

Taxonomic Assignment-Based Genome Reconstruction from Apical Periodontal Metagenomes to Identify Antibiotic Resistance and Virulence Factors


 , ,
, , 
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Selection
2.2. Sampling
2.3. Metagenomic DNA Extraction, High-Throughput Sequencing and Bioinformatics Analysis
2.4. Quality Filtering, Co-Assembly and Extractions of MAGs
2.5. Phylogenetic and Functional Analysis
3. Results
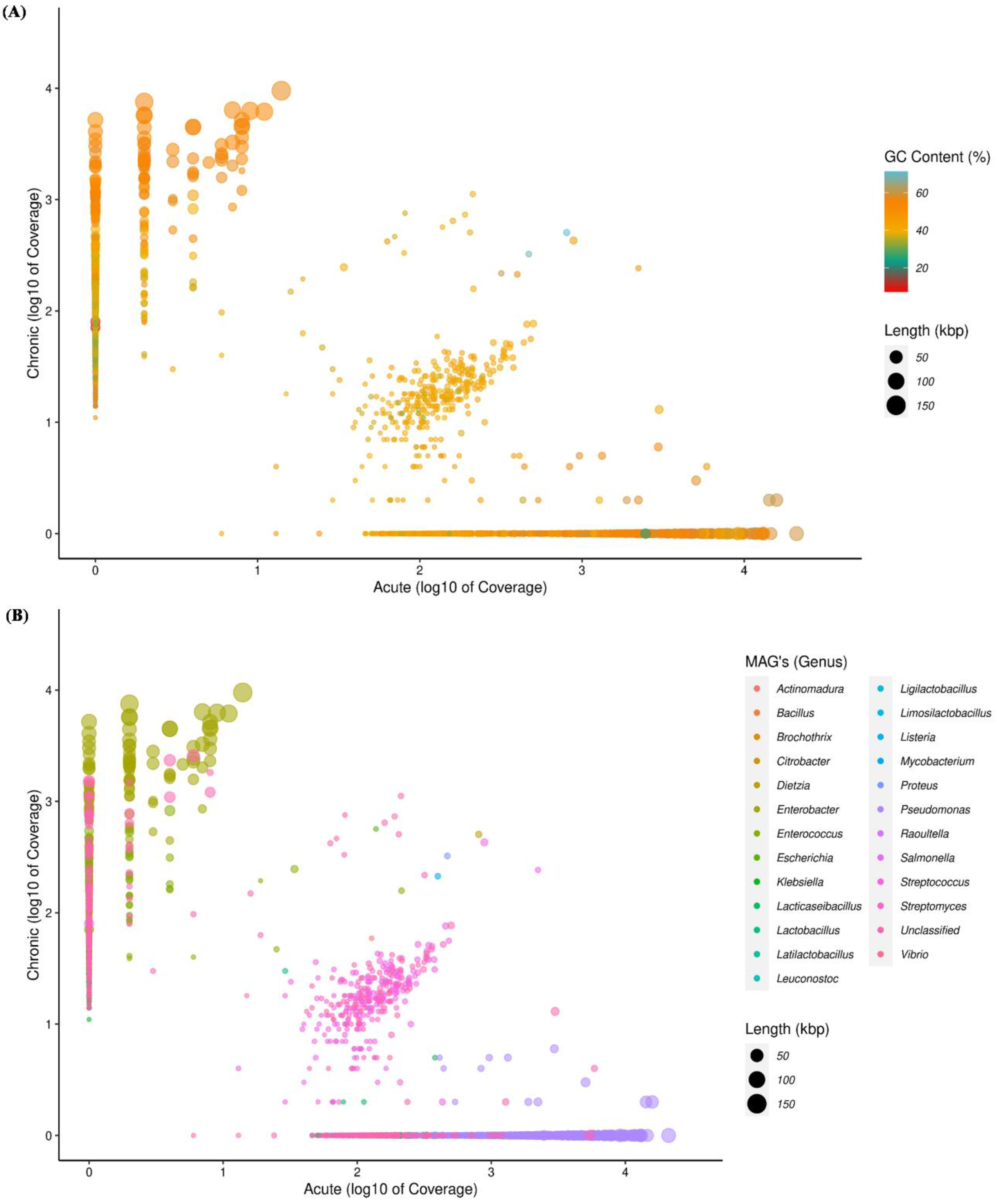
3.1. Metagenomic Assembly of Apical Periodontal Microbiota
3.2. Distribution of Contigs at Different Levels of the Taxonomy
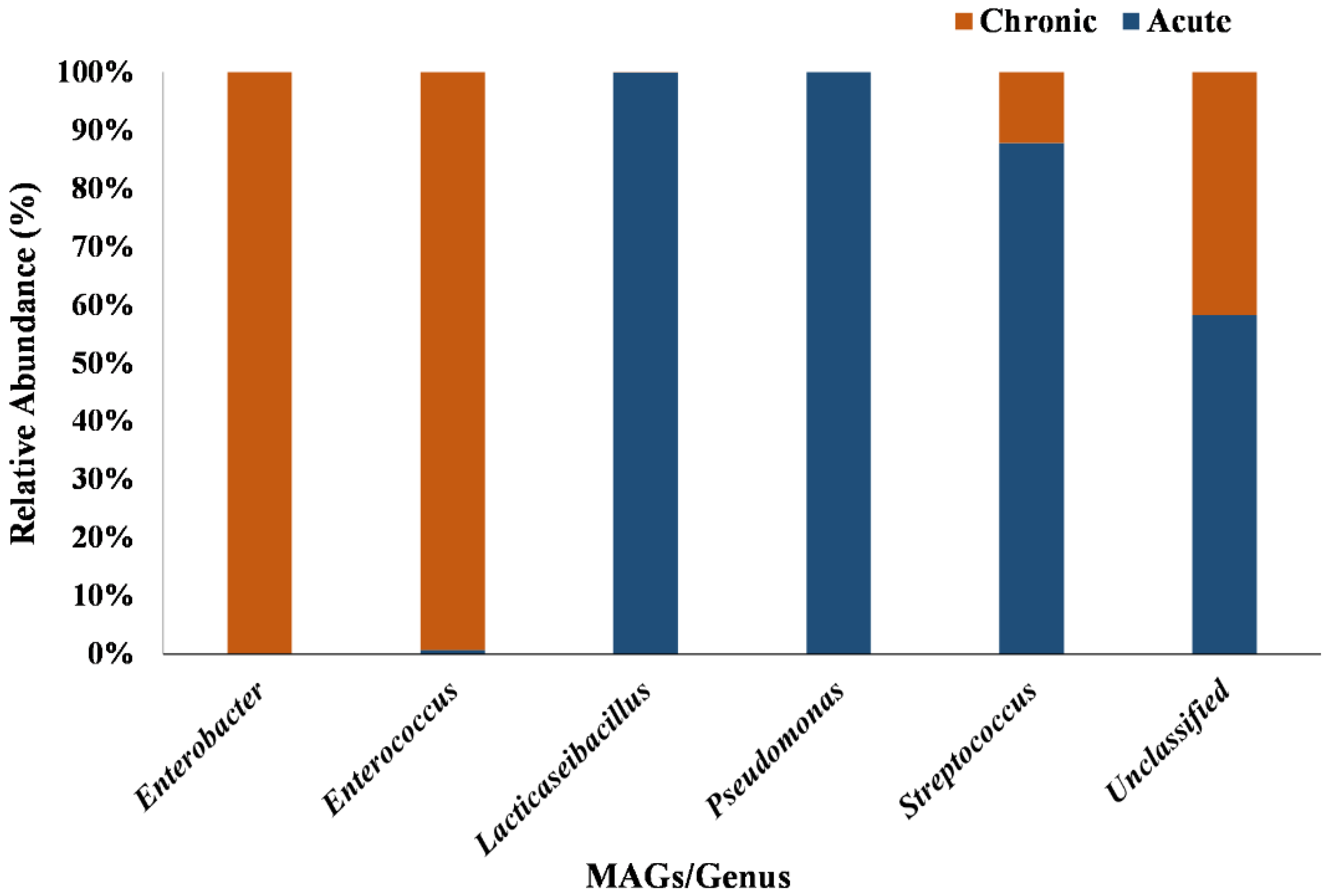
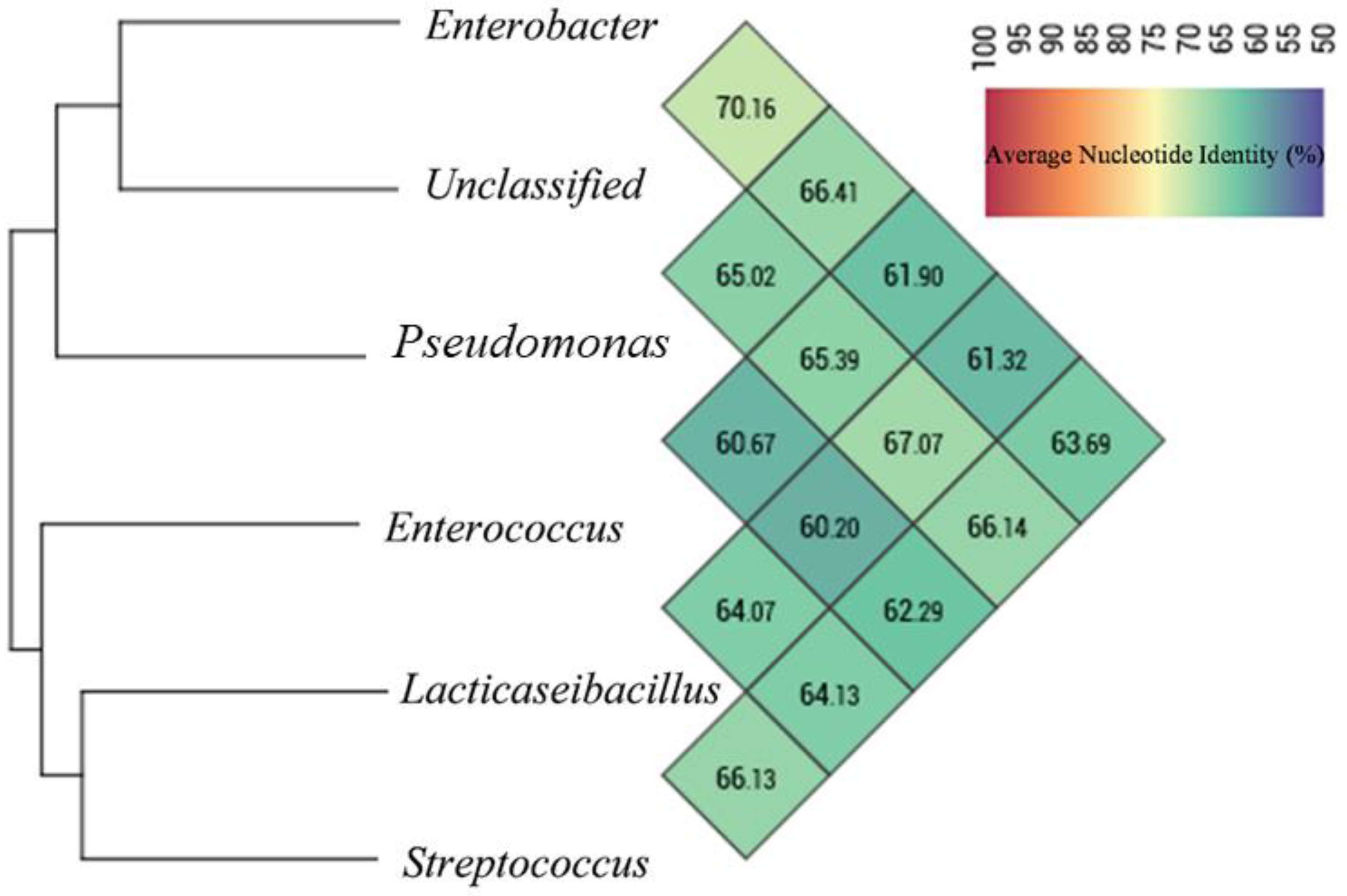
3.3. Extraction of MAGs and Phylogenetic Analysis
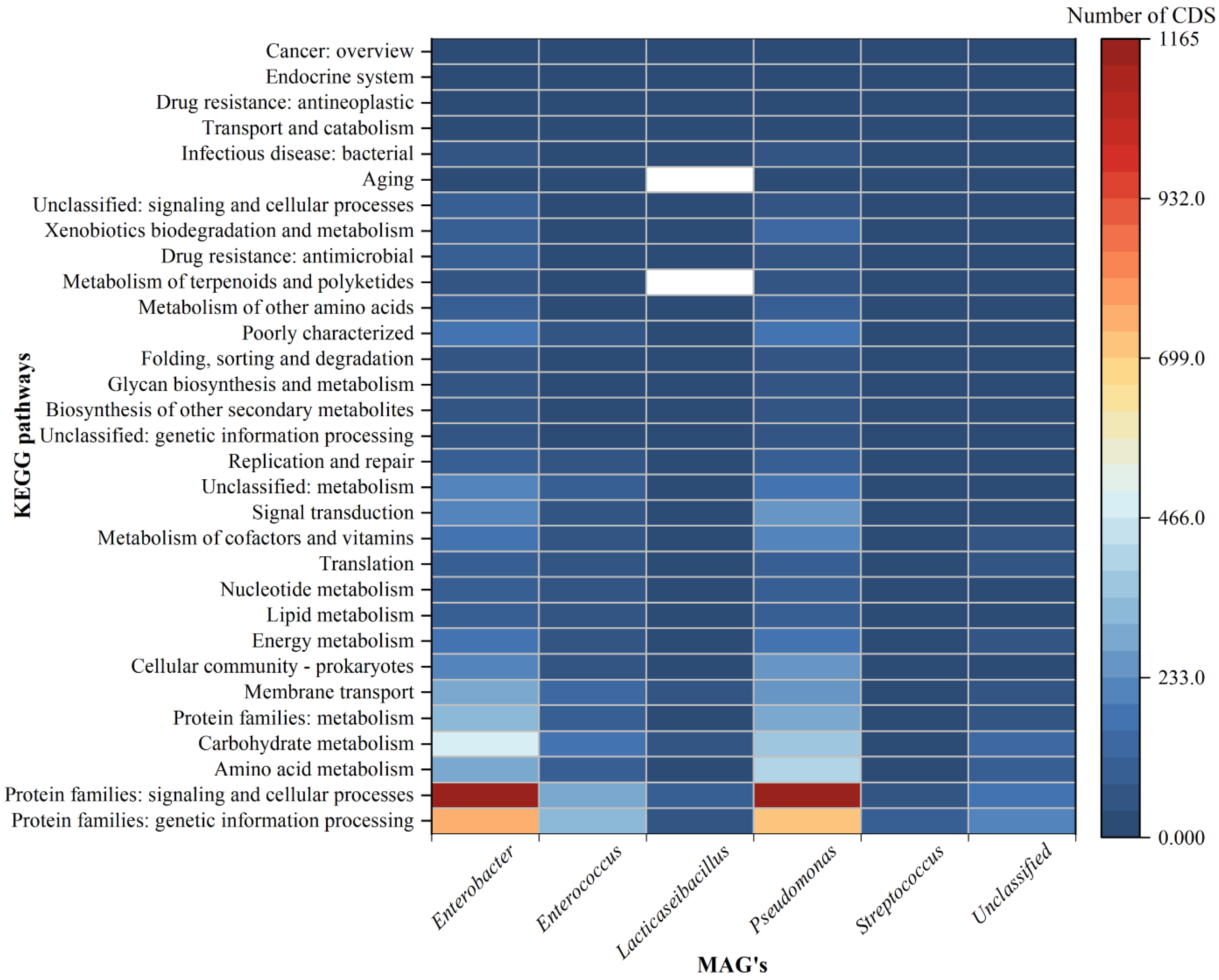
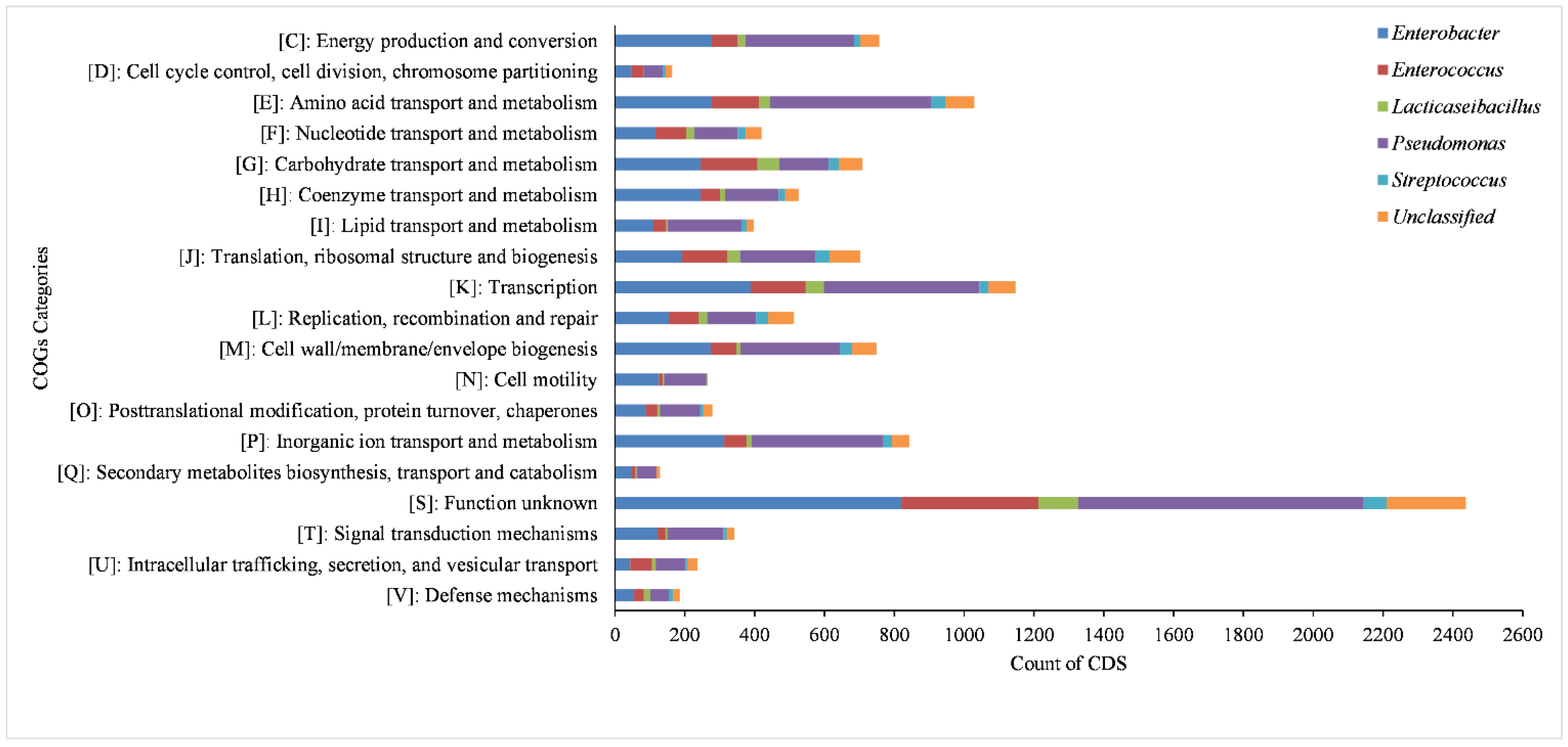
3.4. Functional Annotation of All the MAGs
3.5. Antibiotic Resistome of MAGs
3.6. Virulome of MAGs
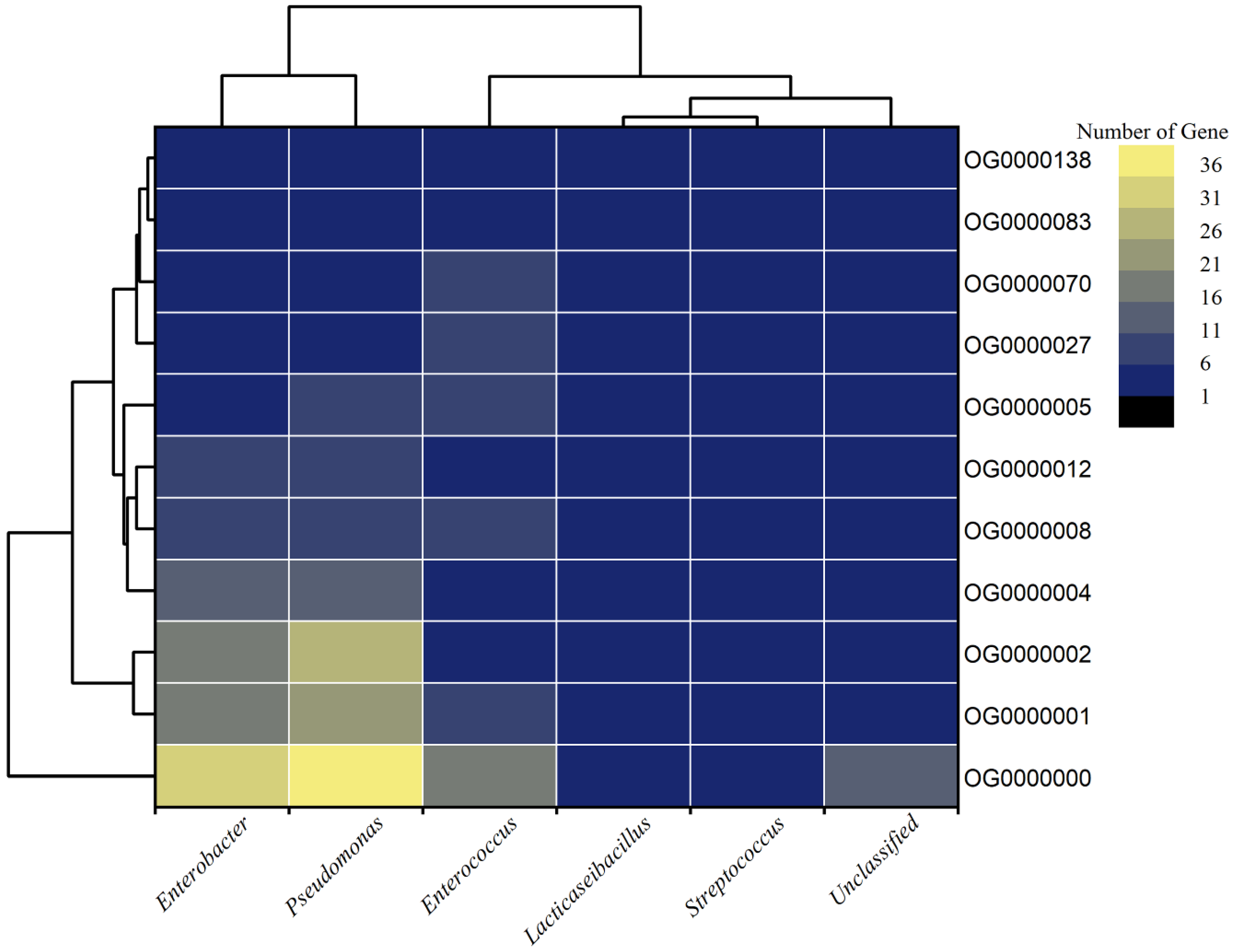
3.7. Orthologous and Core Proteome of MAGs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nair, P.N.R. Apical Periodontitis: A Dynamic Encounter between Root Canal Infection and Host Response. Periodontology 2000 1997, 13, 121–148. [Google Scholar] [CrossRef]
- Graunaite, I.; Lodiene, G.; Maciulskiene, V. Pathogenesis of Apical Periodontitis: A Literature Review. J. Oral Maxillofac. Res. 2011, 2, 630–659. [Google Scholar] [CrossRef] [PubMed]
- Nair, P.N.R. Pathogenesis of Apical Periodontitis and the Causes of Endodontic Failures. Crit. Rev. Oral Biol. Med. 2004, 15, 348–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siqueira, J.F. Endodontic Infections: Concepts, Paradigms, and Perspectives. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2002, 94, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, J.J.; Rôças, I.N. Exploiting Molecular Methods to Explore Endodontic Infections: Part 2—Redefining the Endodontic Microbiota. J. Endod. 2005, 31, 488–498. [Google Scholar] [CrossRef]
- Clemente, J.C.; Ursell, L.K.; Parfrey, L.W.; Knight, R. The Impact of the Gut Microbiota on Human Health: An Integrative View. Cell 2012, 148, 1258–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nüsslein, K.; Tiedje, J.M. Soil Bacterial Community Shift Correlated with Change from Forest to Pasture Vegetation in a Tropical Soil. Appl. Environ. Microbiol. 1999, 65, 3622–3626. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Chowdhury, D.; Zhang, Z.; Cheung, W.K.; Lu, A.; Bian, Z.; Zhang, L. A Review of Computational Tools for Generating Metagenome-Assembled Genomes from Metagenomic Sequencing Data. Comput. Struct. Biotechnol. J. 2021, 19, 6301–6314. [Google Scholar] [CrossRef]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of Age: Ten Years of next-Generation Sequencing Technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef]
- Yarza, P.; Yilmaz, P.; Pruesse, E.; Glöckner, F.O.; Ludwig, W.; Schleifer, K.-H.; Whitman, W.B.; Euzéby, J.; Amann, R.; Rosselló-Móra, R. Uniting the Classification of Cultured and Uncultured Bacteria and Archaea Using 16S RRNA Gene Sequences. Nat. Rev. Microbiol. 2014, 12, 635–645. [Google Scholar] [CrossRef]
- Hasan, N.A.; Young, B.A.; Minard-Smith, A.T.; Saeed, K.; Li, H.; Heizer, E.M.; McMillan, N.J.; Isom, R.; Abdullah, A.S.; Bornman, D.M.; et al. Microbial Community Profiling of Human Saliva Using Shotgun Metagenomic Sequencing. PLoS ONE 2014, 9, e97699. [Google Scholar] [CrossRef]
- Methé, B.A.; Nelson, K.E.; Pop, M.; Creasy, H.H.; Giglio, M.G.; Huttenhower, C.; Gevers, D.; Petrosino, J.F.; Abubucker, S.; Badger, J.H.; et al. A Framework for Human Microbiome Research. Nature 2012, 486, 215–221. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Phillips, S.; Gail, M.H.; Goedert, J.J.; Humphrys, M.; Ravel, J.; Ren, Y.; Caporaso, N.E. Evaluation of Buccal Cell Samples for Studies of Oral Microbiota. Cancer Epidemiol. Biomark. Prev. 2017, 26, 249–253. [Google Scholar] [CrossRef] [Green Version]
- Caselli, E.; Fabbri, C.; D’Accolti, M.; Soffritti, I.; Bassi, C.; Mazzacane, S.; Franchi, M. Defining the Oral Microbiome by Whole-Genome Sequencing and Resistome Analysis: The Complexity of the Healthy Picture. BMC Microbiol. 2020, 20, 120. [Google Scholar] [CrossRef] [PubMed]
- Sogin, M.L.; Morrison, H.G.; Huber, J.A.; Welch, D.M.; Huse, S.M.; Neal, P.R.; Arrieta, J.M.; Herndl, G.J. Microbial Diversity in the Deep Sea and the Underexplored “Rare Biosphere”. Proc. Natl. Acad. Sci. USA 2006, 103, 12115–12120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huse, S.M.; Dethlefsen, L.; Huber, J.A.; Welch, D.M.; Relman, D.A.; Sogin, M.L. Exploring Microbial Diversity and Taxonomy Using SSU RRNA Hypervariable Tag Sequencing. PLoS Genet. 2008, 4, e1000255. [Google Scholar] [CrossRef]
- Schirrmeister, J.F.; Liebenow, A.-L.; Pelz, K.; Wittmer, A.; Serr, A.; Hellwig, E.; Al-Ahmad, A. New Bacterial Compositions in Root-Filled Teeth with Periradicular Lesions. J. Endod. 2009, 35, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Gloor, G.B.; Hummelen, R.; Macklaim, J.M.; Dickson, R.J.; Fernandes, A.D.; MacPhee, R.; Reid, G. Microbiome Profiling by Illumina Sequencing of Combinatorial Sequence-Tagged PCR Products. PLoS ONE 2010, 5, e15406. [Google Scholar] [CrossRef] [Green Version]
- Diaz, P.I.; Dupuy, A.K.; Abusleme, L.; Reese, B.; Obergfell, C.; Choquette, L.; Dongari-Bagtzoglou, A.; Peterson, D.E.; Terzi, E.; Strausbaugh, L.D. Using High Throughput Sequencing to Explore the Biodiversity in Oral Bacterial Communities. Mol. Oral Microbiol. 2012, 27, 182–201. [Google Scholar] [CrossRef] [PubMed]
- Di Bella, J.M.; Bao, Y.; Gloor, G.B.; Burton, J.P.; Reid, G. High Throughput Sequencing Methods and Analysis for Microbiome Research. J. Microbiol. Methods 2013, 95, 401–414. [Google Scholar] [CrossRef]
- Dröge, J.; Mchardy, A.C. Taxonomic Binning of Metagenome Samples Generated by Next-Generation Sequencing Technologies. Brief. Bioinform. 2012, 13, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Zanet, J.; Benrabah, E.; Li, T.; Pélissier-Monier, A.; Chanut-Delalande, H.; Ronsin, B.; Bellen, H.J.; Payre, F.; Plaza, S. Pri SORF Peptides Induce Selective Proteasome-Mediated Protein Processing. Science 2015, 349, 1356–1358. [Google Scholar] [CrossRef]
- Rôças, I.N.; Siqueira, J.F. Detection of Antibiotic Resistance Genes in Samples from Acute and Chronic Endodontic Infections and after Treatment. Arch. Oral Biol. 2013, 58, 1123–1128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schirrmeister, J.F.; Liebenow, A.-L.; Braun, G.; Wittmer, A.; Hellwig, E.; Al-Ahmad, A. Detection and Eradication of Microorganisms in Root-Filled Teeth Associated with Periradicular Lesions: An In Vivo Study. J. Endod. 2007, 33, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Sogo, Y.; Doi, H.; Yamanaka, H. Usefulness and Limitations of Sample Pooling for Environmental DNA Metabarcoding of Freshwater Fish Communities. Sci. Rep. 2017, 7, 14860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sham, P.; Bader, J.S.; Craig, I.; O’Donovan, M.; Owen, M. DNA Pooling: A Tool for Large-Scale Association Studies. Nat. Rev. Genet. 2002, 3, 862–871. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. Others FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/ (accessed on 22 September 2022).
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. MetaSPAdes: A New Versatile Metagenomic Assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, I.J.; Rees, E.R.; Ross, J.; Miller, I.; Baxa, J.; Lopera, J.; Kerby, R.L.; Rey, F.E.; Kwan, J.C. Autometa: Automated Extraction of Microbial Genomes from Individual Shotgun Metagenomes. Nucleic Acids Res. 2019, 47, e57. [Google Scholar] [CrossRef] [Green Version]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness with Single-Copy Orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, I.; Kim, Y.O.; Park, S.C.; Chun, J. OrthoANI: An Improved Algorithm and Software for Calculating Average Nucleotide Identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [Green Version]
- Aramaki, T.; Blanc-Mathieu, R.; Endo, H.; Ohkubo, K.; Kanehisa, M.; Goto, S.; Ogata, H. KofamKOALA: KEGG Ortholog Assignment Based on Profile HMM and Adaptive Score Threshold. Bioinformatics 2019, 36, 2251–2252. [Google Scholar] [CrossRef] [Green Version]
- Cantalapiedra, C.P.; Hern̗andez-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. EggNOG-Mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic Resistome Surveillance with the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2020, 8, D517–D525. [Google Scholar] [CrossRef]
- Feldgarden, M.; Brover, V.; Gonzalez-Escalona, N.; Frye, J.G.; Haendiges, J.; Haft, D.H.; Hoffmann, M.; Pettengill, J.B.; Prasad, A.B.; Tillman, G.E.; et al. AMRFinderPlus and the Reference Gene Catalog Facilitate Examination of the Genomic Links among Antimicrobial Resistance, Stress Response, and Virulence. Sci. Rep. 2021, 11, 1–9. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A Comparative Pathogenomic Platform with an Interactive Web Interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Ye, J.; McGinnis, S.; Madden, T.L. BLAST: Improvements for Better Sequence Analysis. Nucleic Acids Res. 2006, 34, W6–W9. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving Fundamental Biases in Whole Genome Comparisons Dramatically Improves Orthogroup Inference Accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef] [Green Version]
- Pérez, A.; Poza, M.; Aranda, J.; Latasa, C.; Medrano, F.J.; Tomás, M.; Romero, A.; Lasa, I.; Bou, G. Effect of Transcriptional Activators SoxS, RobA, and RamA on Expression of Multidrug Efflux Pump AcrAB-TolC in Enterobacter cloacae. Antimicrob. Agents Chemother. 2012, 56, 6256–6266. [Google Scholar] [CrossRef] [Green Version]
- Borišek, J.; Pintar, S.; Ogrizek, M.; Grdadolnik, S.G.; Hodnik, V.; Turk, D.; Perdih, A.; Novič, M. Discovery of (Phenylureido)Piperidinyl Benzamides as Prospective Inhibitors of Bacterial Autolysin E from Staphylococcus aureus. J. Enzym. Inhib. Med. Chem. 2018, 33, 1239–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, X.; Wan, Z.; Bu, Z.; Wang, X.; Wang, X.; Zhu, L.; Wan, J.; Sun, Y.; Wang, X. Catabolite Control Protein A Is an Important Regulator of Metabolism in Streptococcus suis Type 2. Biomed. Rep. 2014, 2, 709–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manoil, D.; Al-Manei, K.; Belibasakis, G.N. A Systematic Review of the Root Canal Microbiota Associated with Apical Periodontitis: Lessons from Next-Generation Sequencing. Proteom.-Clin. Appl. 2020, 14, 1900060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, A.L.; Siqueira, J.F.; Rôças, I.N.; Jesus, E.C.; Rosado, A.S.; Tiedje, J.M. Comparing the Bacterial Diversity of Acute and Chronic Dental Root Canal Infections. PLoS ONE 2011, 6, e28088. [Google Scholar] [CrossRef] [Green Version]
- Windley, W.; Teixeira, F.; Levin, L.; Sigurdsson, A.; Trope, M. Disinfection of Immature Teeth with a Triple Antibiotic Paste. J. Endod. 2005, 31, 439–443. [Google Scholar] [CrossRef] [Green Version]
- Iwahara, K.; Kuriyama, T.; Shimura, S.; Williams, D.W.; Yanagisawa, M.; Nakagawa, K.; Karasawa, T. Detection of CfxA and CfxA2, the β-Lactamase Genes of Prevotella spp., in Clinical Samples from Dentoalveolar Infection by Real-Time PCR. J. Clin. Microbiol. 2006, 44, 172–176. [Google Scholar] [CrossRef] [Green Version]
- Jungermann, G.B.; Burns, K.; Nandakumar, R.; Tolba, M.; Venezia, R.A.; Fouad, A.F. Antibiotic Resistance in Primary and Persistent Endodontic Infections. J. Endod. 2011, 37, 1337–1344. [Google Scholar] [CrossRef] [Green Version]
- Rosenblum, R.; Khan, E.; Gonzalez, G.; Hasan, R.; Schneiders, T. Genetic Regulation of the RamA Locus and Its Expression in Clinical Isolates of Klebsiella pneumoniae. Int. J. Antimicrob. Agents 2011, 38, 39–45. [Google Scholar] [CrossRef]
- Ku, Y.-H.; Lee, M.-F.; Chuang, Y.-C.; Yu, W.-L. Detection of Plasmid-Mediated β-Lactamase Genes and Emergence of a Novel AmpC (CMH-1) in Enterobacter cloacae at a Medical Center in Southern Taiwan. J. Clin. Med. 2018, 8, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotoh, N.; Tsujimoto, H.; Tsuda, M.; Okamoto, K.; Nomura, A.; Wada, T.; Nakahashi, M.; Nishino, T. Characterization of the MexC-MexD-OprJ Multidrug Efflux System in Δ MexA-MexB-OprM Mutants of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 1998, 42, 1938–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.T.; Tsou, M.F.; Wu, H.J.; Chen, H.E.; Chuang, Y.C.; Yu, W.L. Survey of CTX-M-3 Extended-Spectrum β-Lactamase (ESBL) among Cefotaxime-Resistant Serratia marcescens at a Medical Center in Middle Taiwan. Diagn. Microbiol. Infect. Dis. 2004, 49, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Koolman, L.; Whyte, P.; Burgess, C.; Bolton, D. Distribution of Virulence-Associated Genes in a Selection of Campylobacter Isolates. Foodborne Pathog. Dis. 2015, 12, 424–432. [Google Scholar] [CrossRef] [PubMed]
- Haque, M.M.; Hirata, H.; Tsuyumu, S. SlyA Regulates MotA and MotB, Virulence and Stress-Related Genes under Conditions Induced by the PhoP-PhoQ System in Dickeya dadantii 3937. Res. Microbiol. 2015, 166, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Rudel, T.; Facius, D.; Barten, R.; Scheuerpflug, I.; Nonnenmacher, E.; Meyer, T.F. Role of Pili and the Phase-Variable PilC Protein in Natural Competence for Transformation of Neisseria gonorrhoeae. Proc. Natl. Acad. Sci. USA 1995, 92, 7986–7990. [Google Scholar] [CrossRef] [Green Version]
- Bonneau, A.; Roche, B.; Schalk, I.J. Iron Acquisition in Pseudomonas Aeruginosa by the Siderophore Pyoverdine: An Intricate Interacting Network Including Periplasmic and Membrane Proteins. Sci. Rep. 2020, 10, 120. [Google Scholar] [CrossRef] [Green Version]
- Jurado-Martín, I.; Sainz-Mejías, M.; McClean, S. Pseudomonas aeruginosa: An Audacious Pathogen with an Adaptable Arsenal of Virulence Factors. Int. J. Mol. Sci. 2021, 22, 3128. [Google Scholar] [CrossRef]
- Mosaddad, S.A.; Tahmasebi, E.; Yazdanian, A.; Rezvani, M.B.; Seifalian, A.; Yazdanian, M.; Tebyanian, H. Oral Microbial Biofilms: An Update. Eur. J. Clin. Microbiol. Infect. Dis. 2019, 38, 2005–2019. [Google Scholar] [CrossRef]
- Zhou, K.; Lokate, M.; Deurenberg, R.H.; Arends, J.; Lo-Ten Foe, J.; Grundmann, H.; Rossen, J.W.A.; Friedrich, A.W. Characterization of a CTX-M-15 Producing Klebsiella pneumoniae Outbreak Strain Assigned to a Novel Sequence Type (1427). Front. Microbiol. 2015, 6, 1250. [Google Scholar] [CrossRef]
- Russell, A.B.; Peterson, S.B.; Mougous, J.D. Type VI Secretion System Effectors: Poisons with a Purpose. Nat. Rev. Microbiol. 2014, 12, 137–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, J.; Özkaya, Ö.; Kümmerli, R. Bacterial Siderophores in Community and Host Interactions. Nat. Rev. Microbiol. 2020, 18, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Rehman, A.; Wang, X.; Ahmad, S.; Shahid, F.; Aslam, S.; Ashfaq, U.A.; Alrumaihi, F.; Qasim, M.; Hashem, A.; Al-Hazzani, A.A.; et al. In Silico Core Proteomics and Molecular Docking Approaches for the Identification of Novel Inhibitors against Streptococcus pyogenes. Int. J. Environ. Res. Public Health 2021, 18, 11355. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AMR Genes | Drug Class | Resistant Mechanism | Enterobacter | Enterococcus | Lacticaseibacillus | Pseudomonas | Streptococcus | Unclassified |
|---|---|---|---|---|---|---|---|---|
| blaSRT | Cephalosporin | Srt/sst family class c β-lactamase | - | - | - | 0.033 | - | - |
| oqxB17 | Phenicol; Quinolone | Multidrug efflux RND transporter permease subunit oqxb17 | - | - | - | 0.042 | - | - |
| tmexC1 | Tetracycline | Multidrug efflux RND transporter periplasmic adaptor subunit tmexc1 | - | - | - | 0.029 | - | - |
| AMR Genes | Drug Class | Resistant Mechanism | Enterobacter | Enterococcus | Lacticaseibacillus | Pseudomonas | Streptococcus | Unclassified |
|---|---|---|---|---|---|---|---|---|
| adeF | Fluoroquinolone; Tetracycline | Efflux mediated | 0.119 | - | - | - | - | - |
| baeR | Aminoglycoside; Aminocoumarin | Efflux mediated | 0.011 | - | - | - | - | - |
| blaCMH-6 | β-Lactam | Class c β-lactamase cmh-6 | 0.016 | - | - | - | - | - |
| CRP | Macrolide; Fluoroquinolone; Penam | Efflux mediated | 0.013 | - | - | - | - | - |
| dfrE | Diaminopyrimidine | Target replacement | - | 0.003 | - | - | - | - |
| emrB | Fluoroquinolone | Efflux mediated | 0.009 | - | - | - | - | - |
| emrR | Fluoroquinolone | Efflux mediated | 0.012 | - | - | - | - | - |
| fosA | Fosfomycin | Fosfomycin resistance glutathione transferase | 0.011 | - | - | - | - | - |
| ftsI | Cephalosporin; Cephamycin; Penam | Antibiotic target alteration | - | - | - | - | - | 0.014 |
| H-NS | Macrolide; Fluoroquinolone; Cephalosporin; Cephamycin; Penam; Tetracycline | Efflux mediated | 0.007 | - | - | - | - | - |
| KpnE | Macrolide; Aminoglycoside; Cephalosporin; Tetracycline; Peptide; Rifamycin; Disinfecting Agents and Antiseptics | Efflux mediated | 0.007 | - | - | - | - | - |
| KpnF | Macrolide; Aminoglycoside; Cephalosporin; Tetracycline; Peptide; Rifamycin; Disinfecting Agents and Antiseptics | Antibiotic efflux | 0.008 | - | - | - | - | - |
| lsa(A) | Lincosamide/Streptogramin | Abc-f type ribosomal protection protein lsa(a) | - | 0.004 | - | - | - | - |
| marA | Fluoroquinolone; Monobactam; Carbapenem; Cephalosporin; Glycylcycline; Cephamycin; Penam; Tetracycline; Rifamycin; Phenicol; Penem; Disinfecting Agents and Antiseptics | Antibiotic efflux; reduced permeability to antibiotic | 0.006 | - | - | - | - | - |
| MarR | Fluoroquinolone; Cephalosporin; Glycylcycline; Penam; Tetracycline; Rifamycin; Phenicol; Disinfecting Agents and Antiseptics | Target alteration; efflux mediated | 0.004 | - | - | - | - | - |
| mef(A) | Erythromycin | Macrolide efflux mfs transporter | - | - | - | - | - | 0.007 |
| msbA | Nitroimidazole | Efflux mediated | 0.011 | - | - | - | - | - |
| msr(D) | Erythromycin | Abc-f type ribosomal protection protein | - | - | - | - | - | 0.002 |
| oqxA | Phenicol/Quinolone | Multidrug efflux RND transporter periplasmic adaptor subunit | 0.009 | - | - | - | - | - |
| oqxB | Phenicol/Quinolone | Multidrug efflux RND transporter permease subunit | 0.009 | - | - | - | - | - |
| patA | Fluoroquinolone | Efflux mediated | - | - | - | - | 0.002 | - |
| ramA | Fluoroquinolone; Monobactam; Carbapenem; Cephalosporin; Glycylcycline; Cephamycin; Penam; Tetracycline; Rifamycin; Phenicol; Penem; Disinfecting Agents And Antiseptics | Efflux mediated; reduced permeability | 0.02 | - | - | - | - | - |
| rsmA | Fluoroquinolone; Diaminopyrimidine; Phenicol | Efflux mediated | 0.006 | - | - | - | - | - |
| tet(34) | Tetracycline | Oxytetracycline resistance phosphoribosyltransferase domain-containing protein | 0.012 | - | - | - | - | - |
| tet(M) | Tetracycline | Tetracycline resistance ribosomal protection protein | - | - | - | - | - | 0.002 |
| uhpT | Tetracycline | Target alteration | 14.525 | - | - | - | - | - |
| vanG | Tigecycline | Target alteration | 0.011 | - | - | - | - | - |
| vanT | Phosphonic Acid | Target alteration | - | 0.008 | - | - | - | - |
| VFclass | Virulence Factors | Enterobacter | Enterococcus | Lacticaseibacillus | Pseudomonas | Streptococcus | Unclassified |
|---|---|---|---|---|---|---|---|
| Acid resistance | Urease (Helicobacter) | - | - | - | 2 | - | - |
| Adherence | Acm | - | 1 | - | - | - | - |
| AS | - | 1 | - | - | - | - | |
| Choline binding proteins | - | - | - | - | 1 | - | |
| Curli fibers | 1 | - | - | - | - | - | |
| E. coli common pilus (ECP) | 2 | - | - | - | - | - | |
| Ebp pill | - | 4 | - | - | - | - | |
| EcbA | - | 1 | - | - | - | - | |
| EfaA | - | 1 | - | - | - | - | |
| Flagella | - | - | - | 44 | - | - | |
| GroEL(Clostridium) | - | - | - | - | - | 1 | |
| Hemorrhagic E. coli pilus (HCP) | 3 | - | - | - | - | - | |
| LPS O-antigen (P. aeruginosa) (Pseudomonas) | 1 | - | - | 1 | - | - | |
| P. fimbriae | 1 | - | - | - | - | - | |
| Polar flagella (Aeromonas) | - | - | - | 1 | - | 1 | |
| Streptococcal plasmin receptor/GAPDH (Streptococcus) | - | - | - | - | - | 1 | |
| Type I fimbriae | 3 | - | - | - | - | - | |
| Type IV pili biosynthesis | - | - | - | 13 | - | - | |
| Type IV pili twitching motility related proteins | - | - | - | 7 | - | - | |
| Alginate regulation | Alginate biosynthesis | - | - | - | 2 | - | - |
| Antiphagocytosis | - | - | - | 9 | - | - | |
| Capsular polysaccharide (Vibrio) | 1 | - | - | 3 | - | - | |
| Capsule (Enterococcus) | - | - | - | - | 1 | - | |
| Capsule (Klebsiella) | 2 | - | - | - | - | - | |
| Autotransporter | Contact-dependent inhibition CDI system | 2 | - | - | - | - | - |
| EhaB | 1 | - | - | - | - | - | |
| Biofilm formation | AdeFGH efflux pump/transport autoinducer | 1 | - | - | 1 | - | - |
| BopD | - | 1 | - | - | - | - | |
| PNAG (Polysaccharide poly-N-acetylglucosamine) (Acinetobacter) | 1 | - | - | - | - | - | |
| Efflux pump | AcrAB (Klebsiella) | - | - | - | 3 | - | - |
| Endotoxin | LOS (Haemophilus) | 2 | - | - | - | - | - |
| Enzyme | Gelatinase | - | 1 | - | - | - | - |
| Hyaluronidase | - | 1 | - | - | - | - | |
| SprE | - | 1 | - | - | - | - | |
| Streptococcal enolase (Streptococcus) | - | - | - | - | - | 1 | |
| Fimbrial adherence determinants | Fim (Salmonella) | 5 | - | - | - | - | - |
| Stf (Salmonella) | 1 | - | - | 1 | - | - | |
| Sti (Salmonella) | 1 | - | - | - | - | - | |
| Stj (Salmonella) | 2 | - | - | - | - | - | |
| Stk (Salmonella) | 2 | - | - | - | - | - | |
| Immune evasion | Capsule | - | - | - | 1 | - | - |
| LPS glucosylation (Shigella) | - | - | - | 1 | - | - | |
| LPS(Brucella) | - | - | - | 1 | - | - | |
| Invasion | Flagella (Burkholderia) | 1 | - | - | - | - | - |
| (empty) | 1 | - | - | - | - | - | |
| Iron uptake | Acinetobactin (Acinetobacter) | - | - | - | 5 | - | - |
| Aerobactin siderophore | 5 | - | - | - | - | - | |
| Heme transport (Shigella) | 1 | - | - | - | - | - | |
| Heme uptake | 3 | - | - | - | - | - | |
| Periplasmic binding protein-dependent ABC transport systems (Vibrio) | - | - | - | 1 | - | - | |
| Pyochelin | - | - | - | 2 | - | - | |
| Pyochelin receptor | - | - | - | 1 | - | - | |
| Pyoverdine | - | - | - | 12 | - | - | |
| Pyoverdine receptors | - | - | - | 1 | - | - | |
| Pyoverdine (Pseudomonas) | 1 | - | - | - | - | - | |
| Salmochelin (Klebsiella) | - | - | - | 1 | - | - | |
| Lipid and fatty acid metabolism | Isocitrate lyase (Mycobacterium) | - | - | - | 1 | - | - |
| Pantothenate synthesis (Mycobacterium) | - | - | - | 1 | - | - | |
| Magnesium uptake | Mg2+ transport (Salmonella) | - | - | - | 1 | - | - |
| Nutritional factor | Allantoin utilization (Klebsiella) | - | - | - | - | - | 2 |
| Others | O-antigen (Yersinia) | - | - | - | 1 | - | - |
| Protease | IgA1 protease | - | - | - | - | 1 | - |
| Pla (Yersinia) | 1 | - | - | - | - | - | |
| Quorum sensing | Acylhomoserine lactone synthase | - | - | - | 1 | - | - |
| Regulation | Carbon storage regulator A (Legionella) | - | - | - | 1 | - | - |
| GacS/GacA two-component system | - | - | - | 2 | - | - | |
| Two-component system (Acinetobacter) | - | - | - | 1 | - | - | |
| Two-component system (Bordetella) | - | - | - | 1 | - | - | |
| Secretion system | EPS type II secretion system (Vibrio) | 1 | - | - | 1 | - | - |
| Flagella (cluster I) (Yersinia) | 1 | - | - | - | - | - | |
| Hcp secretion island-1 encoded type VI secretion system (H-T6SS) (Pseudomonas) | 3 | - | - | 7 | - | - | |
| Lsp type II secretion system (Legionella) | - | - | - | 1 | - | - | |
| P. syringae TTSS effectors | - | - | - | 2 | - | - | |
| SCI-I T6SS | 7 | - | - | - | - | 6 | |
| T2SS (Yst1) (Yersinia) | 1 | - | - | - | - | - | |
| T2SS (Aeromonas) | 2 | - | - | - | - | - | |
| T6SS-II (Klebsiella) | - | - | - | - | - | 1 | |
| T6SS-III (Klebsiella) | 1 | - | - | - | - | - | |
| T6SS (Aeromonas) | - | - | - | 3 | - | - | |
| TTSS (SPI-1 encode) (Salmonella) | 1 | - | - | - | - | - | |
| Serum resistance | LPS rfb locus (Klebsiella) | 1 | - | - | - | - | - |
| Stress adaptation | Catalase (Neisseria) | - | - | - | 1 | - | - |
| Manganese transport system (Neisseria) | 1 | - | - | 1 | - | - | |
| Surface protein anchoring | Lipoprotein diacylglyceryl transferase (Listeria) | - | - | 1 | - | - | - |
| Toxin | Phytotoxin syringomycin (Pseudomonas) | 1 | - | - | - | - | - |
| TccC-type insecticidal toxins | - | - | - | 1 | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumari, K.S.; Dixit, S.; Gaur, M.; Behera, D.U.; Dey, S.; Sahoo, R.K.; Dash, P.; Subudhi, E. Taxonomic Assignment-Based Genome Reconstruction from Apical Periodontal Metagenomes to Identify Antibiotic Resistance and Virulence Factors. Life 2023, 13, 194. https://doi.org/10.3390/life13010194
Kumari KS, Dixit S, Gaur M, Behera DU, Dey S, Sahoo RK, Dash P, Subudhi E. Taxonomic Assignment-Based Genome Reconstruction from Apical Periodontal Metagenomes to Identify Antibiotic Resistance and Virulence Factors. Life. 2023; 13(1):194. https://doi.org/10.3390/life13010194
Chicago/Turabian StyleKumari, K. Swapna, Sangita Dixit, Mahendra Gaur, Dibyajyoti Uttameswar Behera, Suchanda Dey, Rajesh Kumar Sahoo, Patitapaban Dash, and Enketeswara Subudhi. 2023. "Taxonomic Assignment-Based Genome Reconstruction from Apical Periodontal Metagenomes to Identify Antibiotic Resistance and Virulence Factors" Life 13, no. 1: 194. https://doi.org/10.3390/life13010194