
Bridging Pyroptosis and Immunity: A Comprehensive Study of the Pyroptosis-Related Long Non-Coding RNA Signature in Breast Cancer
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. Screening Pyroptosis-Related Differentially Expressed Genes (DEGs) and Analyzing Their Functions
2.3. The Identification of Pyroptosis-Related LncRNAs
2.4. The Establishment and Verification of the Pyroptosis-Related LncRNAs Signature
2.5. Screening Risk-Related DEGs and Analyzing Their Functions
2.6. Gene Set Enrichment Analysis (GSEA) of the High- and Low-Risk Groups
2.7. Tumor Mutation Burden (TMB) of the High- and Low-Risk Groups
2.8. The Tumor Immune Microenvironment of the High- and Low-Risk Groups
2.9. Cluster of Breast Cancer Subtypes and Their Associated Functional Analysis
2.10. The Construction of the ceRNA Regulatory Network
2.11. Statistical Analyses
3. Results
3.1. The Establishment and Verification of the Prognostic Pyroptosis-Related LncRNA Signature
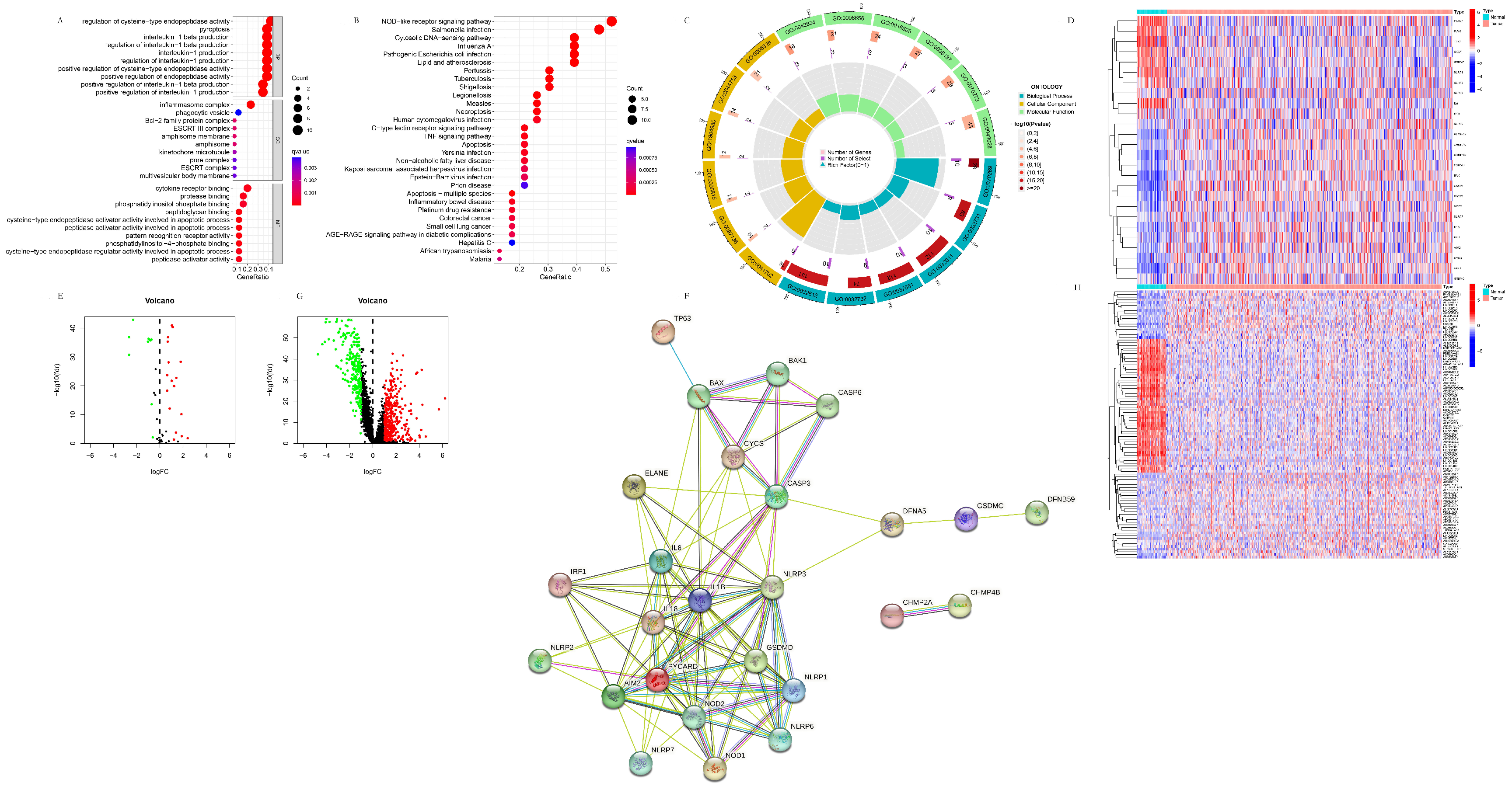
3.1.1. The Identification and Functional Analysis of the Pyroptosis-Related DEGs
3.1.2. The Identification of the Pyroptosis-Related DElncRNAs
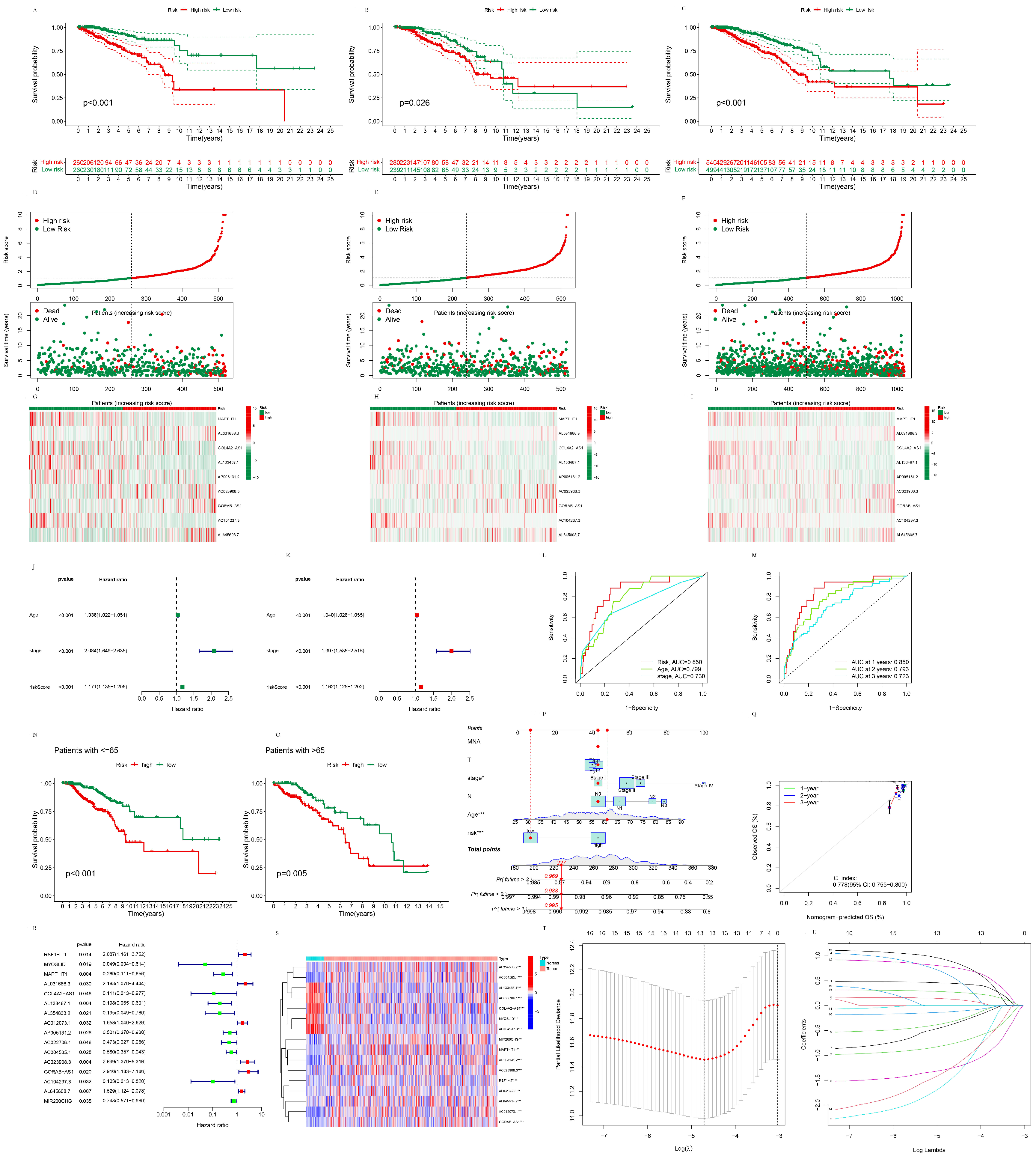
3.1.3. The Establishment of the Prognostic Pyroptosis-Related LncRNA Signature
3.1.4. The Validation of the Prognostic Pyroptosis-Related LncRNA Signature
3.2. The Functional Analysis of the Pyroptosis-Related LncRNA Signature
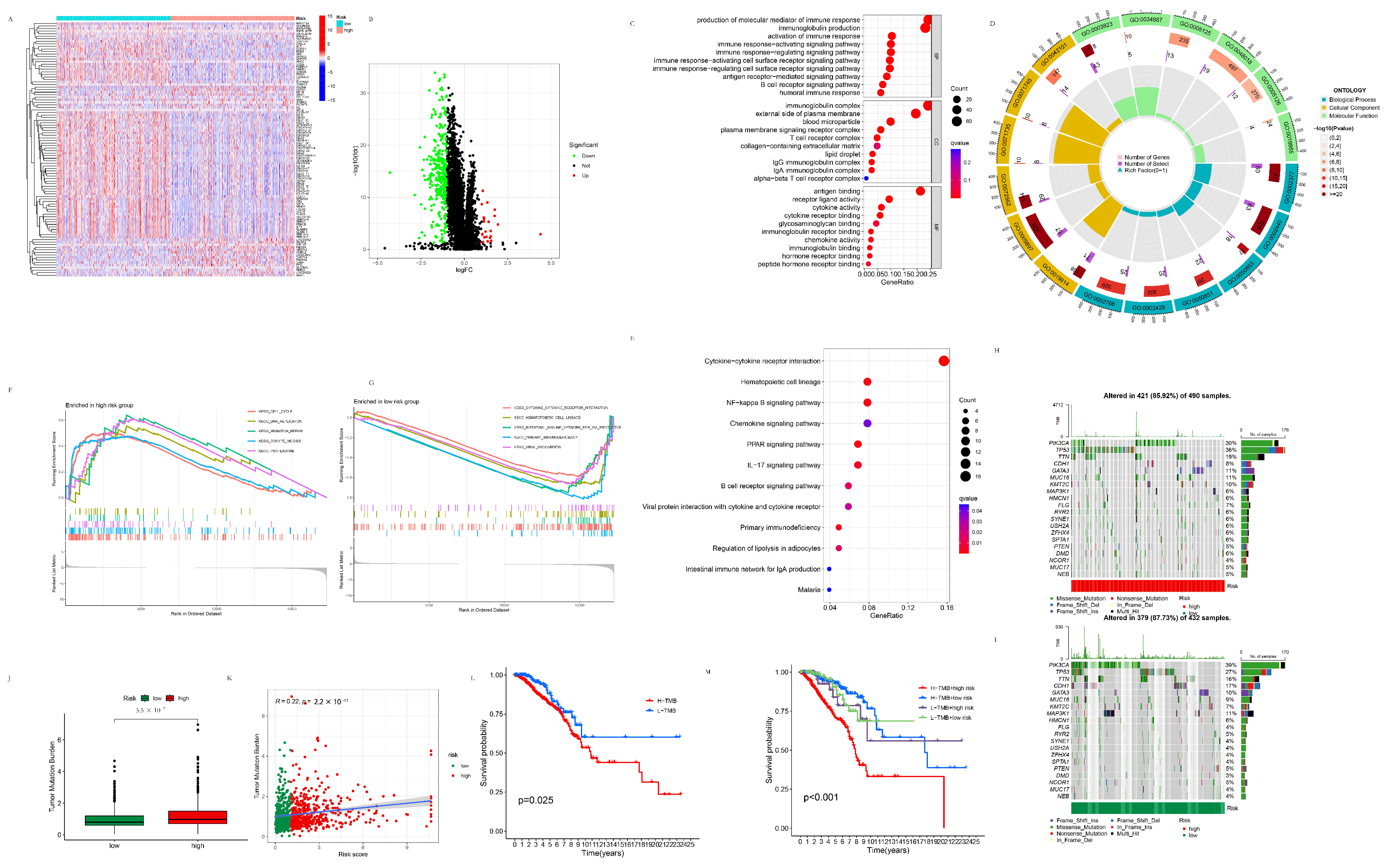
3.2.1. Differentially Expressed Genes between the High- and Low-Risk Groups
3.2.2. GSEA Analysis of the High- and Low-Risk Groups
3.2.3. TMB Analysis of the High- and Low-Risk Groups
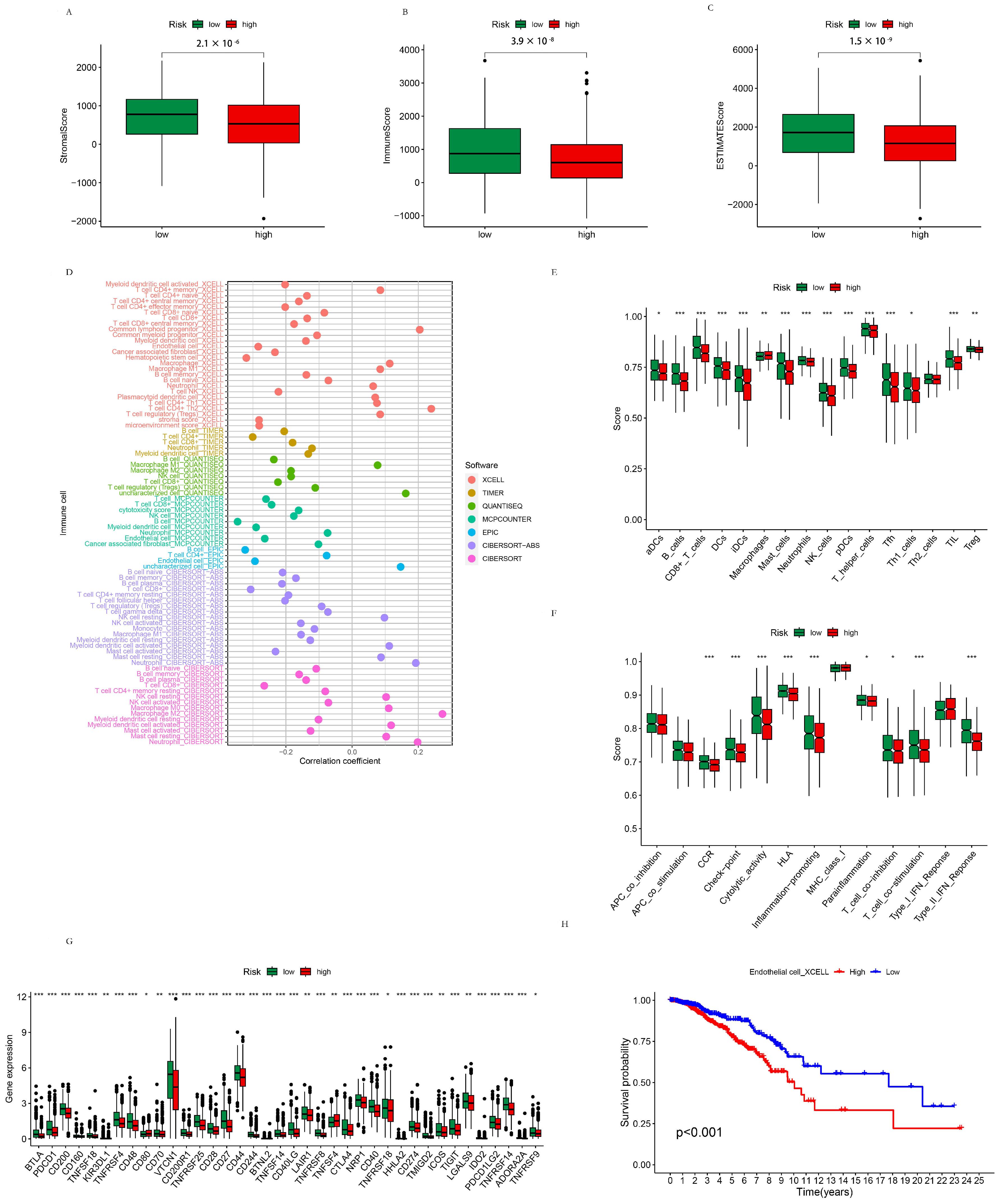
3.2.4. TME Analysis of the High- and Low-Risk Groups
3.3. The Subtypes of Breast Cancer Patients Based on Their Risk Score
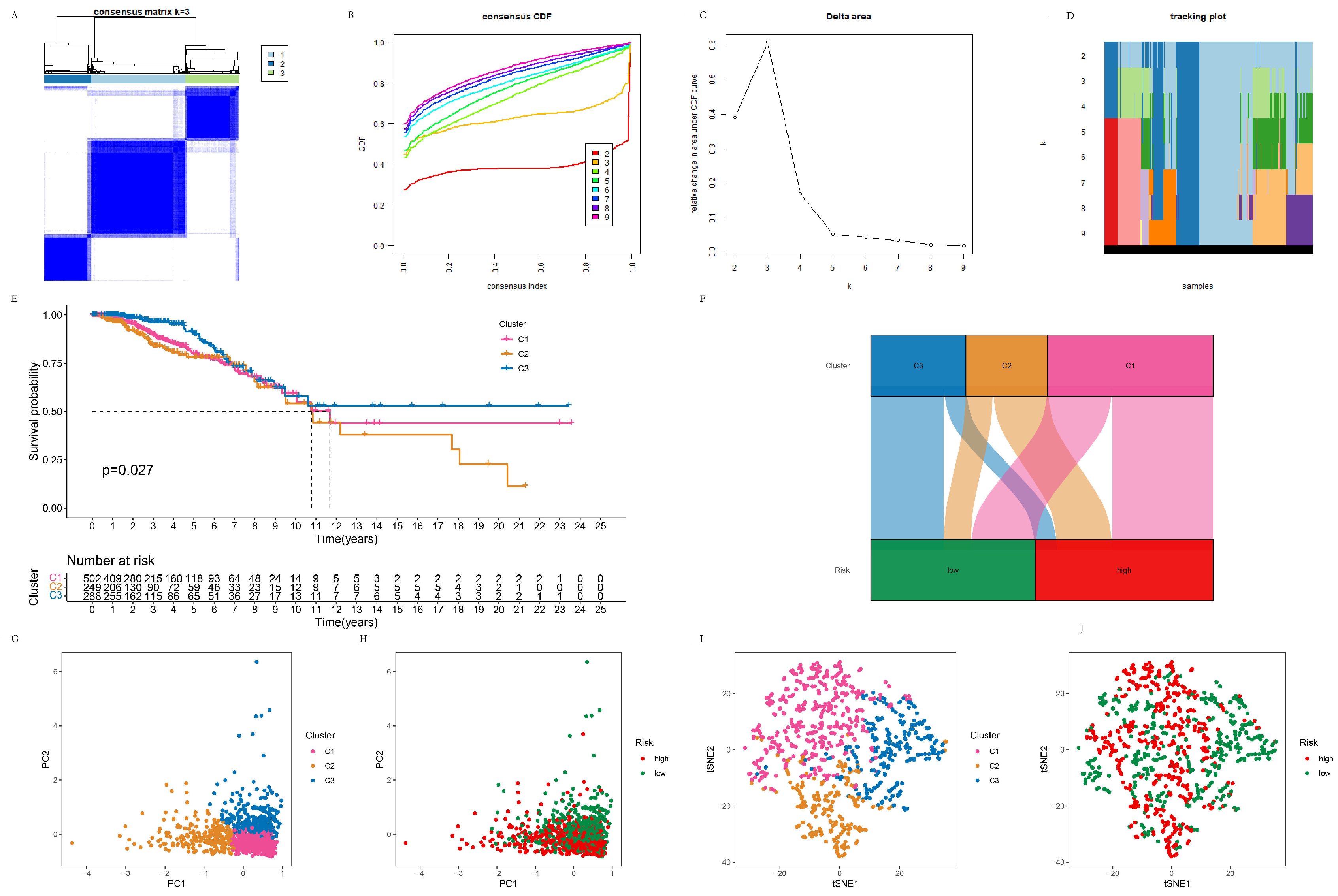
3.3.1. The Clustering of Breast Cancer Patients Using the Consensus Cluster Method
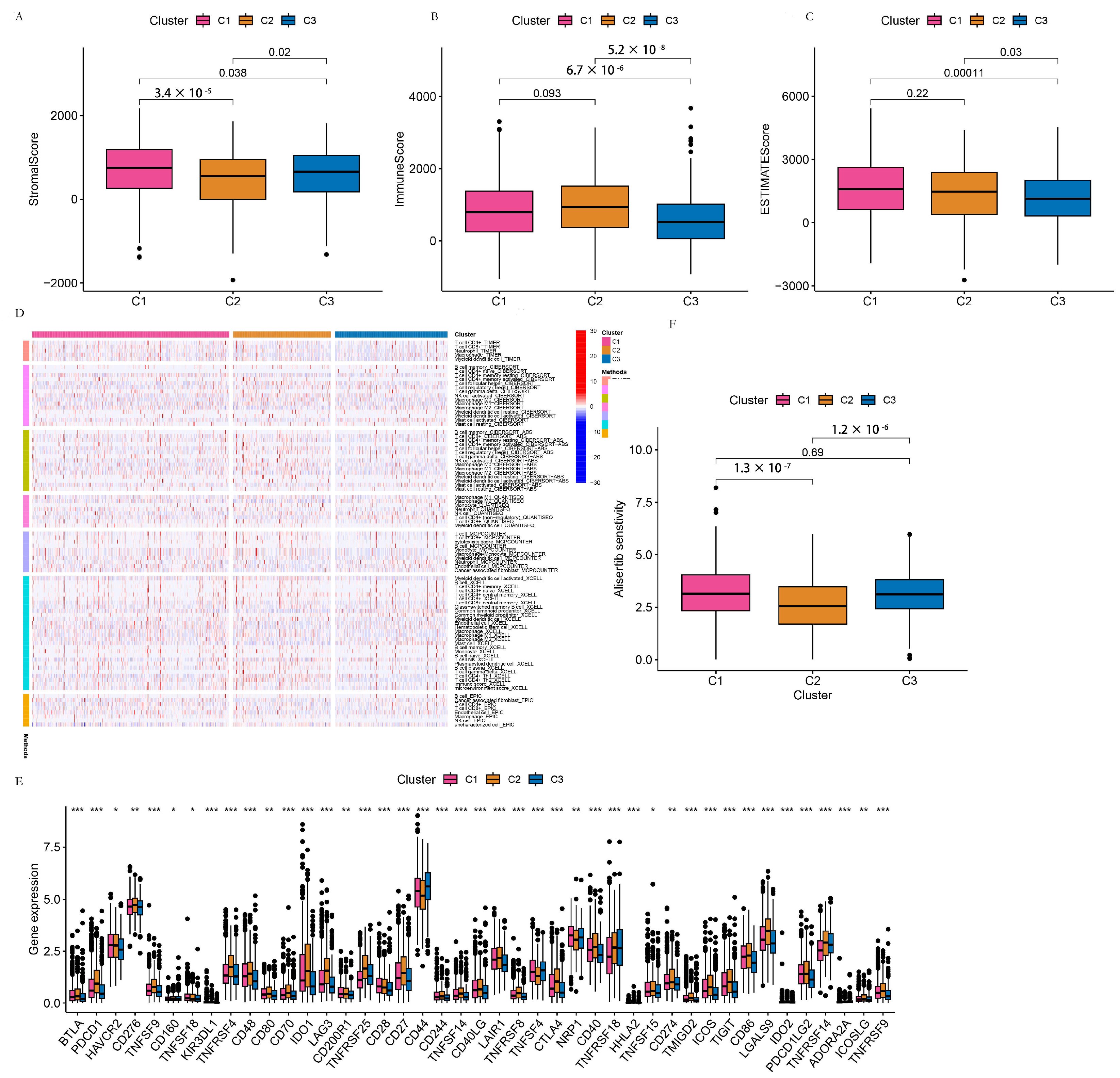
3.3.2. TME Analysis of the C1, C2, and C3 Clusters
3.4. The Construction and Analysis of the Pyroptosis-Related ceRNA Regulatory Network
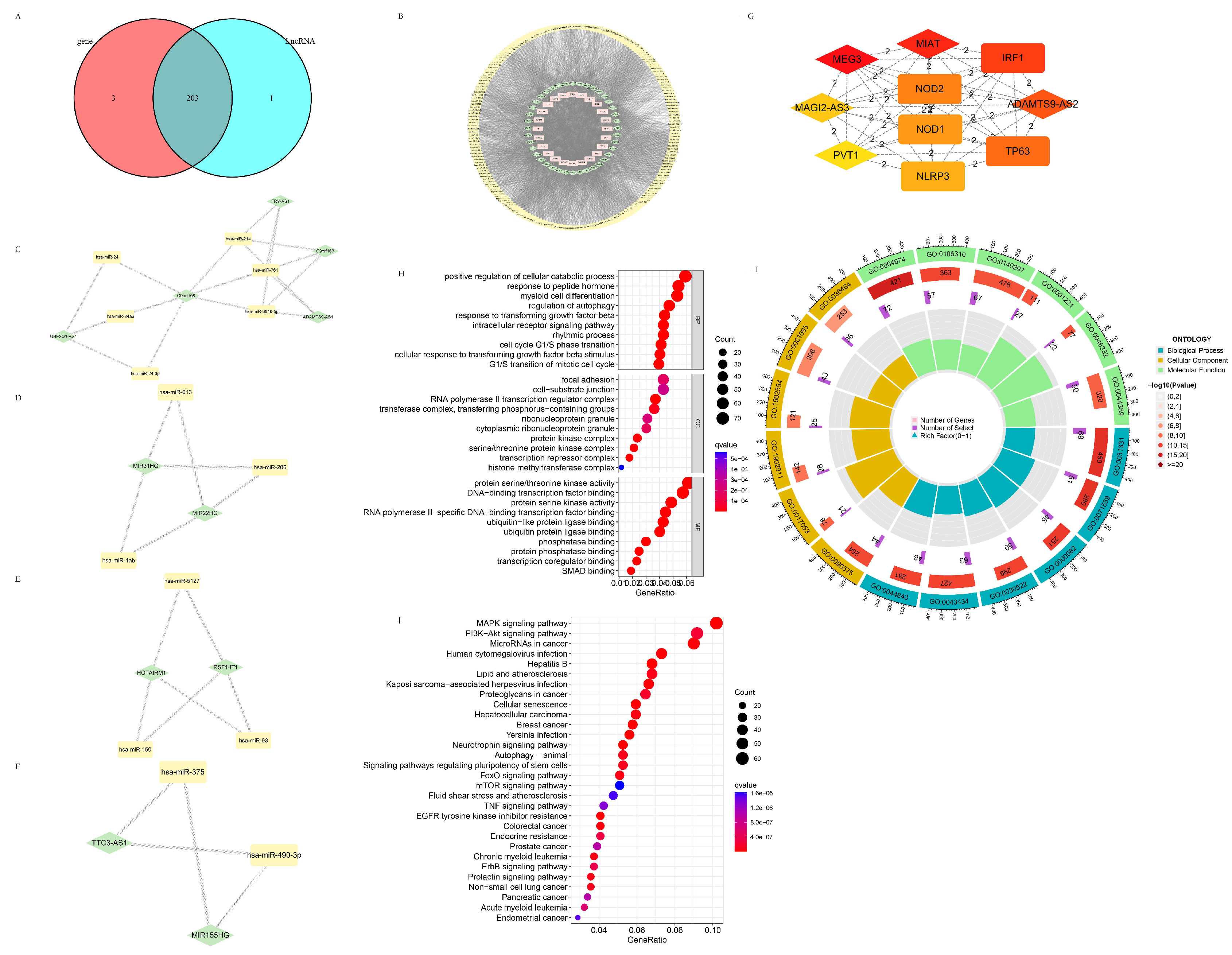
3.4.1. The Establishment of the Pyroptosis-Related ceRNA Regulatory Network
3.4.2. Potential Target Genes of the Intersection Target miRNA Predicted by the Pyroptosis-Related DEGs and DELncRNAs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Yeo, S.K.; Guan, J.L. Breast Cancer: Multiple Subtypes within a Tumor? Trends Cancer 2017, 3, 753–760. [Google Scholar] [CrossRef]
- Loibl, S.; Poortmans, P.; Morrow, M.; Denkert, C.; Curigliano, G. Breast cancer. Lancet 2021, 397, 1750–1769, Erratum in: Lancet 2021, 397, 1710. [Google Scholar] [CrossRef]
- Goldhirsch, A.; Wood, W.C.; Coates, A.S.; Gelber, R.D.; Thürlimann, B.; Senn, H.J.; Panel members. Trategies for subtypes--dealing with the diversity of breast cancer: Highlights of the St. Gallen International Expert Consensus on the Primary Therapy of Early Breast Cancer 2011. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2011, 22, 1736–1747. [Google Scholar] [CrossRef]
- Sarhangi, N.; Hajjari, S.; Heydari, S.F.; Ganjizadeh, M.; Rouhollah, F.; Hasanzad, M. Breast cancer in the era of precision medicine. Mol. Biol. Rep. 2022, 49, 10023–10037. [Google Scholar] [CrossRef]
- Fang, Y.; Tian, S.; Pan, Y.; Li, W.; Wang, Q.; Tang, Y.; Yu, T.; Wu, X.; Shi, Y.; Ma, P.; et al. Pyroptosis: A new frontier in cancer. Biomed. Pharmacother. 2020, 121, 109595. [Google Scholar] [CrossRef]
- Hsu, S.K.; Li, C.Y.; Lin, I.L.; Syue, W.J.; Chen, Y.F.; Cheng, K.C.; Teng, Y.N.; Lin, Y.H.; Yen, C.H.; Chiu, C.C. Inflammation-related pyroptosis, a novel programmed cell death pathway, and its crosstalk with immune therapy in cancer treatment. Theranostics 2021, 11, 8813–8835. [Google Scholar] [CrossRef]
- Hsu, S.K.; Chen, Y.E.; Shu, E.D.; Ko, C.C.; Chang, W.T.; Lin, I.L.; Li, C.Y.; Gallego, R.P.; Chiu, C.C. The Pyroptotic and Nonpyroptotic Roles of Gasdermins in Modulating Cancer Progression and Their Perspectives on Cancer Therapeutics. Arch. Immunol. Ther. Exp. 2023, 71, 14. [Google Scholar] [CrossRef]
- Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [CrossRef]
- Zychlinsky, A.; Prevost, M.C.; Sansonetti, P.J. Shigella flexneri induces apoptosis in infected macrophages. Nature 1992, 358, 167–169. [Google Scholar] [CrossRef]
- Zychlinsky, A.; Fitting, C.; Cavaillon, J.M.; Sansonetti, P.J. Interleukin 1 is released by murine macrophages during apoptosis induced by Shigella flexneri. J. Clin. Investig. 1994, 94, 1328–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hersh, D.; Monack, D.M.; Smith, M.R.; Ghori, N.; Falkow, S.; Zychlinsky, A. The Salmonella invasin SipB induces macrophage apoptosis by binding to caspase-1. Proc. Natl. Acad. Sci. USA 1999, 96, 2396–2401. [Google Scholar] [CrossRef]
- Brennan, M.A.; Cookson, B.T. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol. Microbiol. 2000, 38, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Christgen, S.; Place, D.E.; Kanneganti, T.D. Toward targeting inflammasomes: Insights into their regulation and activation. Cell Res. 2020, 30, 315–327. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [Green Version]
- He, W.T.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.H.; Zhong, C.Q.; Han, J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Vande Walle, L.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Zhou, Z.; He, H.; Wang, K.; Shi, X.; Wang, Y.; Su, Y.; Wang, Y.; Li, D.; Liu, W.; Zhang, Y.; et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 2020, 368, eaaz7548. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Y.; Xia, S.; Kong, Q.; Li, S.; Liu, X.; Junqueira, C.; Meza-Sosa, K.F.; Mok, T.M.Y.; Ansara, J.; et al. Suppresses tumour growth by activating anti-tumour immunity. Nature 2020, 579, 415–420. [Google Scholar] [CrossRef]
- Deng, W.; Bai, Y.; Deng, F.; Pan, Y.; Mei, S.; Zheng, Z.; Min, R.; Wu, Z.; Li, W.; Miao, R.; et al. Streptococcal pyrogenic exotoxin B cleaves GSDMA and triggers pyroptosis. Nature 2022, 602, 496–502. [Google Scholar] [CrossRef]
- Wei, X.; Xie, F.; Zhou, X.; Wu, Y.; Yan, H.; Liu, T.; Huang, J.; Wang, F.; Zhou, F.; Zhang, L. Role of pyroptosis in inflammation and cancer. Cell. Mol. Immunol. 2022, 19, 971–992. [Google Scholar] [CrossRef]
- Xia, X.; Wang, X.; Cheng, Z.; Qin, W.; Lei, L.; Jiang, J.; Hu, J. The role of pyroptosis in cancer: pro-cancer or pro-"host"? Cell Death Dis. 2019, 10, 650. [Google Scholar] [CrossRef] [Green Version]
- An, H.; Heo, J.S.; Kim, P.; Lian, Z.; Lee, S.; Park, J.; Hong, E.; Pang, K.; Park, Y.; Ooshima, A.; et al. Tetraarsenic hexoxide enhances generation of mitochondrial ROS to promote pyroptosis by inducing the activation of caspase-3/GSDME in triple-negative breast cancer cells. Cell Death Dis. 2021, 12, 159. [Google Scholar] [CrossRef]
- Huang, Y.; Xu, W.; Zhou, R. NLRP3 inflammasome activation and cell death. Cell. Mol. Immunol. 2021, 18, 2114–2127. [Google Scholar] [CrossRef]
- Wu, T.C.; Xu, K.; Martinek, J.; Young, R.R.; Banchereau, R.; George, J.; Turner, J.; Kim, K.I.; Zurawski, S.; Wang, X.; et al. IL1 Receptor Antagonist Controls Transcriptional Signature of Inflammation in Patients with Metastatic Breast Cancer. Cancer Res. 2018, 78, 5243–5258. [Google Scholar] [CrossRef] [Green Version]
- Jang, J.H.; Kim, D.H.; Lim, J.M.; Lee, J.W.; Jeong, S.J.; Kim, K.P.; Surh, Y.J. Breast Cancer Cell-Derived Soluble CD44 Promotes Tumor Progression by Triggering Macrophage IL1β Production. Cancer Res. 2020, 80, 1342–1356. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Yuan, R.Q.; Fuchs, A.; Yao, Y.; Joseph, A.; Schwall, R.; Schnitt, S.J.; Guida, A.; Hastings, H.M.; Andres, J.; et al. Expression of interleukin-1beta in human breast carcinoma. Cancer 1997, 80, 421–434. [Google Scholar] [CrossRef]
- Günel, N.; Coskun, U.; Sancak, B.; Hasdemir, O.; Sare, M.; Bayram, O.; Celenkoglu, G.; Ozkan, S. Prognostic value of serum IL-18 and nitric oxide activity in breast cancer patients at operable stage. Am. J. Clin. Oncol. 2003, 26, 416–421. [Google Scholar] [CrossRef]
- El-Deeb, M.M.K.; El-Sheredy, H.G.; Mohammed, A.F. The Possible Role of Interleukin (IL)-18 and Nitrous Oxide and Their Relation to Oxidative Stress in the Development and Progression of Breast Cancer. Asian Pac. J. Cancer Prev. 2019, 20, 2659–2665. [Google Scholar] [CrossRef]
- Hergueta-Redondo, M.; Sarrió, D.; Molina-Crespo, Á.; Megias, D.; Mota, A.; Rojo-Sebastian, A.; García-Sanz, P.; Morales, S.; Abril, S.; Cano, A.; et al. Gasdermin-B promotes invasion and metastasis in breast cancer cells. PLoS ONE 2014, 9, e90099. [Google Scholar] [CrossRef] [Green Version]
- Hergueta-Redondo, M.; Sarrio, D.; Molina-Crespo, Á.; Vicario, R.; Bernadó-Morales, C.; Martínez, L.; Rojo-Sebastián, A.; Serra-Musach, J.; Mota, A.; Martínez-Ramírez, Á.; et al. Gasdermin B expression predicts poor clinical outcome in HER2-positive breast cancer. Oncotarget 2016, 7, 56295–56308. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Mao, X.; Huang, Y.; Zhu, Q.; Guan, J.; Wu, L. Detection of proteins associated with the pyroptosis signaling pathway in breast cancer tissues and their significance. Int. J. Clin. Exp. Pathol. 2020, 13, 1408–1414. [Google Scholar]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef] [Green Version]
- Scarpitta, A.; Hacker, U.T.; Büning, H.; Boyer, O.; Adriouch, S. Pyroptotic and Necroptotic Cell Death in the Tumor Microenvironment and Their Potential to Stimulate Anti-Tumor Immune Responses. Front. Oncol. 2021, 11, 731598. [Google Scholar] [CrossRef]
- Liu, J.; Hong, M.; Li, Y.; Chen, D.; Wu, Y.; Hu, Y. Programmed Cell Death Tunes Tumor Immunity. Front. Immunol. 2022, 13, 847345. [Google Scholar] [CrossRef]
- Voloshin, T.; Alishekevitz, D.; Kaneti, L.; Miller, V.; Isakov, E.; Kaplanov, I.; Voronov, E.; Fremder, E.; Benhar, M.; Machluf, M.; et al. Blocking IL1β Pathway Following Paclitaxel Chemotherapy Slightly Inhibits Primary Tumor Growth but Promotes Spontaneous Metastasis. Mol. Cancer Ther. 2015, 14, 1385–1394. [Google Scholar] [CrossRef] [Green Version]
- Park, I.H.; Yang, H.N.; Lee, K.J.; Kim, T.S.; Lee, E.S.; Jung, S.Y.; Kwon, Y.; Kong, S.Y. Tumor-derived IL-18 induces PD-1 expression on immunosuppressive NK cells in triple-negative breast cancer. Oncotarget 2017, 8, 32722–32730. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Wang, H.; Ding, A.; Golenbock, D.T.; Latz, E.; Czura, C.J.; Fenton, M.J.; Tracey, K.J.; Yang, H. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock 2006, 26, 174–179. [Google Scholar] [CrossRef]
- Peng, W.; Si, S.; Zhang, Q.; Li, C.; Zhao, F.; Wang, F.; Yu, J.; Ma, R. Long non-coding RNA MEG3 functions as a competing endogenous RNA to regulate gastric cancer progression. J. Exp. Clin. Cancer Res. 2015, 34, 79. [Google Scholar] [CrossRef] [Green Version]
- Ma, F.; Wang, S.-H.; Cai, Q.; Zhang, M.-D.; Yang, Y.; Ding, J. Overexpression of LncRNA AFAP1-AS1 predicts poor prognosis and promotes cells proliferation and invasion in gallbladder cancer. Biomed. Pharmacother. 2016, 84, 1249–1255. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Liu, G.-B.; Liu, B.-H.; Chen, G.; Li, K.; Zheng, S.; Dong, K.-R. Targeting long non-coding RNA-TUG1 inhibits tumor growth and angiogenesis in hepatoblastoma. Cell Death Dis. 2016, 7, e2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Ye, C.; Xiong, H.; Shen, Y.; Lu, Y.; Zhou, J.; Wang, L. Dysregulation of long non-coding RNA in breast cancer: An overview of mechanism and clinical implication. Oncotarget 2017, 8, 5508–5522. [Google Scholar] [CrossRef] [Green Version]
- Chi, Y.; Wang, D.; Wang, J.; Yu, W.; Yang, J. Long Non-Coding RNA in the Pathogenesis of Cancers. Cells 2019, 8, 1015. [Google Scholar] [CrossRef] [Green Version]
- Alipoor, F.J.; Asadi, M.H.; Torkzadeh-Mahani, M. MIAT lncRNA is overexpressed in breast cancer and its inhibition triggers senescence and G1 arrest in MCF7 cell line. J. Cell. Biochem. 2018, 119, 6470–6481. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, Y.; Chang, W.; Zhang, L.; Syrigos, K.N.; Li, P. Noncoding RNA-mediated regulation of pyroptotic cell death in cancer. Front Oncol. 2022, 12, 1015587. [Google Scholar] [CrossRef]
- Yan, H.; Luo, B.; Wu, X.; Guan, F.; Yu, X.; Zhao, L.; Ke, X.; Wu, J.; Yuan, J. Cisplatin Induces Pyroptosis via Activation of MEG3/NLRP3/caspase-1/GSDMD Pathway in Triple-Negative Breast Cancer. Int. J. Biol. Sci. 2021, 17, 2606–2621. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Tian, Z.; Hou, H.; Gai, W. The noncoding RNAs regulating pyroptosis in colon adenocarcinoma were derived from the construction of a ceRNA network and used to develop a prognostic model. BMC Med. Genom. 2022, 15, 201. [Google Scholar] [CrossRef]
- Lin, W.; Chen, Y.; Wu, B.; Chen, Y.; Li, Z. Identification of the pyroptosis-related prognostic gene signature and the associated regulation axis in lung adenocarcinoma. Cell Death Discov. 2021, 7, 161. [Google Scholar] [CrossRef]
- Han, J.; Hu, Y.; Ding, S.; Liu, S.; Wang, H. The analysis of the pyroptosis-related genes and hub gene TP63 ceRNA axis in osteosarcoma. Front. Immunol. 2022, 13, 974916. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Wang, L.; Han, Y.; He, Q. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Shi, W.; Chen, Z.; Liu, H.; Miao, C.; Feng, R.; Wang, G.; Chen, G.; Chen, Z.; Fan, P.; Pang, W.; et al. COL11A1 as an novel biomarker for breast cancer with machine learning and immunohistochemistry validation. Front. Immunol. 2022, 13, 937125. [Google Scholar] [CrossRef]
- Yu, G. Enrichplot: Visualization of Functional Enrichment Result. R Package Version 1.20.0. 2023. Available online: https://yulab-smu.top/biomedical-knowledge-mining-book/ (accessed on 15 April 2023).
- Jensen, L.J.; Kuhn, M.; Stark, M.; Chaffron, S.; Creevey, C.; Muller, J.; Doerks, T.; Julien, P.; Roth, A.; Simonovic, M.; et al. STRING 8—A global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res. 2009, 37, D412-6. [Google Scholar] [CrossRef] [Green Version]
- Kolde, R. Pheatmap: Pretty Heatmaps. R Package Version 1.0.12. 2019. Available online: https://CRAN.R-project.org/package=pheatmap (accessed on 15 April 2023).
- Therneau, T.M.; Grambsch, P.M. Modeling Survival Data: Extending the Cox Model; Springer: New York, NY, USA, 2000; ISBN 0-387-98784-3. [Google Scholar]
- Kuhn, M. Building Predictive Models in R Using the caret Package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.; Tibshirani, R.; Hastie, T. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef]
- Kassambara, A.; Kosinski, M.; Biecek, P. Survminer: Drawin‘Surviv’l Curves Using ‘ggplot2’. R Package Version 0.4.9. 2021. Available online: https://CRAN.R-project.org/package=survminer (accessed on 15 April 2023).
- Blanche, P.; Dartigues, J.-F.; Jacqmin, H. Gadda Estimating and Comparing time-dependent areas under receiver operating characteristic curves for censored event times with competing risks. Stat. Med. 2013, 32, 5381–5397. Available online: http://onlinelibrary.wiley.com/doi/10.1002/sim.5958/full (accessed on 18 April 2023). [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassambara, A. Ggpubr: ‘ggplot2’ Based Publication Ready Plots. R Package Version 0.6.0. 2023. Available online: https://CRAN.R-project.org/package=ggpubr (accessed on 18 April 2023).
- Wilke, C.; Wiernik, B. Ggtext: Improved Text Rendering Support for ‘ggplot2’. R Package Version 0.1.2. 2022. Available online: https://CRAN.R-project.org/package=ggtext (accessed on 18 April 2023).
- Wickham, H.; Seidel, D. Scales: Scale Functions for Visualization. R Package Version 1.2.1. 2022. Available online: https://CRAN.R-project.org/package=scales (accessed on 18 April 2023).
- Hadley Wickham Reshaping Data with the reshape Package. J. Stat. Softw. 2007, 21, 1–20. Available online: http://www.jstatsoft.org/v21/i12/ (accessed on 18 April 2023).
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef] [Green Version]
- Hänzelmann, S.; Castelo, R.; Guinney, A. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.; Falcon, S.; Gentleman, R. GSEA Base: Gene Set Enrichment Data Structures and Methods. R Package Version 1.62.0. 2023. Available online: https://bioconductor.org/packages/GSEABase (accessed on 20 April 2023).
- Maeser, D. oncoPredict: Drug and Biomarker Discovery. R Package Version 0.2. 2021. Available online: https://CRAN.R-project.org/package=oncoPredict (accessed on 20 April 2023).
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2023; Available online: https://www.R-project.org/ (accessed on 20 April 2023).
- Yoshihara, K.; Kim, H.; Verhaak, R.G. estimate: Estimate of Stromal and Immune Cells in Malignant Tumor Tissues from Expression Data. R Package Version 1.0.13/r21. 2016. Available online: https://R-Forge.R-project.org/projects/estimate/ (accessed on 20 April 2023).
- Chen, C.; Ye, Q.; Wang, L.; Zhou, J.; Xiang, A.; Lin, X.; Guo, J.; Hu, S.; Rui, T.; Liu, J. Targeting pyroptosis in breast cancer: Biological functions and therapeutic potentials on It. Cell Death Discov. 2023, 9, 75. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Hayes, D.N. Consensus Cluster Plus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef] [Green Version]
- Brunson, J.C.; Quentin, D. Read Ggalluvial: Alluvial Plots in ‘ggplot2’. R Package Version 0.12.5. 2023. Available online: http://corybrunson.github.io/ggalluvial/ (accessed on 20 April 2023).
- Wickham, H.; François, R.; Henry, L.; Müller, K.; Vaughan, D. dplyr: A Grammar of Data Manipulation. R Package Version 1.1.2. 2023. Available online: https://CRAN.R-project.org/package=dplyr (accessed on 20 April 2023).
- Krijthe, J.H. Rtsne: T-Distributed Stochastic Neighbor Embedding using a Barnes-Hut Implementation.2015. Available online: https://github.com/jkrijthe/Rtsne (accessed on 20 April 2023).
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Bader, G.D.; Hogue, C.W. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014, 8 (Suppl. S4), S11. [Google Scholar] [CrossRef] [Green Version]
- Tuncer, M.; Alcan, S. Pyroptosis: A new therapeutic strategy in cancer. Mol. Biol. Rep. 2023, 50, 6191–6200. [Google Scholar] [CrossRef]
- Zhou, Y.; Zheng, J.; Bai, M.; Gao, Y.; Lin, N. Effect of Pyroptosis-Related Genes on the Prognosis of Breast Cancer. Front. Oncol. 2022, 12, 948169. [Google Scholar] [CrossRef]
- Sharma, B.R.; Karki, R.; Kanneganti, T.D. Role of AIM2 inflammasome in inflammatory diseases, cancer and infection. Eur. J. Immunol. 2019, 49, 1998–2011. [Google Scholar] [CrossRef] [Green Version]
- Cui, C.; Chakraborty, K.; Tang, X.A.; Zhou, G.; Schoenfelt, K.Q.; Becker, K.M.; Hoffman, A.; Chang, Y.F.; Blank, A.; Reardon, C.A.; et al. Neutrophil elastase selectively kills cancer cells and attenuates tumorigenesis. Cell 2021, 184, 3163–3177.e21. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Wei, D.; Xu, X.; Mao, X.; Mo, D.; Yan, L.; Xu, W.; Yan, F. Long non-coding RNA MIR200CHG promotes breast cancer proliferation, invasion, and drug resistance by interacting with and stabilizing YB-1. NPJ Breast Cancer 2021, 7, 94. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yu, Y.; Zhang, P.; Ma, G.; Zhang, M.; Liang, Y.; Jiao, W.; Niu, H. Identification of NTRK3 as a potential prognostic biomarker associated with tumor mutation burden and immune infiltration in bladder cancer. BMC Cancer 2021, 21, 458. [Google Scholar] [CrossRef]
- Zhang, L.; Li, B.; Peng, Y.; Wu, F.; Li, Q.; Lin, Z.; Xie, S.; Xiao, L.; Lin, X.; Ou, Z.; et al. The prognostic value of TMB and the relationship between TMB and immune infiltration in head and neck squamous cell carcinoma: A gene expression-based study. Oral Oncol. 2020, 110, 104943. [Google Scholar] [CrossRef] [PubMed]
- Park, E.G.; Pyo, S.J.; Cui, Y.; Yoon, S.H.; Nam, J.W. Tumor immune microenvironment lncRNAs. Brief Bioinform. 2022, 23, bbab504. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, A.; Turco, C.; Fontemaggi, G.; Fazi, F. Non-Coding RNAs in the Crosstalk between Breast Cancer Cells and Tumor-Associated Macrophages. Non-Coding RNA 2022, 8, 16. [Google Scholar] [CrossRef]
- Yin, Q.; Ma, H.; Bamunuarachchi, G.; Zheng, X.; Ma, Y. Long Non-Coding RNAs, Cell Cycle, and Human Breast Cancer. Hum. Gene Ther. 2023, 34, 481–494. [Google Scholar] [CrossRef]
- de Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 2023, 41, 374–403. [Google Scholar] [CrossRef]
- Dieci, M.V.; Miglietta, F.; Guarneri, V. Immune Infiltrates in Breast Cancer: Recent Updates and Clinical Implications. Cells 2021, 10, 223. [Google Scholar] [CrossRef] [PubMed]
- Heeke, A.L.; Tan, A.R. Checkpoint inhibitor therapy for metastatic triple-negative breast cancer. Cancer Metastasis Rev. 2021, 40, 537–547. [Google Scholar] [CrossRef]
- Pellegrino, B.; Tommasi, C.; Cursio, O.E.; Musolino, A.; Migliori, E.; De Silva, P.; Senevirathne, T.H.; Schena, M.; Scartozzi, M.; Farci, D.; et al. A review of immune checkpoint blockade in breast cancer. Semin. Oncol. 2021, 48, 208–225. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting Tumor Microenvironment for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef] [Green Version]
- Loi, S.; Michiels, S.; Adams, S.; Loibl, S.; Budczies, J.; Denkert, C.; Salgado, R. The journey of tumor-infiltrating lymphocytes as a biomarker in breast cancer: Clinical utility in an era of checkpoint inhibition. Ann. Oncol. 2021, 32, 1236–1244. [Google Scholar] [CrossRef]
- El Bairi, K.; Haynes, H.R.; Blackley, E.; Fineberg, S.; Shear, J.; Turner, S.; de Freitas, J.R.; Sur, D.; Amendola, L.C.; Gharib, M.; et al. International Immuno-Oncology Biomarker Working Group. The tale of TILs in breast cancer: A report from The International Immuno-Oncology Biomarker Working Group. NPJ Breast Cancer 2021, 7, 150. [Google Scholar] [CrossRef] [PubMed]
- Karn, T.; Jiang, T.; Hatzis, C.; Sänger, N.; El-Balat, A.; Rody, A.; Holtrich, U.; Becker, S.; Bianchini, G.; Pusztai, L. Association Between Genomic Metrics and Immune Infiltration in Triple-Negative Breast Cancer. JAMA Oncol. 2017, 3, 1707–1711. [Google Scholar] [CrossRef]
- Silwal-Pandit, L.; Langerød, A.; Børresen-Dale, A.L. TP53 Mutations in Breast and Ovarian Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a026252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miricescu, D.; Totan, A.; Stanescu-Spinu, I.I.; Badoiu, S.C.; Stefani, C.; Greabu, M. PI3K/AKT/mTOR Signaling Pathway in Breast Cancer: From Molecular Landscape to Clinical Aspects. Int. J. Mol. Sci. 2020, 22, 173. [Google Scholar] [CrossRef]
- Ghafouri-Fard, S.; Hussen, B.M.; Gharebaghi, A.; Eghtedarian, R.; Taheri, M. LncRNA signature in colorectal cancer. Pathol. Res. Pract. 2021, 222, 153432. [Google Scholar] [CrossRef] [PubMed]
- Qi, X.; Chen, X.; Zhao, Y.; Chen, J.; Niu, B.; Shen, B. Prognostic Roles of ceRNA Network-Based Signatures in Gastrointestinal Cancers. Front. Oncol. 2022, 12, 921194. [Google Scholar] [CrossRef]
- Chen, J.; Cheng, L.; Zou, W.; Wang, R.; Wang, X.; Chen, Z. ADAMTS9-AS1 Constrains Breast Cancer Cell Invasion and Proliferation via Sequestering miR-301b-3p. Front. Cell Dev. Biol. 2021, 9, 719993. [Google Scholar] [CrossRef]
- Xin, C.; Bi, X.; Xiao, C.; Dong, L. MIR31HG regulates the proliferation, migration and invasion of breast cancer by regulating the expression of POLDIP2. J. BUON. 2021, 26, 459–465. [Google Scholar]
- Deng, X.; Ye, D.; Hua, K.; Song, H.; Luo, Q.; Munankarmy, A.; Liu, D.; Zhou, B.; Zheng, W.; Zhou, X.; et al. MIR22HG inhibits breast cancer progression by stabilizing LATS2 tumor suppressor. Cell Death Dis. 2021, 12, 810. [Google Scholar] [CrossRef]
- Kim, C.Y.; Oh, J.H.; Lee, J.Y.; Kim, M.H. The LncRNA HOTAIRM1 Promotes Tamoxifen Resistance by Mediating HOXA1 Expression in ER+ Breast Cancer Cells. J. Cancer 2020, 11, 3416–3423. [Google Scholar] [CrossRef]
- Ye, T.; Feng, J.; Cui, M.; Yang, J.; Wan, X.; Xie, D.; Liu, J. LncRNA MIAT Services as a Noninvasive Biomarker for Diagnosis and Correlated with Immune Infiltrates in Breast Cancer. Int. J. Women’s Health 2021, 13, 991–1004. [Google Scholar] [CrossRef]
- Li, Y.; Jiang, B.; Wu, X.; Huang, Q.; Chen, W.; Zhu, H.; Qu, X.; Xie, L.; Ma, X.; Huang, G. Long non-coding RNA MIAT is estrogen-responsive and promotes estrogen-induced proliferation in ER-positive breast cancer cells. Biochem. Biophys. Res. Commun. 2018, 503, 45–50. [Google Scholar] [CrossRef]
- Fridrichova, I.; Zmetakova, I. MicroRNAs Contribute to Breast Cancer Invasiveness. Cells 2019, 8, 1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noorolyai, S.; Shajari, N.; Baghbani, E.; Sadreddini, S.; Baradaran, B. The relation between PI3K/AKT signalling pathway and cancer. Gene 2019, 698, 120–128. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tian, Y.; Dong, J.; Li, L. Bridging Pyroptosis and Immunity: A Comprehensive Study of the Pyroptosis-Related Long Non-Coding RNA Signature in Breast Cancer. Life 2023, 13, 1599. https://doi.org/10.3390/life13071599
Tian Y, Dong J, Li L. Bridging Pyroptosis and Immunity: A Comprehensive Study of the Pyroptosis-Related Long Non-Coding RNA Signature in Breast Cancer. Life. 2023; 13(7):1599. https://doi.org/10.3390/life13071599
Chicago/Turabian StyleTian, Ye, Jing Dong, and Lin Li. 2023. "Bridging Pyroptosis and Immunity: A Comprehensive Study of the Pyroptosis-Related Long Non-Coding RNA Signature in Breast Cancer" Life 13, no. 7: 1599. https://doi.org/10.3390/life13071599