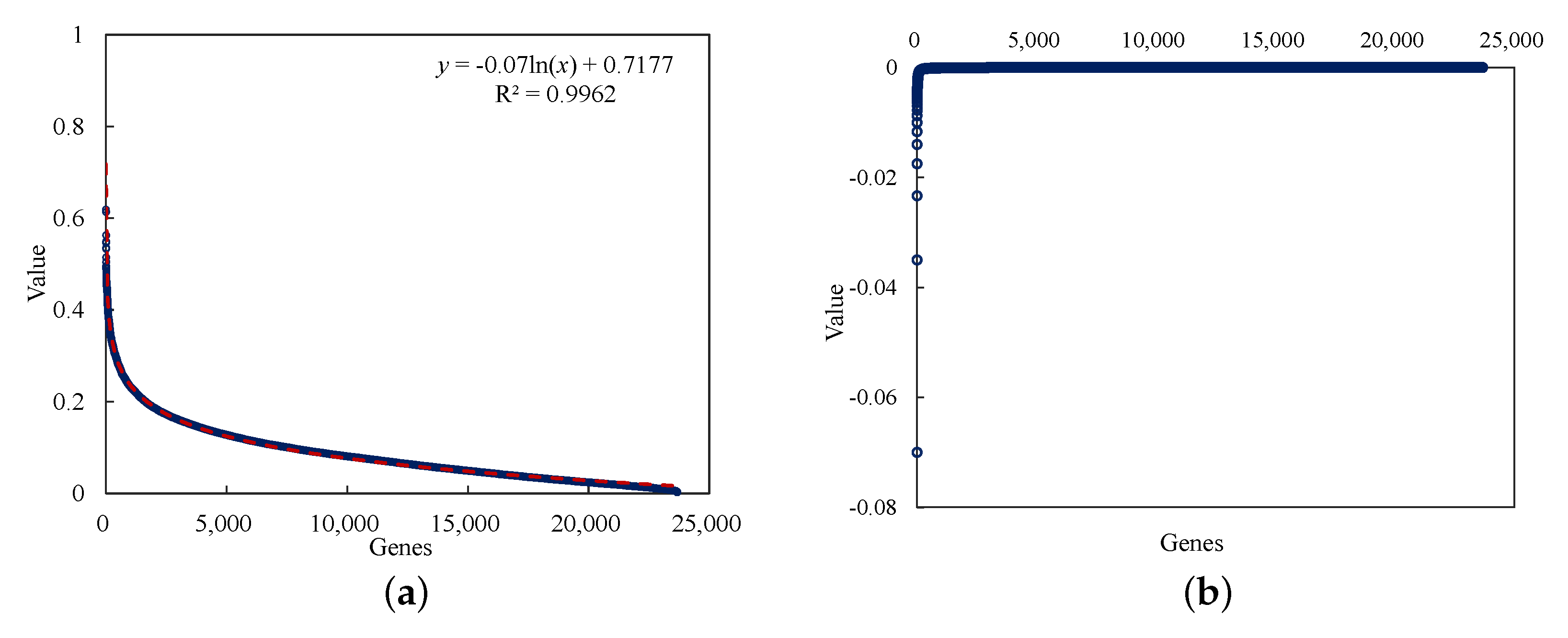
- The mutual information method is used for the gene selection step, which selects breast cancer-relevant genes from the whole genome. Using this method, we selected 230 genes as the relevant genes for breast cancer.
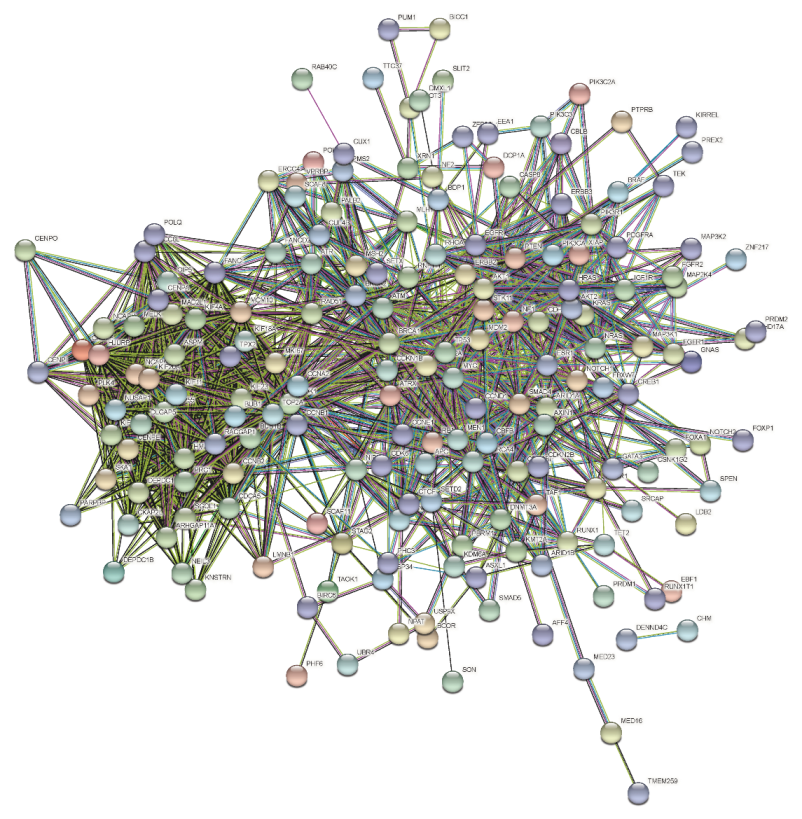
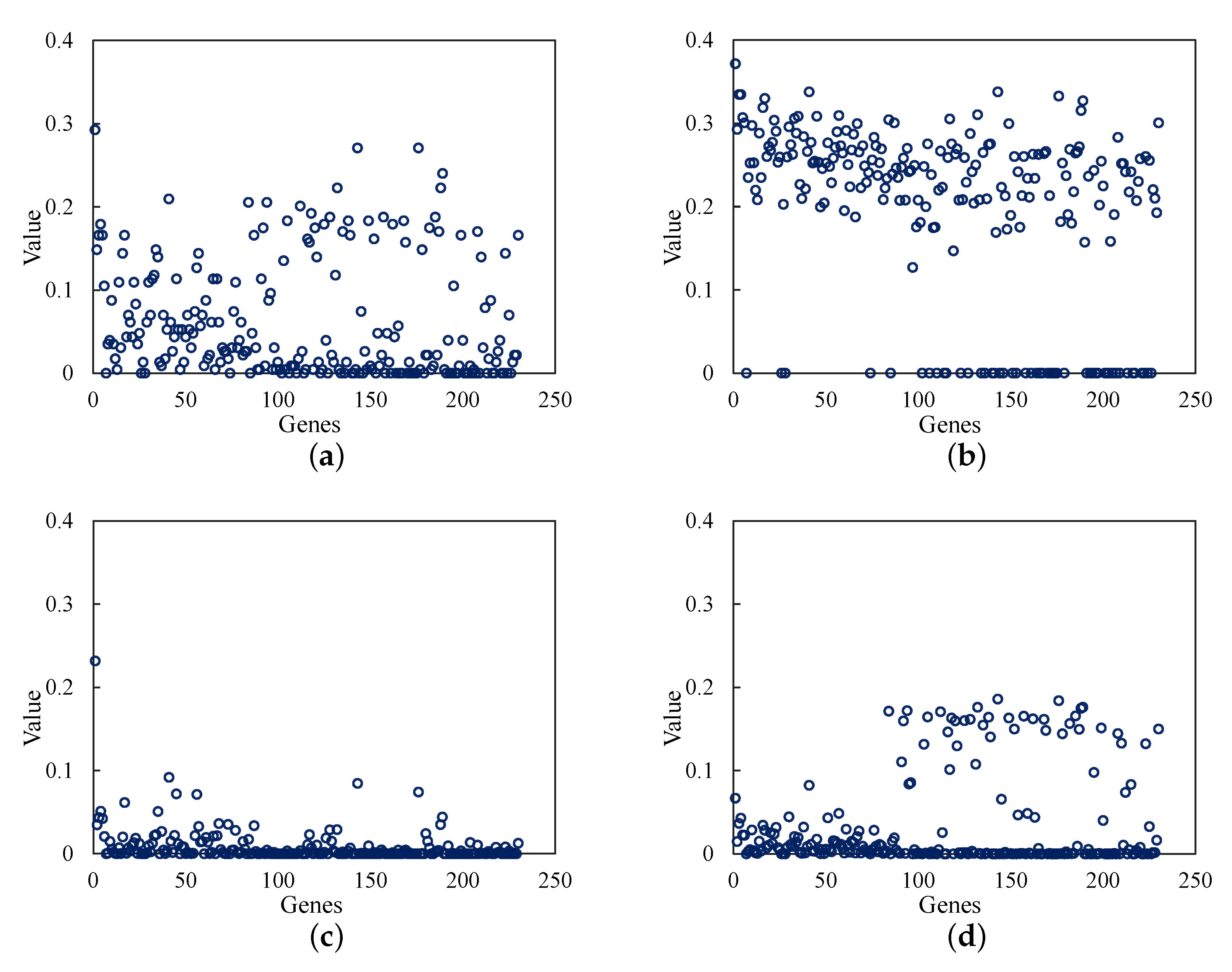
- The protein–protein interaction network is built and analyzed based on the selected genes from the mutual information method. By analyzing the node centrality of the protein–protein interaction network, we obtained the important genes with important positions and connectivity in the network.
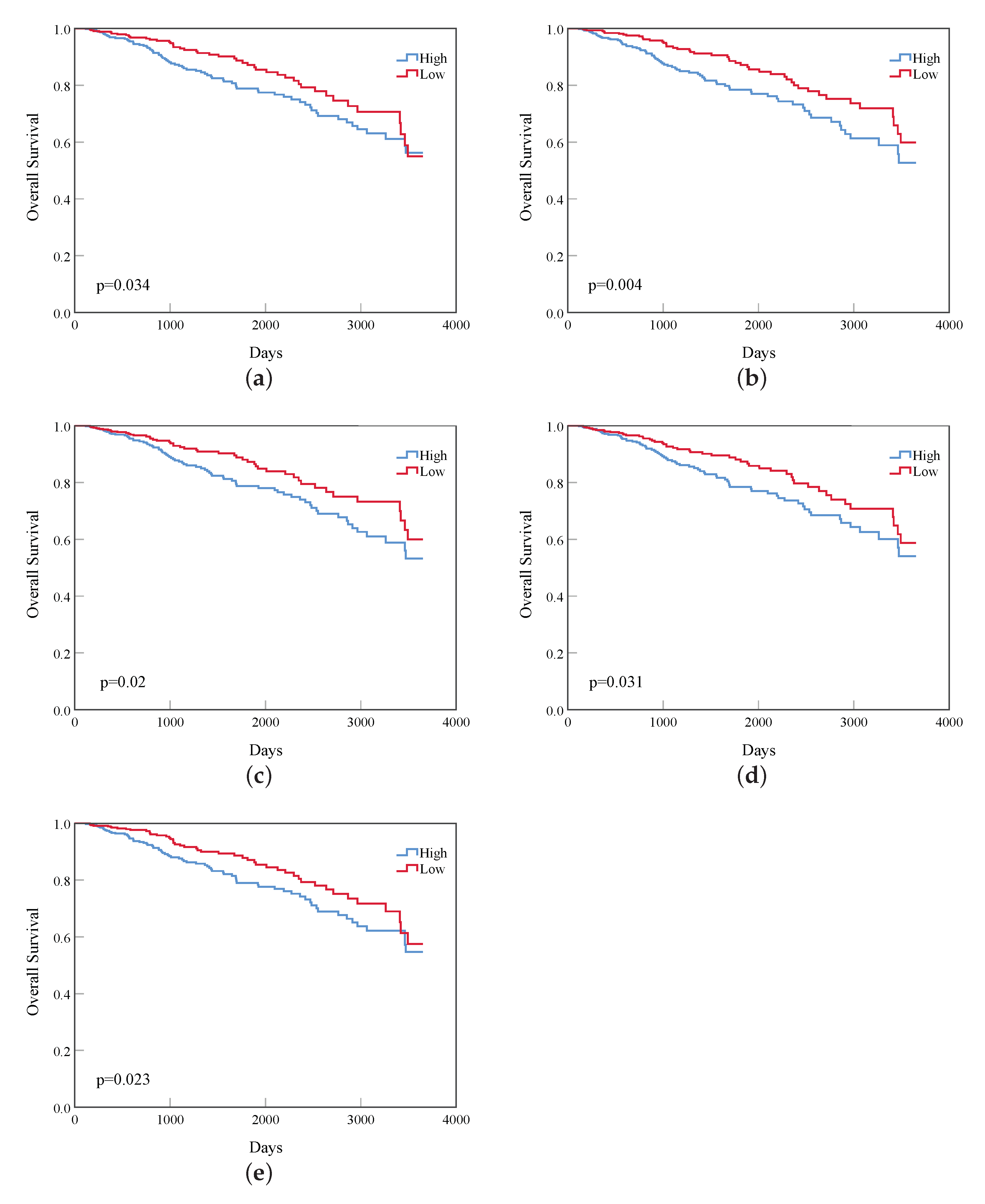
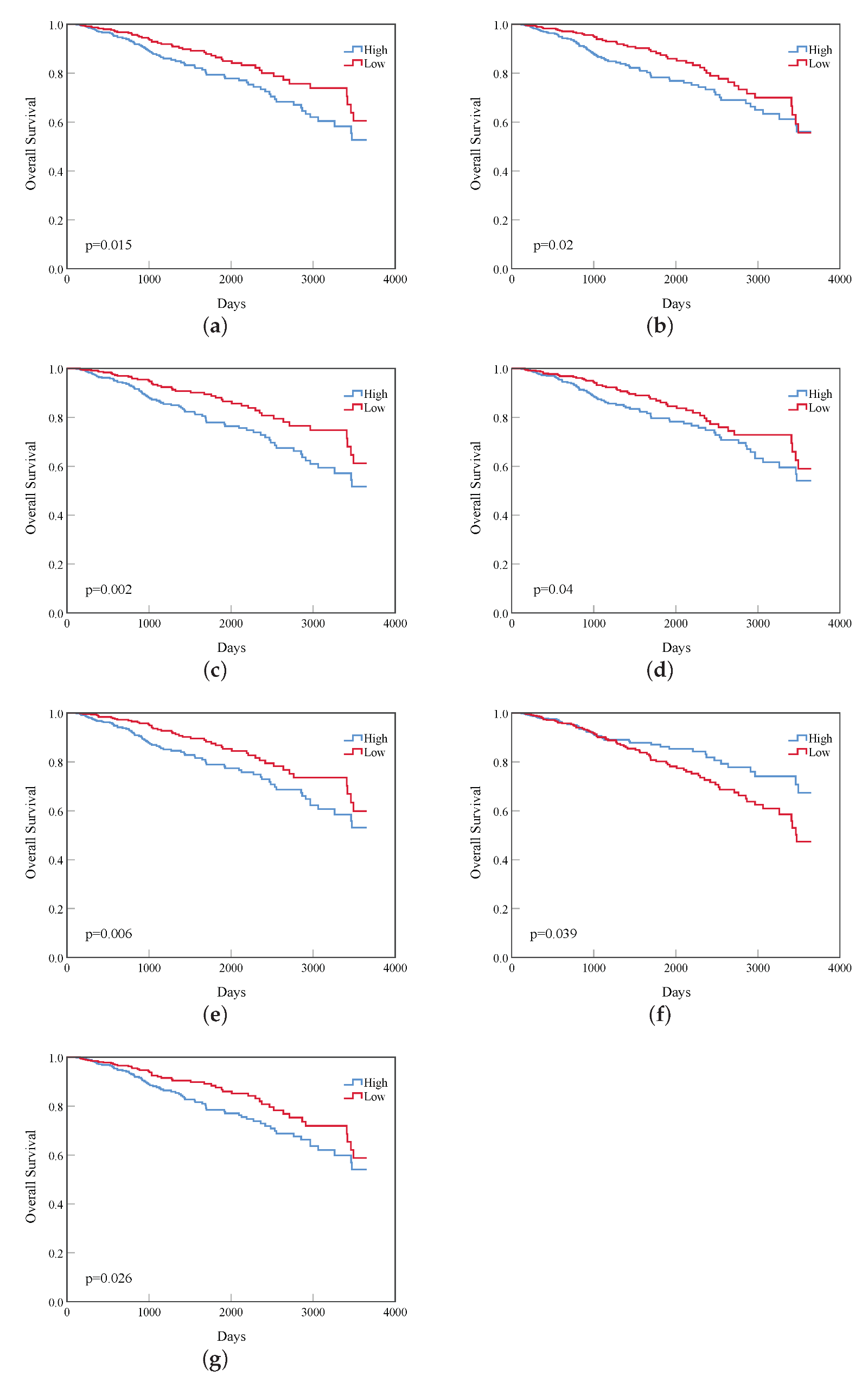
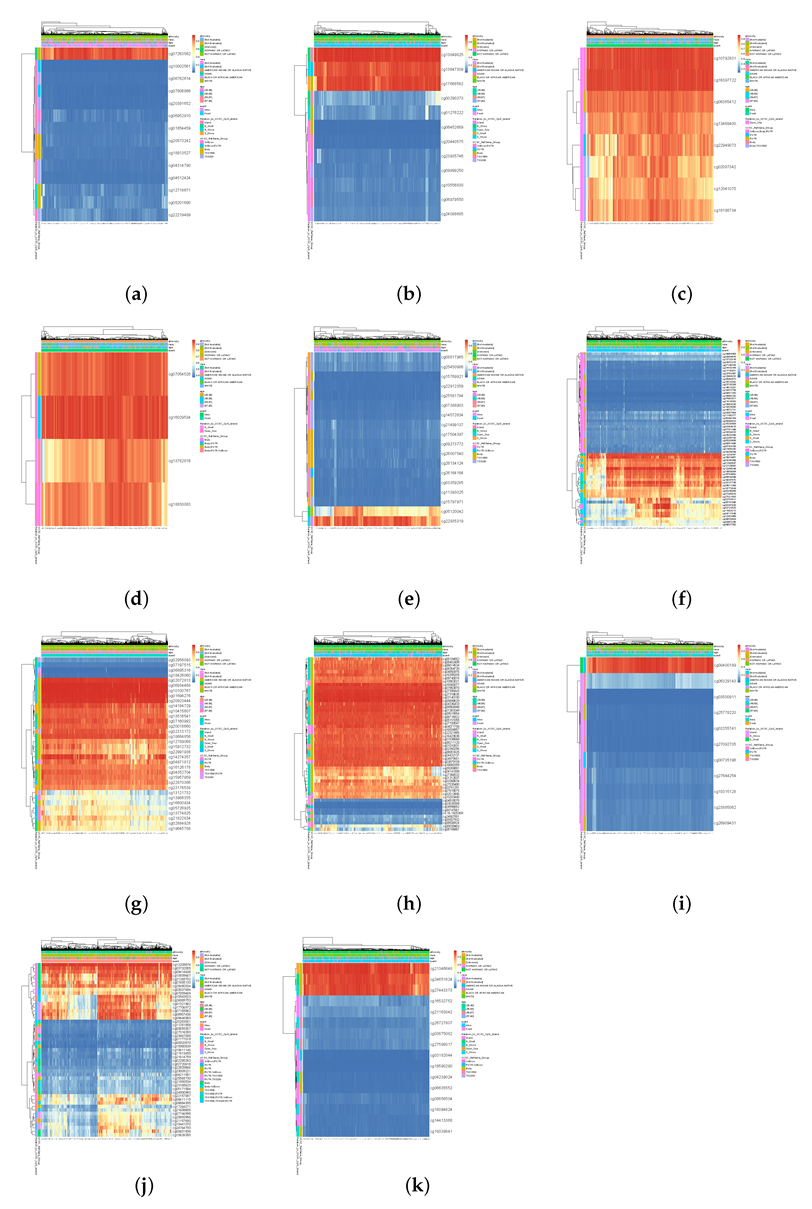
- Based on the vital genes, through survival analysis, DNA methylation analysis, and RNA expression level in breast cancer and the prognosis in other cancers, we found some genes that reduce the survival rate of breast cancer patients due to different expression levels and confirmed their biological significance.
2. Methods
2.1. The General Framework
Mutual Information Method
| Algorithm 1 Gene selection based on Mutual Information. |
Input: Gene expression data ; Output: MI values and the according Genes 1 for to n do 2for to m do 3 calculate , , ; 4 calculate using Equation (6); 5 if is the max do 6 save to ; 7
end if 8 save several according to the maximum values of ; 9
return |
2.2. Node Centrality
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of Age: Ten Years of Next-Generation Sequencing Technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; Fitzhugh, W. Initial sequencing and analysis of the human genome. Methods Inf. Med. Suppl. 2001, 409, 860–921. [Google Scholar]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [Green Version]
- Parikshak, N.N.; Gandal, M.J.; Geschwind, D.H. Systems Biology and Gene Networks in Neurodevelopmental and Neurodegenerative Disorders. Nat. Rev. Genet. 2015, 16, 441–458. [Google Scholar] [CrossRef]
- Hermeking, H. MicroRNAs in the p53 network: Micromanagement of tumour suppression. Nat. Rev. Cancer 2012, 12, 613–626. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; Liedtke, C.; Tutt, A.; Minckwitz, G.V. Molecular alterations in triple-negative breast cancer—The road to new treatment strategies. Lancet 2016, 389, 2430–2442. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.W.; Dvinge, H.; Kim, E.; Cho, H.; Micol, J.B.; Chung, Y.R.; Durham, B.H.; Yoshimi, A.; Kim, Y.J.; Thomas, M. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat. Med. 2016, 22, 672–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ring, B.Z.; Hout, D.R.; Morris, S.W.; Lawrence, K.; Schweitzer, B.L.; Bailey, D.B.; Lehmann, B.D.; Pietenpol, J.A.; Seitz, R.S. Generation of an algorithm based on minimal gene sets to clinically subtype triple negative breast cancer patients. BMC Cancer 2016, 16, 143. [Google Scholar]
- Andersson, Y.; Bergkvist, L.; Frisell, J.; Boniface, J.D. Long-term breast cancer survival in relation to the metastatic tumor burden in axillary lymph nodes. Breast Cancer Res. Treat. 2018, 171, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Henna, H.; Tatiana, L.; Biswajyoti, S.; Henna, P.; Paivi, P.; Riku, L.; Ping, G.; Wei, G.; Sampsa, H.; Janne, O.A. Identification of several potential chromatin binding sites of HOXB7 and its downstream target genes in breast cancer. Int. J. Cancer 2015, 137, 2374–2383. [Google Scholar]
- Nikdelfaz, O.; Jalili, S. Disease genes prediction by HMM based PU-learning using gene expression profiles. J. Biomed. Inform. 2018, 81, 102–111. [Google Scholar] [CrossRef]
- Wang, D.; Haley, J.D.; Thompson, P. Comparative gene co-expression network analysis of epithelial to mesenchymal transition reveals lung cancer progression stages. BMC Cancer 2017, 17, 830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Treloar, A.E.; Lupien, M. Emergence of the Noncoding Cancer Genome: A Target of Genetic and Epigenetic Alterations. Cancer Discov. 2016, 6, 1215–1229. [Google Scholar] [CrossRef]
- Kim, J.J.; Kim, J.Y.; Kang, H.J.; Shin, J.K.; Kang, T.; Lee, S.W.; Bae, Y.T. Computer-aided Diagnosis-generated Kinetic Features of Breast Cancer at Preoperative MR Imaging: Association with Disease-free Survival of Patients with Primary Operable Invasive Breast Cancer. Radiology 2017, 284, 45–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, W.; Clarke, L.P.; Song, M.D.; Clark, R.A. Digital mammography: Hybrid four-channel wavelet transform for microcalcification segmentation. Acad. Radiol. 1998, 5, 354–364. [Google Scholar] [CrossRef]
- Wang, Z.; Qu, Q.; Yu, G.; Kang, Y. Breast Tumor Detection in Double Views Mammography Based on Extreme Learning Machine. Neural Comput. Appl. 2016, 27, 227–240. [Google Scholar] [CrossRef]
- Wang, Z.; Yu, G.; Kang, Y.; Zhao, Y.; Qu, Q. Breast Tumor Detection in Digital Mammography Based on Extreme Learning Machine. Neurocomputing 2014, 128, 175–184. [Google Scholar] [CrossRef]
- Alkawaa, F.M.; Chaudhary, K.; Garmire, L.X. Deep Learning Accurately Predicts Estrogen Receptor Status in Breast Cancer Metabolomics Data. J. Proteome Res. 2018, 17, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Saha, M.; Chakraborty, C. Her2Net: A Deep Framework for Semantic Segmentation and Classification of Cell Membranes and Nuclei in Breast Cancer Evaluation. IEEE Trans. Image Process. 2018, 27, 2189–2200. [Google Scholar] [CrossRef]
- Stephens, P.J.; Tarpey, P.S.; Davies, H.; Loo, P.V.; Greenman, C.; Wedge, D.C.; Nik-Zainal, S.; Martin, S.; Varela, I.; Bignell, G.R.; et al. The Landscape of Cancer Genes and Mutational Processes in Breast Cancer. Nature 2012, 486, 400–404. [Google Scholar] [CrossRef] [Green Version]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nuss, P.; Chen, W.Q.; Ohno, H.; Graedel, T.E. Structural Investigation of Aluminum in the U.S. Economy using Network Analysis. Environ. Sci. Technol. 2016, 50, 4091–4101. [Google Scholar] [CrossRef] [PubMed]
- Brandes, U.; Borgatti, S.P.; Freeman, L.C. Maintaining the duality of closeness and betweenness centrality. Soc. Netw. 2016, 44, 153–159. [Google Scholar] [CrossRef]
- Riondato, M.; Kornaropoulos, E.M. Fast approximation of betweenness centrality through sampling. Data Min. Knowl. Discov. 2016, 30, 438–475. [Google Scholar] [CrossRef]
- Binnewijzend, M.A.; Adriaanse, S.M.; Flier, W.M.; Teunissen, C.E.; Munck, J.C.; Stam, C.J.; Scheltens, P.; Berckel, B.N.; Barkhof, F.; Wink, A.M. Brain network alterations in Alzheimer’s disease measured by Eigenvector centrality in fMRI are related to cognition and CSF biomarkers. Hum. Brain Mapp. 2014, 35, 2383–2393. [Google Scholar] [CrossRef]
- Küffner, R.; Petri, T.; Tavakkolkhah, P.; Windhager, L.; Zimmer, R. Inferring gene regulatory networks by ANOVA. Bioinformatics 2012, 28, 1376–1382. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhao, X.M.; He, K.; Lu, L.; Cao, Y.; Liu, J.; Hao, J.K.; Liu, Z.P.; Chen, L. Inferring gene regulatory networks from gene expression data by path consistency algorithm based on conditional mutual information. Bioinformatics 2012, 28, 98–104. [Google Scholar] [CrossRef]
- Freeman, L.C. Centrality in social networks conceptual clarification. Soc. Netw. 1978, 1, 215–239. [Google Scholar] [CrossRef] [Green Version]
- Modhukur, V.; Iljasenko, T.; Metsalu, T.; Lokk, K.; Laisk-Podar, T.; Vilo, J. MethSurv: A web tool to perform multivariable survival analysis using DNA methylation data. Epigenomics 2018, 10, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Anuraga, G.; Wang, W.J.; Phan, N.N.; Ton, N.T.A.; Ta, H.D.K.; Berenice, P.F.; Minh, X.D.T.; Ku, S.C.; Wu, Y.F.; Andriani, V.; et al. Potential prognostic biomarkers of NIMA (never in mitosis, gene a)-related kinase (NEK) family members in breast cancer. J. Pers. Med. 2021, 11, 1089. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Bathia, B.B.; Pande, B.P. Giant eimerian schizonts in the Indian water-buffalo. Acta Vet. Acad. Sci. Hung. 1967, 17, 351–357. [Google Scholar] [PubMed]
- Wang, C.Y.; Chiao, C.C.; Phan, N.N.; Li, C.Y.; Sun, Z.D.; Jiang, J.Z.; Hung, J.H.; Chen, Y.L.; Yen, M.C.; Weng, T.Y.; et al. Gene signatures and potential therapeutic targets of amino acid metabolism in estrogen receptor-positive breast cancer. Am. J. Cancer Res. 2020, 10, 95–113. [Google Scholar] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metrics | Top 10 Genes |
|---|---|
| Degree Centrality | TP53, CCNA2, CDK1, CCNB1, BUB1, TOP2A, BRCA1, BUB1B, KIF11, NCAPG |
| Closeness Centrality | TP53, BRCA1, CCNA2, MYC, CCND1, CDK1, AKT1, CCNB1, CDKN2A, TOP2A |
| Betweenness Centrality | TP53, BRCA1, CCNA2, CDK1, CREBBP, SMAD4, AKT1, CCND1, ESR1, CCNB1 |
| Eigenvector Centrality | CCNA2, CDK1, BUB1, CCNB1, TOP2A, KIF11, BUB1B, NCAPG, KIF20A, CENPE |
| Metrics | Top 20 Genes |
|---|---|
| Degree Centrality | TP53, CCNA2 CDK1, CCNB1, BUB1, TOP2A, BRCA1, BUB1B, KIF11, NCAPG, CCNB2, KIF23, CENPE, KIF20A, KIF4A, ASPM, TPX2, DLGAP5, CCND1, KIF15 |
| Closeness Centrality | TP53, BRCA1, CCNA2, MYC, CCND1, CDK1, AKT1, CCNB1, CDKN2A, TOP2A, BUB1, ATM, ESR1, CREBBP, PTEN, EGFR, RAD51, BUB1B, MDM2, ERBB2 |
| Betweenness Centrality | TP53, BRCA1, CCNA2, CDK1, REBBP, SMAD4, AKT1, CCND1, ESR1, CCNB1, MYC, PTEN, STAG2, CNOT3, TOP2A, PIK3CA, HRAS, ATM, BUB1, KIF23 |
| Eigenvector Centrality | CCNA2, CDK1, BUB1, CCNB1, TOP2A, KIF11, BUB1B, NCAPG, KIF20A, CENPE, KIF4A, ASPM, CCNB2, TPX2, MELK, DLGAP5, KIF23, KIF15, CEP55, NUSAP1 |
| Gene | Prognostic Marker in Cancer |
|---|---|
| CCNA2 | renal cancer(−); pancreatic cancer(−); liver cancer(−); lung cancer(−); endometrial cancer(−) |
| CDK1 | renal cancer(−); liver cancer(−); pancreatic cancer(−); lung cancer(−); cervical cancer(+) |
| CCNB1 | renal cancer(−); liver cancer(−); lung cancer(−) |
| TOP2A | renal cancer(−); liver cancer(−); pancreatic cancer(−); lung cancer(−) |
| BUB1 | liver cancer(−); pancreatic cancer(−); endometrial cancer(−); lung cancer(−) |
| TP53 | endometrial cancer(+); prostate cancer(−) |
| BUB1B | liver cancer(-); pancreatic cancer(−); lung cancer(−) |
| CCND1 | pancreatic cancer(−); head and neck cancer(−) |
| KIF23 | liver cancer(−); pancreatic cancer(−); endometrial cancer(−) |
| MYC | renal cancer(−); urothelial cancer(−); ovarian cancer(−) |
| KIF11 | renal cancer(−); liver cancer(−); pancreatic cancer(−); lung cancer(−) |
| NCAPG | liver cancer(−); pancreatic cancer(−); endometrial cancer(−) |
| CCNB2 | renal cancer(−); pancreatic cancer(−); melanoma(-); liver cancer(−); lung cancer(−) |
| KIF20A | renal cancer(−); liver cancer(−); pancreatic cancer(−); lung cancer(−) |
| KIF4A | liver cancer(−); pancreatic cancer(−); |
| ASPM | liver cancer(−); endometrial cancer(−); pancreatic cancer(−); lung cancer(−) |
| TPX2 | renal cancer(−); liver cancer(−); endometrial cancer(−); pancreatic cancer(−); lung cancer(−) |
| DLGAP5 | liver cancer(−); pancreatic cancer (−); endometrial cancer(−); lung cancer(−) |
| KIF15 | colorectal cancer(+) |
| ESR1 | endometrial cancer(+) |
| CREBBP | renal cancer(+) |
| PTEN | renal cancer(−) |
| AKT1 | renal cancer(+); ovarian cancer(+) |
| SMAD4 | renal cancer(+) |
| CDKN2A | endometrial cancer(−); renal cancer(−); liver cancer(−); head and neck cancer(+) |
| CNOT3 | liver cancer(−); renal cancer(−) |
| MELK | renal cancer(−); liver cancer(−); lung cancer(−); pancreatic cancer(−) |
| EGFR | urothelial cancer(−) |
| RAD51 | breast cancer(−); liver cancer(−) |
| HRAS | liver cancer(−) |
| MDM2 | endometrial cancer(+); cervical cancer(+) |
| CEP55 | renal cancer(−); liver cancer(−); pancreatic cancer(−); lung cancer(−); stomach cancer(+) |
| ERBB2 | renal cancer(+); endometrial cancer(−); pancreatic cancer(−); |
| NUSAP1 | renal cancer(−); pancreatic cancer(−) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qu, L.; Wang, Z.; Zhang, H.; Wang, Z.; Liu, C.; Qian, W.; Xin, J. The Analysis of Relevant Gene Networks Based on Driver Genes in Breast Cancer. Diagnostics 2022, 12, 2882. https://doi.org/10.3390/diagnostics12112882
Qu L, Wang Z, Zhang H, Wang Z, Liu C, Qian W, Xin J. The Analysis of Relevant Gene Networks Based on Driver Genes in Breast Cancer. Diagnostics. 2022; 12(11):2882. https://doi.org/10.3390/diagnostics12112882
Chicago/Turabian StyleQu, Luxuan, Zhiqiong Wang, Hao Zhang, Zhongyang Wang, Caigang Liu, Wei Qian, and Junchang Xin. 2022. "The Analysis of Relevant Gene Networks Based on Driver Genes in Breast Cancer" Diagnostics 12, no. 11: 2882. https://doi.org/10.3390/diagnostics12112882