
Challenges in the Timely Diagnosis of Behcet’s Disease

Abstract
:1. Introduction
2. The International Study Group Criteria
3. The International Criteria for Behcet’s Disease
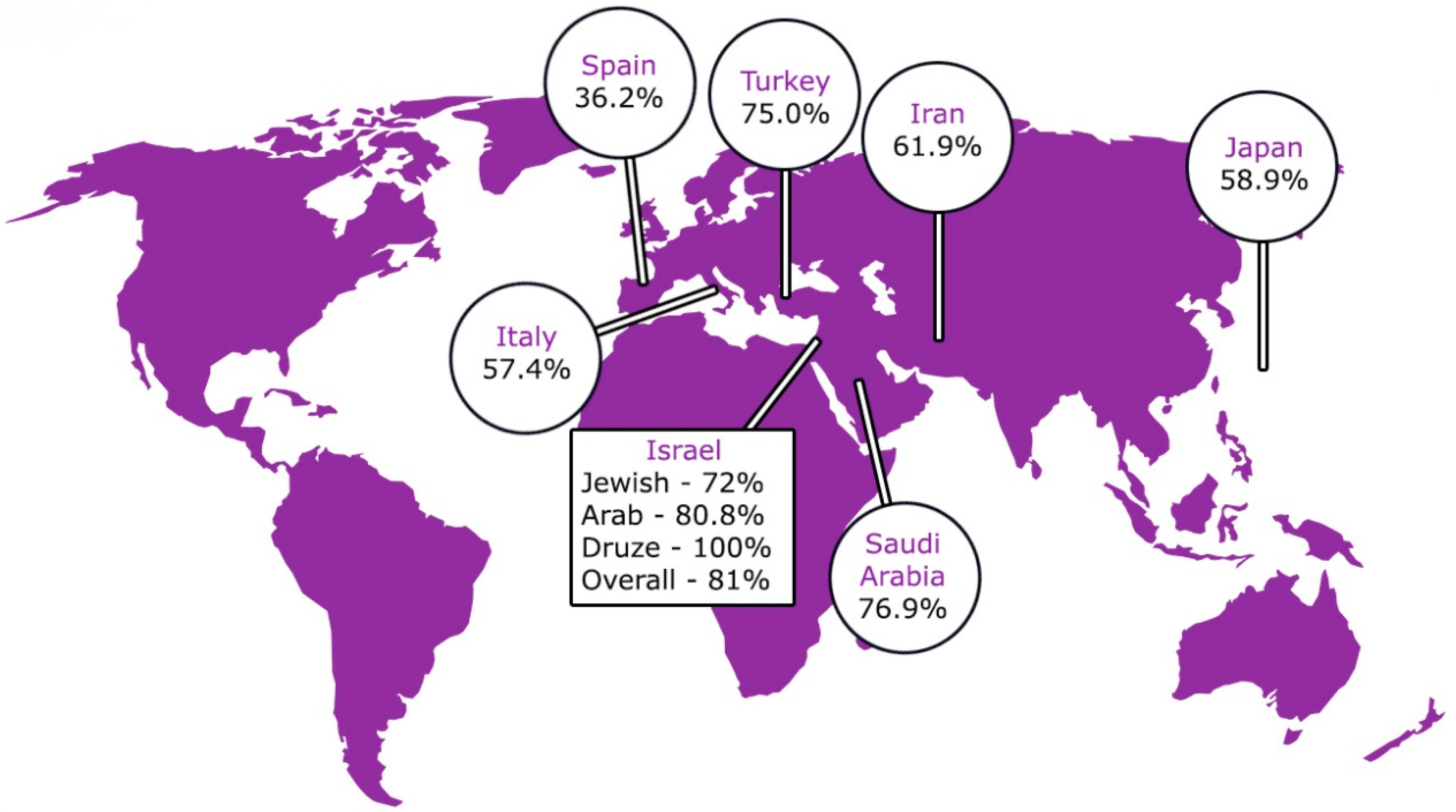
4. Looking to the Future
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Al-Mutawa, S.A.; Hegab, S.M. Behcet’s disease. Clin. Exp. Med. 2004, 4, 103–131. [Google Scholar] [CrossRef] [PubMed]
- Akman-Demir, G.; Serdaroglu, P.; Tasçi, B.; The Neuro-Behçet Study Group. Clinical patterns of neurological involvement in Behçet’s disease: Evaluation of 200 patients. Brain 1999, 122 Pt 11, 2171–2182. [Google Scholar] [CrossRef] [PubMed]
- Serdaroğlu, P. Behçet’s disease and the nervous system. J. Neurol. 1998, 245, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Siva, A.; Saip, S. The spectrum of nervous system involvement in Behçet’s syndrome and its differential diagnosis. J. Neurol. 2009, 256, 513–529. [Google Scholar] [CrossRef]
- Al-Araji, A.; Kidd, D.P. Neuro-Behçet’s disease: Epidemiology, clinical characteristics, and management. Lancet Neurol. 2009, 8, 192–204. [Google Scholar] [CrossRef]
- Sarica-Kucukoglu, R.; Akdag-Kose, A.; KayabalI, M.; Yazganoglu, K.D.; Disci, R.; Erzengin, D.; Azizlerli, G. Vascular involvement in Behçet’s disease: A retrospective analysis of 2319 cases. Int. J. Dermatol. 2006, 45, 919–921. [Google Scholar] [CrossRef]
- Gürgün, C.; Ercan, E.; Ceyhan, C.; Yavuzgil, O.; Zoghi, M.; Aksu, K.; Cinar, C.S.; Türkoglu, C. Cardiovascular involvement in Behçet’s disease. Jpn. Heart J. 2002, 43, 389–398. [Google Scholar] [CrossRef]
- Yurdakul, S.; Hamuryudan, V.; Yazici, H. Behçet syndrome. Curr. Opin. Rheumatol. 2004, 16, 38–42. [Google Scholar] [CrossRef]
- Yazici, H.; Fresko, I.; Yurdakul, S. Behçet’s syndrome: Disease manifestations, management, and advances in treatment. Nat. Clin. Pract. Rheumatol. 2007, 3, 148–155. [Google Scholar] [CrossRef]
- Krause, I.; Yankevich, A.; Fraser, A.; Rosner, I.; Mader, R.; Zisman, D.; Boulman, N.; Rozenbaum, M.; Weinberger, A. Prevalence and clinical aspects of Behcet’s disease in the north of Israel. Clin. Rheumatol. 2007, 26, 555–560. [Google Scholar] [CrossRef]
- Krause, I.; Mader, R.; Sulkes, J.; Paul, M.; Uziel, Y.; Adawi, M.; Weinberger, A. Behçet’s disease in Israel: The influence of ethnic origin on disease expression and severity. J. Rheumatol. 2001, 28, 1033–1036. [Google Scholar]
- Nussenblatt, R.B. Uveitis in Behçet’s disease. Int. Rev. Immunol. 1997, 14, 67–79. [Google Scholar] [CrossRef]
- Vaiopoulos, A.G.; Sfikakis, P.P.; Kanakis, M.A.; Vaiopoulos, G.; Kaklamanis, P.G. Gastrointestinal manifestations of Behçet’s disease: Advances in evaluation and management. Clin. Exp. Rheumatol. 2014, 32 (Suppl. S84), S140–S148. [Google Scholar] [PubMed]
- Kalra, S.; Silman, A.; Akman-Demir, G.; Bohlega, S.; Borhani-Haghighi, A.; Constantinescu, C.S.; Houman, H.; Mahr, A.; Salvarani, C.; Sfikakis, P.P.; et al. Diagnosis and management of Neuro-Behçet’s disease: International consensus recommendations. J. Neurol. 2014, 261, 1662–1676. [Google Scholar] [CrossRef] [PubMed]
- Davatchi, F.; Sadeghi Abdollahi, B.; Chams-Davatchi, C.; Shahram, F.; Shams, H.; Nadji, A.; Faezi, T.; Akhlaghi, M.; Ghodsi, Z.; Mohtasham, N.; et al. The saga of diagnostic/classification criteria in Behcet’s disease. Int. J. Rheum. Dis. 2015, 18, 594–605. [Google Scholar] [CrossRef] [PubMed]
- Lee, S. Diagnostic criteria of Behçet’s disease: Problems and suggestions. Yonsei Med. J. 1997, 38, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Curth, H.O. Recurrent genito-oral aphthosis and uveitis with hypopyon (Behcet’s syndrome). A report of 2 cases. Arch. Dermatol. Res. 1946, 54, 179–196. [Google Scholar]
- Mason, R.M.; Barnes, C.G. Behcet’s syndrome with arthritis. Ann. Rheum. Dis. 1969, 28, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, J.; Escande, J.P.; Maness, S. Revision des criteres diagnostiques du syndrome de Behcet. Presse Med. 1971, 79, 901. [Google Scholar]
- Behcet’s Disease Research Committee of Japan. Behcet’s disease guide to diagnosis of Behcet’s disease (1972). Jpn. J. Ophthalmol. 1974, 18, 291–294. [Google Scholar]
- Hubault, A.; Hamza, M. La maladie de Behcet en 1974. In L’actualite Rhumatologique; de Seze, S., Ryckewaert, A., Kahn, M.-F., Vitale, C., Eds.; Expension Scientifique: Paris, France, 1974; Volume 15, pp. 43–55. [Google Scholar]
- O’Duffy, J.D. Criteres proposes pour le diagnostique de la maladie de Behcet et notes therapeutiques. Rev. Med. 1974, 36, 2371–2379. [Google Scholar]
- Cheng, S.P.; Zhang, X.Q. Some special clinical manifestations of Behcet’s disease and review of literature. Chin. J. Intern. Med. 1980, 19, 15–22. (In Chinese) [Google Scholar]
- Dilsen, N.; Konice, M.; Aral, O. Our Diagnostic Criteria of Behcet’s Disease—An Overview. In Recent Advances in Behcet’s Disease. London Royal Society of Medicine Services; Lehner, T., Barnes, C.G., Eds.; International Congress and Symposium Series 103; London Royal Society of Medicine Services: London, UK, 1986; pp. 177–180. [Google Scholar]
- Mizushima, Y. Recent research into Behcet’s disease in Japan. Int. J. Tissue React. 1988, 10, 59–65. [Google Scholar] [PubMed]
- Criteria for Diagnosis of Behçet’s Disease. International Study Group for Behçet’s Disease. Lancet 1990, 335, 1078–1080. [Google Scholar]
- Davatchi, F.; Shahram, F.; Akbarian, M. Accuracy of Existing Diagnosis Criteria for Behcet’s Disease. In Behcet’s Disease; Wechsler, B., Godeau, P., Eds.; Excerpta Medica International Congress Series 1037; Excerpta Medica: Amsterdam, The Netherlands, 1993; pp. 225–228. [Google Scholar]
- Davatchi, F.; Shahram, F.; Akbarian, M.; Gharibdoost, F.; Chams, C.; Chams, H. Classification Tree for the Diagnosis of Behcet’s Disease. In Behcet’s Disease; Wechsler, B., Godeau, P., Eds.; Excerpta Medica International Congress Series 1037; Excerpta Medica: Amsterdam, The Netherlands, 1993; pp. 245–248. [Google Scholar]
- Dilsen, N. About Diagnostic Criteria for Behcet’s Disease: Our New Proposal. In Behcet’s Disease; Bang, D., Lee, E.S., Lee, S., Eds.; Design Mecca Publishing Co.: Seoul, Republic of Korea, 2003; pp. 101–104. [Google Scholar]
- Chang, H.K.; Lee, S.S.; Bai, H.J.; Lee, Y.W.; Yoon, B.Y.; Lee, C.H.; Lee, Y.H.; Song, G.G.; Chung, W.T.; Lee, S.W.; et al. Validation of the classification criteria commonly used in Korea and a modified set of preliminary criteria for Behcet’s disease: A multi-center study. Clin. Exp. Rheumatol. 2004, 22 (Suppl. S34), S21–S26. [Google Scholar]
- International Team for the Revision of the International Criteria for Behçet’s Disease (ITR-ICBD). The International Criteria for Behçet’s Disease (ICBD): A collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J. Eur. Acad. Dermatol. Venereol. 2014, 28, 338–347. [Google Scholar] [CrossRef]
- Amer-Alshiek, J.; Alshiek, T.; Amir Levy, Y.; Azem, F.; Amit, A.; Amir, H. Israeli Druze women’s sex preferences when choosing obstetricians and gynecologists. Isr. J. Health Policy Res. 2015, 4, 13. [Google Scholar] [CrossRef] [PubMed]
- Zidan, J.; Ben-Avraham, D.; Carmi, S.; Maray, T.; Friedman, E.; Atzmon, G. Genotyping of geographically diverse Druze trios reveals substructure and a recent bottleneck. Eur. J. Hum. Genet. 2015, 23, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Emmi, G.; Vitale, A.; Silvestri, E.; Boddi, M.; Becatti, M.; Fiorillo, C.; Fabiani, C.; Frediani, B.; Emmi, L.; Di Scala, G.; et al. Adalimumab-Based Treatment Versus Disease-Modifying Antirheumatic Drugs for Venous Thrombosis in Behçet’s Syndrome: A Retrospective Study of Seventy Patients with Vascular Involvement. Arthritis Rheumatol. 2018, 70, 1500–1507. [Google Scholar] [CrossRef]
- Yazici, H.; Yazici, Y. Criteria for Behçet’s disease with reflections on all disease criteria. J. Autoimmun. 2014, 48, 104–107. [Google Scholar] [CrossRef]
- Davatchi, F.; Schirmer, M.; Zouboulis, C.; on behalf of the International Team for the Revision of the International Study Group Criteria for Bechet’s disease. Evaluation and Revision of the International Study Group Criteria for Behcet’s disease. In Proceedings of the American College of Rheumatology Meeting, Boston, MA, USA, 6–11 November 2007. Abstract 1233. [Google Scholar]
- Boyvat, A.; Ekmekçi, P.; Gürgey, E. Bipolar aphthosis. A forme fruste of Behçet’s disease. Long term follow-up of 26 cases. Adv. Exp. Med. Biol. 2003, 528, 321–322. [Google Scholar] [PubMed]
- Gupta, S.; Ajith, C.; Malhotra, S.; Kumar, B. Bipolar aphthosis presenting as mutilating genital ulcers in women. Indian J. Dermatol. Venereol. Leprol. 2004, 70, 357–360. [Google Scholar] [PubMed]
- Aeschlimann, F.A.; Batu, E.D.; Canna, S.W.; Go, E.; Gül, A.; Hoffmann, P.; Leavis, H.L.; Ozen, S.; Schwartz, D.M.; Stone, D.L.; et al. A20 haploinsufficiency (HA20): Clinical phenotypes and disease course of patients with a newly recognised NF-kB-mediated autoinflammatory disease. Ann. Rheum. Dis. 2018, 77, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Remmers, E.F.; Cosan, F.; Kirino, Y.; Ombrello, M.J.; Abaci, N.; Satorius, C.; Le, J.M.; Yang, B.; Korman, B.D.; Cakiris, A.; et al. Genome-wide association study identifies variants in the MHC class I, IL10, and IL23R-IL12RB2 regions associated with Behcet’s disease. Nat. Genet. 2010, 42, 698–702. [Google Scholar] [CrossRef]
- Kirino, Y.; Bertsias, G.; Ishigatsubo, Y.; Mizuki, N.; Tugal-Tutkun, I.; Seyahi, E.; Ozyazgan, Y.; Sacli, F.S.; Erer, B.; Inoko, H.; et al. Genomewide association analysis identifies new susceptibility loci for Behcet’s disease and epistasis between HLA-B*51 and ERAP1. Nat. Genet. 2013, 45, 202–207. [Google Scholar] [CrossRef]
- Takeuchi, M.; Ombrello, M.J.; Kirino, Y.; Erer, B.; Tugal-Tutkun, I.; Seyahi, E.; Özyazgan, Y.; Watts, N.R.; Gül, A.; Kastner, D.L.; et al. A single endoplasmic reticulum aminopeptidase-1 protein allotype is a strong risk factor for Behcet’s disease in HLA-B*51 carriers. Ann. Rheum. Dis. 2016, 75, 2208–2211. [Google Scholar] [CrossRef]
- Maldini, C.; LaValley, M.P.; Cheminant, M.; de Menthon, M.; Mahr, A. Relationships of HLA-B51 or B5 genotype with Behcet’s disease clinical characteristics: Systematic review and meta-analyses of observational studies. Rheumatology 2012, 51, 887–900. [Google Scholar] [CrossRef]
- Kirino, Y.; Ideguchi, H.; Takeno, M.; Suda, A.; Higashitani, K.; Kunishita, Y.; Takase-Minegishi, K.; Tamura, M.; Watanabe, T.; Asami, Y.; et al. Continuous evolution of clinical phenotype in 578 Japanese patients with Behcet’s disease: A retrospective observational study. Arthritis Res. Ther. 2016, 18, 217. [Google Scholar] [CrossRef]
- Ombrello, M.J.; Kirino, Y.; de Bakker, P.I.; Gül, A.; Kastner, D.L.; Remmers, E.F. Behçet disease-associated MHC class I residues implicate antigen binding and regulation of cell-mediated cytotoxicity. Proc. Natl. Acad. Sci. USA 2014, 111, 8867–8872. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, M.; Horita, N.; Ishido, T.; Mizuki, R.; Kawagoe, T.; Shibuya, E.; Yuda, K.; Ishido, M.; Mizuki, Y.; et al. HLA-A26 is a risk factor for Behçet’s disease ocular lesions. Mod. Rheumatol. 2020, 31, 214–218. [Google Scholar] [CrossRef]
- Takeno, M. The association of Behçet’s syndrome with HLA-B51 as understood in 2021. Curr. Opin. Rheumatol. 2022, 34, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Leonardo, N.M.; McNeil, J. Behcet’s Disease: Is There Geographical Variation? A Review Far from the Silk Road. Int. J. Rheumatol. 2015, 2015, 945262. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, M.J.; Saadoun, D.; Garrido, M.; Klatzmann, D.; Six, A.; Cacoub, P. Behçet’s disease physiopathology: A contemporary review. Autoimmun. Highlights 2016, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Joseph, F.G.; Scolding, N.J. Neuro-Behçet’s disease in Caucasians: A study of 22 patients. Eur. J. Neurol. 2007, 14, 174–180. [Google Scholar] [CrossRef]
- Davatchi, F.; Shahram, F.; Chams-Davatchi, C.; Shams, H.; Nadji, A.; Akhlaghi, M.; Faezi, T.; Ghodsi, Z.; Faridar, A.; Ashofteh, F.; et al. Behcet’s disease: From East to West. Clin. Rheumatol. 2010, 29, 823–833. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Criteria | Sensitivity | Specificity |
|---|---|---|
| Curth [17] | 99% | 82% |
| Mason [18] | 82% | 95% |
| Hewitt [19] | 57% | 96% |
| Japan Original [20] | 88% | 92% |
| Hubault and Hamza [21] | 58% | 97% |
| O’Duffy [22] | 88% | 90% |
| Cheng and Zhang [23] | 98% | 83% |
| Dilsen (original) [24] | 88% | 91% |
| Japan (revised) [25] | 91% | 91% |
| International Study Group [26] | 85% | 96% |
| Iran traditional [27] | 90% | 92% |
| Iran—Classification Tree [28] | 96% | 90% |
| Dilsen revised [29] | 87% | 96% |
| Korea [30] | 92% | 92% |
| International Criteria for Behcet’s Disease [31] | 95% | 91% |
| For a clinical diagnosis of Behcet’s disease, the patient must have recurrent oral ulceration (minor [<10 mm] or major aphthous [>10 mm], or herpetiform ulcers observed by the physician or reliably described by the patient, which recurred at least three times over a 12-month period) in addition to at least two of the following additional findings in the absence of any other clinical explanation: |
|
|
|
|
| Sensitivity of 91–95% and a specificity of 85–98% in the international study group cohort compared to 81.2% sensitivity and 95.9% specificity in a multinational cohort. |
| Point score system: scoring 4 indicates diagnosis of Behcet’s disease | |
| Sign or Symptom | Points |
| Ocular lesions | 2 |
| Genital ulcers | 2 |
| Oral ulcers | 2 |
| Skin lesions | 1 |
| Neurologic manifestations | 1 |
| Vascular manifestations | 1 |
| Positive pathergy test result | 1 |
| Sensitivity of 93.9–94.8% and a specificity of 90.5–92.1% in the international criteria for Behcet’s disease cohort. | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassan, F.; Jeries, H.; Naffaa, M.E. Challenges in the Timely Diagnosis of Behcet’s Disease. Life 2023, 13, 1157. https://doi.org/10.3390/life13051157
Hassan F, Jeries H, Naffaa ME. Challenges in the Timely Diagnosis of Behcet’s Disease. Life. 2023; 13(5):1157. https://doi.org/10.3390/life13051157
Chicago/Turabian StyleHassan, Fadi, Helana Jeries, and Mohammad E. Naffaa. 2023. "Challenges in the Timely Diagnosis of Behcet’s Disease" Life 13, no. 5: 1157. https://doi.org/10.3390/life13051157