
Preimplantation Genetic Diagnosis in Hereditary Hearing Impairment

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects
2.2. PGD and Prenatal Confirmation Process
3. Results
3.1. Overall Outcomes
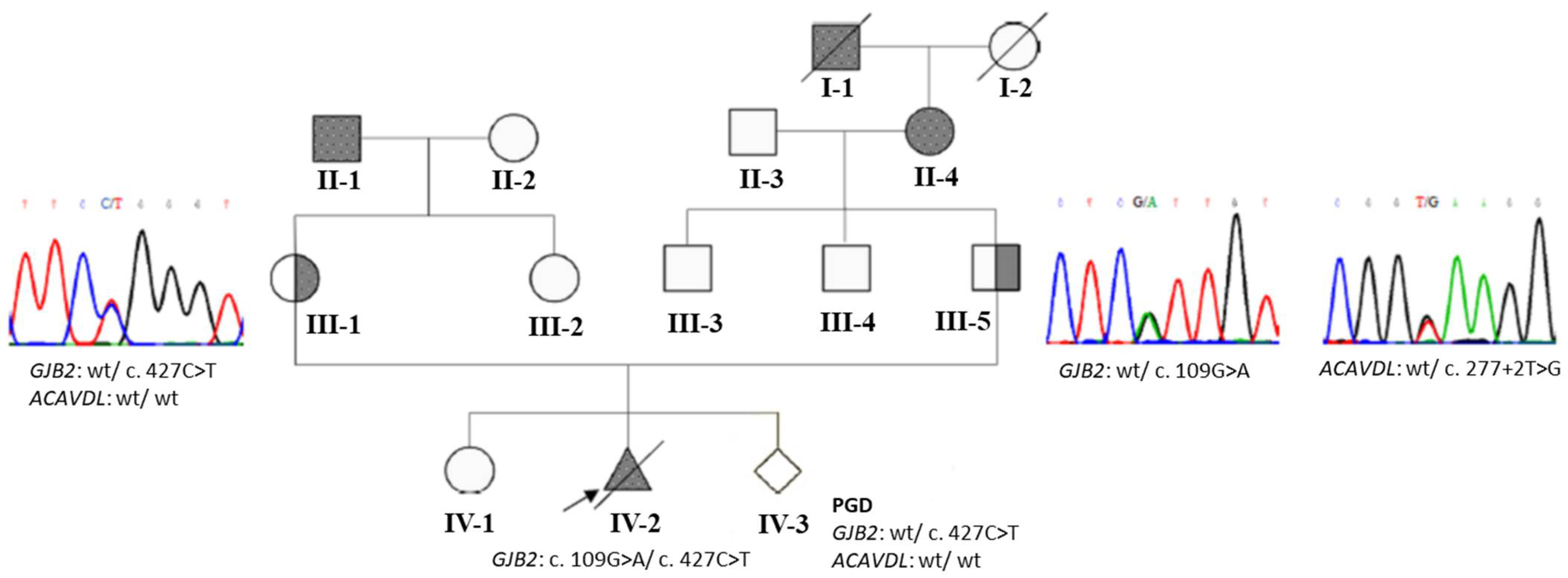
3.2. An Example with GJB2 and ACADVL Variants
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
Appendix A

{kind=link}
| . | Forward Primer | Sequence | Tm | Reverse Primer | Sequence | Tm | Size (bp) |
|---|---|---|---|---|---|---|---|
| Duplex-nested PCR for GJB2 PGD | |||||||
| GJB2-1st-F | CCTGTTTTGGTGAGGTTGTG | 50.2 | GJB2-1st-R | GGACACAAAGCAGTCCACAG | 50.2 | 710 | |
| GJB2-2nd-F | TTGGTGTTTGCTCAGGAAGA | 50.6 | GJB2-2nd-R | AGAAGCCGTCGTACATGACA | 50.5 | 619 | |
| ARMS-qPCR | |||||||
| for c. 79G>A | |||||||
| GJB2-wt-c.79G | TTGGAAAGATCTGGCTCAGCG | 56.8 | GJB2-c.79-R | GATCGTAGCACACGTTCTTGCAG | 56.4 | 141 | |
| GJB2-mu-c.79A | TTGGAAAGATCTGGCTCAGCA | 54.9 | |||||
| for c. 109G>A | |||||||
| GJB2-c.109-F | CACTCCACCAGCATTGGAAA | 53.3 | GJB2-wt-c.109G | CCTCCTTTGCAGCCACAGC | 55.6 | 82 | |
| GJB2-mu-c.109A | CCTCCTTTGCAGCCACAGT | 52.5 | |||||
| for c.235 delC | |||||||
| GJB2-wt-c.235 | CACATCCGGCTATGGGCTC | 55.2 | GJB2-c.235/257-R | GCGGACCTTCTGGGTTTTGAT | 56.8 | 165 | |
| GJB2-mu-c.235delC | CACATCCGGCTATGGGCTT | 54.8 | |||||
| for c. 257C>G | |||||||
| GJB2-wt-c.257C | CAGCTGATCTTCGTGTCCGC | 55.6 | GJB2-c.235/257-R | GCGGACCTTCTGGGTTTTGAT | 144 | ||
| GJB2-mu-c.257G | CAGCTGATCTTCGTGTCCGC | 55.5 | |||||
| for c. 341A>G | |||||||
| GJB2-c.341-F | GGCCTACCGGAGACATGAGAAG | 56.6 | GJB2-wt-c.341A | TTGATCTCCTCGATGTCCTTAAACT | 54.4 | 82 | |
| GJB2-mu-c.341G | TTGATCTCCTCGATGTCCTTAAACC | 56.7 | |||||
| for c. 427C>T | |||||||
| GJB2-c.427-F | GCCTACCGGAGACATGAGAAGA | 54.8 | GJB2-wt-c.427C | GCGGCTTGCAAGATGACCTG | 57.1 | 161 | |
| GJB2-mu-c.427T | GCGGCTTGCAAGATGACCTA | 54.4 | |||||
| Duplex-nested PCR for SLC26A4 PGD | |||||||
| SLC26A4-1st-9F | GCTGATTTCACACTGCTTGC | 50.8 | SLC26A4-1st-9R | CCCACATGCAGAACTTCAAC | 50.2 | 1127 | |
| SLC26A4-2nd-9F | AGTAATCAAGCAGAATAACAGCACT | 50.8 | SLC26A4-2nd-9R | CTCAGGAAGAAATGCCAAAA | 49.6 | 1006 | |
| SLC26A4-1st-18F | CGACCAGTTATGGGATAACCA | 51.6 | SLC26A4-1st-18R | GGCTTGTTTGTGGCTTGATT | 51.1 | 965 | |
| SLC26A4-2nd-18F | CTGTCATTTCAAATCTGGGTCA | 51.7 | SLC26A4-2nd-18R | GCATTATAGCTAATGCCCACTTT | 51.7 | 746 | |
| ARMS-qPCR | |||||||
| for c.1229C>T | |||||||
| SLC26A4-wt-c.1229C | ACCACTGCTCTTTCCCGTGC | 56.4 | SLC26A4-3rd-9R | GTGCGAGCCTTCCTCTGTTG | 54.8 | 126 | |
| SLC26A4-mu-c.1229T | ACCACTGCTCTTTCCCGTGT | 53.5 | |||||
| for c.2168A>G | |||||||
| SLC26A4-wt-c.2168A | GGACACATTCTTTTTGACGGTCTA | 54.1 | SLC26A4-3Rd-18R | AAAACTGAGGCTCCATGAAGTTA | 51.8 | 224 | |
| SLC26A4-mu-c.2168G | GGACACATTCTTTTTGACGGTCTG | 56.5 | |||||
| Duplex-nested PCR for OTOF PGD | |||||||
| OTOF-40F-1st | ACTTTCAGGTGCTGGGACAG | 51.5 | OTOF-40R-1st | AACATGTCCACCCACAGCTC | 52 | 872 | |
| OTOF-40F-2nd | CAGGGCTCTCCAGTCAACTT | 50.8 | OTOF-40R-2nd | TCTGTCAAGGACCCAGTTCA | 50 | 698 | |
| OTOF-42F-1st | CAGGAGAGCCATGCTCAGAT | 51.6 | OTOF-42FR-1st | CAGGTAGTCGAAGGGGAACA | 51.6 | 794 | |
| OTOF-42F-2nd | GAGGGGAAAATCCTCTTTGG | 51.8 | OTOF-42R-2nd | ACAGGTAGCGCCAGTTGAAG | 52 | 680 | |
| ARMS-qPCR | |||||||
| for c.5098G>C | |||||||
| OTOF-40F-3rd | CTGAGGCACTGGGAGGACAT | 53.8 | OTOF-wt-c.5098G | ATGCAAGTGTCACCTGCCC | 53 | 115 | |
| OTOF-mu-c.5098C | ATGCAAGTGTCACCTGCCG | 54.2 | |||||
| for c.5197G>A | |||||||
| OTOF-42F-3rd | AGACCCCAGGGCTTCTCTCC | 55.9 | OTOF-wt-c.5197G | CAGATGATGACCCGCAGCAC | 56.2 | 123 | |
| OTOF-mu-c.5197A | CAGATGATGACCCGCAGCAT | 55.7 | |||||
| Duplex-nested PCR for ACADVL PGD | |||||||
| ACADVL-1st-5F | CCTGTTCTCCCCTTGACACA | 52.1 | ACADVL-1st-5R | CACCATCTCCAGAGCGTCAT | 52.2 | 689 | |
| ACADVL-2nd-5F | CTCTTTTCCCAGCTGGCTCT | 52.8 | ACADVL-2nd-5R | TACTGGGATGTGGCGATAGG | 52.4 | 514 | |
| ARMS-qPCR | |||||||
| for c.277+2T>G | |||||||
| ACADVL-c.277+2T-wt | TGTTCCCATACCCGTCCGAT | 56.2 | ACADVL-3rd-5R | ACCTCGAAGAAACGGGACACAG | 56.9 | 174 | |
| ACADVL-c.277+2G-m | TGTTCCCATACCCGTCCGAG | 56.9 | |||||
References
- Prosser, J.D.; Cohen, A.P.; Greinwald, J.H. Diagnostic Evaluation of Children with Sensorineural Hearing Loss. Otolaryngol. Clin. N. Am. 2015, 48, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Lieu, J.E.C.; Kenna, M.; Anne, S.; Davidson, L. Hearing Loss in Children. JAMA 2020, 324, 2195–2205. [Google Scholar] [CrossRef]
- Korver, A.M.H.; Smith, R.J.H.; Van Camp, G.; Schleiss, M.R.; Bitner-Glindzicz, M.A.K.; Lustig, L.R.; Usami, S.-I.; Boudewyns, A.N. Congenital hearing loss. Nat. Rev. Dis. Prim. 2017, 3, 16094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roizen, N.J. Nongenetic causes of hearing loss. Ment. Retard. Dev. Disabil. Res. Rev. 2003, 9, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.J.; Bale, J.F.; White, K.R. Sensorineural hearing loss in children. Lancet 2005, 365, 879–890. [Google Scholar] [CrossRef]
- Handyside, A.H.; Kontogianni, E.H.; Hardy, K.; Winston, R.M.L. Pregnancies from biopsied human preimplantation embryos sexed by Y-specific DNA amplification. Nature 1990, 344, 768–770. [Google Scholar] [CrossRef]
- Brezina, P.R.; Brezina, D.S.; Kearns, W.G. Preimplantation genetic testing. BMJ 2012, 345, e5908. [Google Scholar] [CrossRef]
- Harton, G.L.; De Rycke, M.; Fiorentino, F.; Moutou, C.; Sengupta, S.; Traeger-Synodinos, J.; Harper, J.C. ESHRE PGD consortium best practice guidelines for amplification-based PGD. Hum. Reprod. 2010, 26, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.-F.; Chang, S.-P.; Wu, S.-H.; Lin, W.-H.; Lee, Y.-C.; Ni, Y.-H.; Chen, C.-A.; Ma, G.-C.; Ginsberg, N.A.; You, E.-M.; et al. Validating a rapid, real-time, PCR-based direct mutation detection assay for preimplantation genetic diagnosis. Gene 2014, 548, 299–305. [Google Scholar] [CrossRef]
- Kuo, S.-J.; Ma, G.-C.; Chang, S.-P.; Wu, H.-H.; Chen, C.-P.; Chang, T.-M.; Lin, W.-H.; Wu, S.-H.; Lee, M.-H.; Hwu, W.-L.; et al. Preimplantation and prenatal genetic diagnosis of aromatic L-amino acid decarboxylase deficiency with an amplification refractory mutation system-quantitative polymerase chain reaction. Taiwan. J. Obstet. Gynecol. 2011, 50, 468–473. [Google Scholar] [CrossRef] [Green Version]
- Andresen, B.S.; Olpin, S.; Poorthuis, B.J.; Scholte, H.R.; Vianey-Saban, C.; Wanders, R.; Ijlst, L.; Morris, A.; Pourfarzam, M.; Bartlett, K.; et al. Clear Correlation of Genotype with Disease Phenotype in Very–Long-Chain Acyl-CoA Dehydrogenase Deficiency. Am. J. Hum. Genet. 1999, 64, 479–494. [Google Scholar] [CrossRef] [Green Version]
- Altarescu, G.; Eldar-Geva, T.; Brooks, B.; Zylber-Haran, E.; Varshaver, I.; Margalioth, E.J.; Levy-Lahad, E.; Renbaum, P. Preimplantation genetic diagnosis (PGD) for nonsyndromic deafness by polar body and blastomere biopsy. J. Assist. Reprod. Genet. 2009, 26, 391–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.-C.; Lin, S.-Y.; Su, Y.-N.; Fang, M.-Y.; Chen, S.-U.; Hsu, C.-J. Preimplantation Genetic Diagnosis (Embryo Screening) for Enlarged Vestibular Aqueduct due toSLC26A4Mutation. Audiol. Neurotol. 2010, 15, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Yazdi, A.K.; Davoudi-Dehaghani, E.; Anari, M.R.; Fouladi, P.; Ebrahimi, E.; Sabeghi, S.; Eftekharian, A.; Fatemi, K.S.; Emami, H.; Sharifi, Z.; et al. The first successful application of preimplantation genetic diagnosis for hearing loss in Iran. Cell. Mol. Biol. 2018, 64, 1708. [Google Scholar] [CrossRef]
- Hao, Y.; Chen, D.; Zhang, Z.; Zhou, P.; Cao, Y.; Wei, Z.; Xu, X.; Chen, B.; Zou, W.; Lv, M.; et al. Successful preimplantation genetic diagnosis by targeted next-generation sequencing on an ion torrent personal genome machine platform. Oncol. Lett. 2018, 15, 4296–4302. [Google Scholar] [CrossRef]
- Giménez, C.; Sarasa, J.; Arjona, C.; Vilamajó, E.; Martínez-Pasarell, O.; Wheeler, K.; Valls, G.; Garcia-Guixé, E.; Wells, D. Karyomapping allows preimplantation genetic diagnosis of a de-novo deletion undetectable using conventional PGD technology. Reprod. Biomed. Online 2015, 31, 770–775. [Google Scholar] [CrossRef] [Green Version]
- Robertson, J.A. Extending preimplantation genetic diagnosis: The ethical debate: Ethical issues in new uses of preimplantation genetic diagnosis. Hum. Reprod. 2003, 18, 465–471. [Google Scholar] [CrossRef] [Green Version]
- Camporesi, S. Choosing Deafness with Preimplantation Genetic Diagnosis: An Ethical Way to Carry on a Cultural Bloodline? Camb. Q. Health Ethics 2009, 19, 86–96. [Google Scholar] [CrossRef] [Green Version]
- Mellon, N.K.; Ouellette, M.; Greer, T.; Gates-Ulanet, P. Achieving Developmental Synchrony in Young Children with Hearing Loss. Trends Amplif. 2009, 13, 223–240. [Google Scholar] [CrossRef]
- Kenneson, A.; Braun, K.V.N.; Boyle, C. GJB2 (connexin 26) variants and nonsyndromic sensorineural hearing loss: A HuGE review. Genet. Med. 2002, 4, 258–274. [Google Scholar] [CrossRef] [Green Version]
- Cryns, K.; Orzan, E.; Murgia, A.; Huygen, P.L.M.; Moreno, F.; del Castillo, I.; Chamberlin, G.P.; Azaiez, H.; Prasad, S.; Cucci, R.; et al. A genotype-phenotype correlation for GJB2 (connexin 26) deafness. J. Med. Genet. 2004, 41, 147–154. [Google Scholar] [CrossRef]
- Kenna, M.A.; Feldman, H.A.; Neault, M.W.; Frangulov, A.; Wu, B.-L.; Fligor, B.; Rehm, H.L. Audiologic phenotype and progression in GJB2 (Connexin 26) hearing loss. Arch. Otolaryngol. Head Neck Surg. 2010, 136, 81–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, D.K.; Chang, K. GJB2-associated hearing loss: Systematic review of worldwide prevalence, genotype, and auditory phenotype. Laryngoscope 2013, 124, E34–E53. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-Y.; Lin, Y.-H.; Liu, T.-C.; Lin, Y.-H.; Tseng, L.-H.; Yang, T.-H.; Chen, P.-L.; Wu, C.-C.; Hsu, C.-J. Prediction Model for Audiological Outcomes in Patients with GJB2 Mutations. Ear Hear. 2020, 41, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; Lee, I.G. Genetic tests by next-generation sequencing in children with developmental delay and/or intellectual disability. Clin. Exp. Pediatr. 2020, 63, 195–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaLonde, E.; Rentas, S.; Lin, F.; Dulik, M.C.; Skraban, C.M.; Spinner, N.B. Genomic Diagnosis for Pediatric Disorders: Revolution and Evolution. Front. Pediatr. 2020, 8, 373. [Google Scholar] [CrossRef]
- Wu, C.-C.; Tsai, C.-Y.; Lin, Y.-H.; Chen, P.-Y.; Lin, P.-H.; Cheng, Y.-F.; Wu, C.-M.; Lin, Y.-H.; Lee, C.-Y.; Erdenechuluun, J.; et al. Genetic Epidemiology and Clinical Features of Hereditary Hearing Impairment in the Taiwanese Population. Genes 2019, 10, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman-Andrews, L. The known unknown: The challenges of genetic variants of uncertain significance in clinical practice. J. Law Biosci. 2017, 4, 648–657. [Google Scholar] [CrossRef] [Green Version]
- Vears, D.F.; Sénécal, K.; Borry, P. Reporting practices for variants of uncertain significance from next generation sequencing technologies. Eur. J. Med. Genet. 2017, 60, 553–558. [Google Scholar] [CrossRef]
- Makhnoon, S.; Shirts, B.H.; Bowen, D.J. Patients’ perspectives of variants of uncertain significance and strategies for uncertainty management. J. Genet. Couns. 2019, 28, 313–325. [Google Scholar] [CrossRef]
- Van Der Aa, N.; Esteki, M.Z.; Vermeesch, J.R.; Voet, T. Preimplantation genetic diagnosis guided by single-cell genomics. Genome Med. 2013, 5, 71. [Google Scholar] [CrossRef]
- Liss, J.; Chromik, I.; Szczyglińska, J.; Jagiełło, M.; Łukaszuk, A.; Lukaszuk, K. Current methods for preimplantation genetic diagnosis. Ginekol. Pol. 2016, 87, 522–526. [Google Scholar] [CrossRef] [Green Version]

| Case | Maternal Age | Maternal Disease Status | Maternal Genotype | Paternal Disease Status | Paternal Genotype | Disease Gene | Inheritance Mode | Mutation Type | Diagnostic Methodology | Oocyte Retrievals | Embryos Diagnosed | Diagnostic Results (Unaffected/ Affected) | Embryos Transferred | Pregnancy Outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 34 | C | c.2168A>G (p.H723R) | C | c.1229C>T (p.T410M) | SLC26A4 | AR | S | ARMS qPCR | 6 | 6 | 3/3 | 3 | failed |
| 2 | 30 | C | c.109G>A (p.V37I) | C | c.235delC (p.L79fs) | GJB2 | AR | S + D | ARMS qPCR | 13 | 2 | 1/1 | 1 | failed |
| 3 | 32 | C | c.109G>A (p.V37I) | C | c.109G>A (p.V37I) | GJB2 | AR | S | ARMS qPCR | 7 | 7 | 1/6 | 1 | failed |
| 4 | 31 | C | c.109G>A (p.V37I) | C | c.235delC (p.L79fs) | GJB2 | AR | S + D | ARMS qPCR | 9 | 5 | 5/0 | 2 | 1 boy |
| 5 | 33 | C | c.235delC (p.L79fs) | C | c.109G>A (p.V37I) | GJB2 | AR | S + D | ARMS qPCR | 11 | 5 | 2/3 | 1 | 1 boy |
| 6 | 33 | C | c.109G>A (p.V37I) | A | homozygous c.109G>A (p.V37I) | GJB2 | AR | S | ARMS qPCR | 28 | 11 | 7/4 | 1 | failed |
| 7 | 35 | C | c.5098G>C (p.E1700Q) | C | c.5197G>A (p.E1733K) | OTOF | AR | S | ARMS qPCR | 20 | 3 | 3/0 | 1 | failed |
| 8 | 33 | C | c.109G>A (p.V37I) | C | c.235delC (p.L79fs) | GJB2 | AR | S + D | ARMS qPCR | 11 | 8 | 6/2 | 2 | 1 girl |
| 9 | 39 | C | GJB2 c.427C>T (p.R143W) | C | GJB2 c.109G>A (p.V37I) heterozygous ACADVL c.277+2T>G | GJB2/ ACADVL | AR | S | ARMS qPCR | 14 | 3 | 1/2 | 1 | 1 boy |
| 10 | 39 | C | c.235delC | C | c.299_300delAT(p.His100fs) | GJB2 | AR | D | STR analysis | 2 | 1 | 1/0 | NI | NI |
| 11 | 31 | A | MYO15A c.3524dup; 6956+1G>A/GJB2 c.299_300delAT carrier | A | MITF c.1052C>T | MITF | AD | S + D | STR analysis | 15 | 9 | 6/3 | NI | NI |
| Year | Disease Genes | PGD Method | Strategy | Success Case No. | Place | Reference |
|---|---|---|---|---|---|---|
| 2009 | GJB2/GJB6 | Polar body and blastomere PCR | Direct diagnosis | 8 | Israel | [12] |
| 2010 | SLC26A4 | Single-cell mini-sequencing | Direct diagnosis | 1 | Taiwan | [13] |
| 2018 | GJB2 | Single-cell Sanger sequencing | Direct diagnosis | 1 | Iran | [14] |
| 2018 | SLC26A4 | Next-generation sequencing | Direct diagnosis | 1 | China | [15] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.-L.; Lin, P.-H.; Chiang, Y.-T.; Huang, W.-J.; Lin, C.-F.; Ma, G.-C.; Chang, S.-P.; Fan, J.-Y.; Lin, S.-Y.; Wu, C.-C.; et al. Preimplantation Genetic Diagnosis in Hereditary Hearing Impairment. Diagnostics 2021, 11, 2395. https://doi.org/10.3390/diagnostics11122395
Chen H-L, Lin P-H, Chiang Y-T, Huang W-J, Lin C-F, Ma G-C, Chang S-P, Fan J-Y, Lin S-Y, Wu C-C, et al. Preimplantation Genetic Diagnosis in Hereditary Hearing Impairment. Diagnostics. 2021; 11(12):2395. https://doi.org/10.3390/diagnostics11122395
Chicago/Turabian StyleChen, Hsin-Lin, Pei-Hsuan Lin, Yu-Ting Chiang, Wen-Jie Huang, Chi-Fang Lin, Gwo-Chin Ma, Shun-Ping Chang, Jun-Yang Fan, Shin-Yu Lin, Chen-Chi Wu, and et al. 2021. "Preimplantation Genetic Diagnosis in Hereditary Hearing Impairment" Diagnostics 11, no. 12: 2395. https://doi.org/10.3390/diagnostics11122395