
Characterization of Bacterial Communities from the Surface and Adjacent Bottom Layers of Water in the Billings Reservoir

 , ,
, ,
Abstract
:1. Introduction
2. Material and Methods
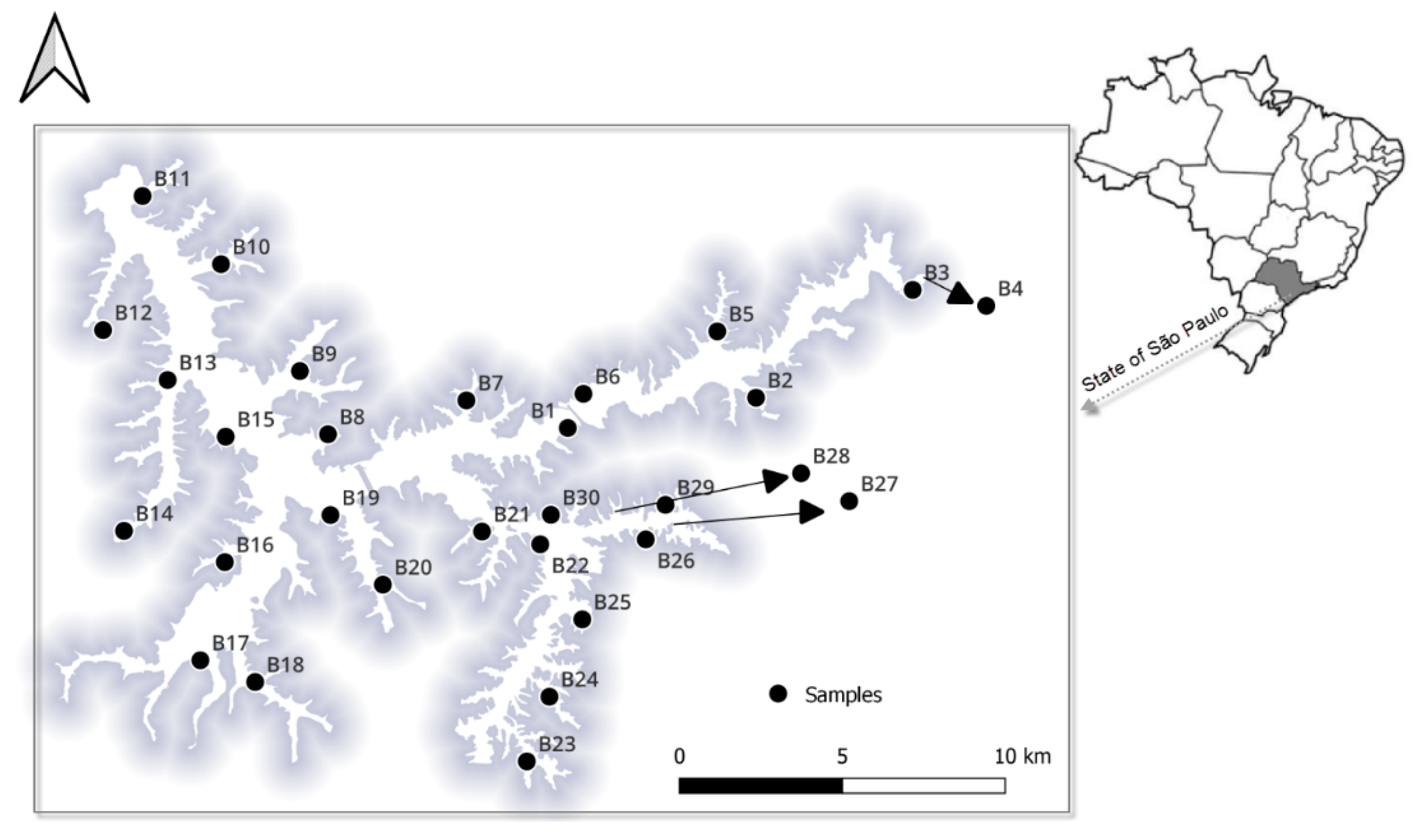
2.1. Study Sites and Sample Collection
2.2. DNA Isolation and Library Preparation
2.3. Detection of Toxin-Producing Cyanobacterial Genes
2.4. Bioinformatics and Statistical Analysis
3. Results
3.1. Physicochemical Characteristics of Water Samples
3.2. Bacterial Community Structure
3.3. Identification of the Billings Bacteriome in Surface and Bottom Layer Samples
3.4. Prediction of Functional Biomarkers
3.5. Search for Predefined Bacterial Groups, Pathogens, and Cyanotoxin Genes
4. Discussion
4.1. Distribution and Diversities of Bacterial Communities along the Billings Reservoir
4.2. Dominant Taxonomic Groups in the Bacterioplankton Community of the Reservoir
4.3. Bacterioplankton Community Diversity between the Surface and Bottom Layers
4.4. Potential Functions and Pathways of Bacterial Communities in the Reservoir
4.5. Occurrence of Pathogenic Bacteria
5. Limitations and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Connor, R. The United Nations World Water Development Report 2015: Water for a Sustainable World; WWAP-United Nations World Water Assessment Programme; United Nations Educational, Scientific and Cultural Organization: Paris, France, 2015; ISBN 978-92-3-100071-3.2. [Google Scholar]
- Mishra, A.K.E. (Ed.) Application of Nanotechnology in Water Research; John Wiley & Sons: Hoboken, NJ, USA, 2014; ISBN 978-1-118-49630-5. [Google Scholar]
- Shi, X.; Ma, W.; Li, X.; Cheng, Y.; Huang, T. Typical Reservoir Pollution Source Analysis. In Water Pollution and Water Quality Control of Selected Chinese Reservoir Basins; Huang, T., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 95–130. [Google Scholar]
- Gonçalves, S.P.; Lucheta, F.; Souza, V.K.D.; Terra, N.R. The influence of xenobiotics in river sediment on the reproduction and survival of Daphnia magna, 1820, Straus. Acta Limnol. Bras. 2012, 24, 220–234. [Google Scholar] [CrossRef] [Green Version]
- Godoy, R.G.; Marcondes, M.A.; Pessoa, R.; Nascimento, A.; Victor, J.R.; Duarte, A.; Clissa, P.B.; Sanabani, S.S. Bacterial community composition and potential pathogens along the Pinheiros River in the southeast of Brazil. Sci. Rep. 2020, 10, 9331. [Google Scholar] [CrossRef] [PubMed]
- Edokpayi, J.N.; Odiyo, J.O.; Durowoju, O.S. Impact of Wastewater on Surface Water Quality in Developing Countries: A Case Study of South Africa. In Water Quality; INTECH: Vienna, Austria, 2017; pp. 401–416. [Google Scholar]
- Park, H.-K.; Byeon, M.-S.; Shin, Y.-N.; Jung, D.-I. Sources and spatial and temporal characteristics of organic carbon in two large reservoirs with contrasting hydrologic characteristics. Water Resour. Res. 2009, 45. [Google Scholar] [CrossRef]
- Simek, K.; Hornák, K.; Jezbera, J.; Nedoma, J.; Znachor, P.; Hejzlar, J.; Sed’a, J. Spatio-temporal patterns of bacterioplankton production and community composition related to phytoplankton composition and protistan bacterivory in a dam reservoir. Aquat. Microb. Ecol. 2008, 51, 249–262. [Google Scholar] [CrossRef] [Green Version]
- Preheim, S.P.; Olesen, S.W.; Spencer, S.J.; Materna, A.; Varadharajan, C.; Blackburn, M.; Friedman, J.; Rodriguez, J.; Hemond, H.; Alm, E.J. Surveys, simulation and single-cell assays relate function and phylogeny in a lake ecosystem. Nat. Microbiol. 2016, 1, 16130. [Google Scholar] [CrossRef]
- Yan, M.; Chen, S.; Huang, T.; Li, B.; Li, N.; Liu, K.; Zong, R.; Miao, Y.; Huang, X. Community Compositions of Phytoplankton and Eukaryotes during the Mixing Periods of a Drinking Water Reservoir: Dynamics and Interactions. Int. J. Environ. Res. Public Health 2020, 17, 1128. [Google Scholar] [CrossRef] [Green Version]
- Xue, Y.; Chen, H.; Yang, J.R.; Liu, M.; Huang, B.; Yang, J. Distinct patterns and processes of abundant and rare eukaryotic plankton communities following a reservoir cyanobacterial bloom. ISME J. 2018, 12, 2263–2277. [Google Scholar] [CrossRef] [Green Version]
- Vila-Costa, M.; Barberan, A.; Auguet, J.C.; Sharma, S.; Moran, M.A.; Casamayor, E.O. Bacterial and archaeal community structure in the surface microlayer of high mountain lakes examined under two atmospheric aerosol loading scenarios. FEMS Microbiol. Ecol. 2013, 84, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Aguirre-Garrido, J.F.; Ramirez-Saad, H.C.; Toro, N.; Martinez-Abarca, F. Bacterial Diversity in the Soda Saline Crater Lake from Isabel Island, Mexico. Microb. Ecol. 2016, 71, 68–77. [Google Scholar] [CrossRef]
- Baricz, A.; Chiriac, C.M.; Andrei, A.S.; Bulzu, P.A.; Levei, E.A.; Cadar, O.; Battes, K.P.; Cimpean, M.; Senila, M.; Cristea, A.; et al. Spatio-temporal insights into microbiology of the freshwater-to-hypersaline, oxic-hypoxic-euxinic waters of Ursu Lake. Environ. Microbiol. 2021, 23, 3523–3540. [Google Scholar] [CrossRef] [Green Version]
- dos Anjos, F.M.; Bittencourt-Oliveira Mdo, C.; Zajac, M.P.; Hiller, S.; Christian, B.; Erler, K.; Luckas, B.; Pinto, E. Detection of harmful cyanobacteria and their toxins by both PCR amplification and LC-MS during a bloom event. Toxicon 2006, 48, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; Kong, X.; Cui, B.; Jin, S.; Xie, Y.; Wang, X.; Deng, Y. Bacterial communities and potential waterborne pathogens within the typical urban surface waters. Sci. Rep. 2018, 8, 13368. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Li, Q.; Ren, M.; Lin, Y.; Wang, J.; Chen, L.; Li, T.; Zhao, J. Trophic Status Is Associated With Community Structure and Metabolic Potential of Planktonic Microbiota in Plateau Lakes. Front. Microbiol. 2019, 10, 2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, K.; Zhao, W.; Wang, Q.; Yang, X.; Zhu, L.; Shen, J.; Cheng, X.; Wang, J. The Relative Abundance of Benthic Bacterial Phyla Along a Water-Depth Gradient in a Plateau Lake: Physical, Chemical, and Biotic Drivers. Front. Microbiol. 2019, 10, 1521. [Google Scholar] [CrossRef]
- De Wever, A.; Muylaert, K.; Van der Gucht, K.; Pirlot, S.; Cocquyt, C.; Descy, J.P.; Plisnier, P.D.; Vyverman, W. Bacterial community composition in Lake Tanganyika: Vertical and horizontal heterogeneity. Appl. Environ. Microbiol. 2005, 71, 5029–5037. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, J.A.; Steele, J.A.; Caporaso, J.G.; Steinbruck, L.; Reeder, J.; Temperton, B.; Huse, S.; McHardy, A.C.; Knight, R.; Joint, I.; et al. Defining seasonal marine microbial community dynamics. ISME J. 2012, 6, 298–308. [Google Scholar] [CrossRef] [Green Version]
- Yung, C.M.; Vereen, M.K.; Herbert, A.; Davis, K.M.; Yang, J.; Kantorowska, A.; Ward, C.S.; Wernegreen, J.J.; Johnson, Z.I.; Hunt, D.E. Thermally adaptive tradeoffs in closely related marine bacterial strains. Environ. Microbiol. 2015, 17, 2421–2429. [Google Scholar] [CrossRef]
- Shade, A.; Jones, S.E.; McMahon, K.D. The influence of habitat heterogeneity on freshwater bacterial community composition and dynamics. Environ. Microbiol. 2008, 10, 1057–1067. [Google Scholar] [CrossRef]
- Wetzel, R.G. lake and river ecosystems. In Limnology, 3rd ed.; Elsevier Academic Press: San Diego, CA, USA, 2001. [Google Scholar]
- Petrere, M., Jr.; Walter, T.; Minte-Vera, C.V. Income evaluation of small—Scale fishers in two Brazilian urban reservoirs: Represa Billings (SP) and Lago Paranoa (DF). Braz. J. Biol. Rev. Brasleira De Biol. 2006, 66, 817–828. [Google Scholar] [CrossRef] [Green Version]
- Rezende, K.F.; Santos, R.M.; Borges, J.C.; Salvo, L.M.; da Silva, J.R. Histopathological and genotoxic effects of pollution on Nile Tilapia (Oreochromis niloticus, Linnaeus, 1758) in the Billings Reservoir (Brazil). Toxicol. Mech. Methods 2014, 24, 404–411. [Google Scholar] [CrossRef]
- Wengrat, S.; Bicudo, D.D.C. Spatial evaluation of water quality in an urban reservoir (Billings Complex, southeastern Brazil). Acta Limnol. Bras. 2011, 23, 200–216. [Google Scholar] [CrossRef] [Green Version]
- Mariani, C.F.; Moschini-Carlos, V.; Brandimarte, A.L.; Nishimura, P.Y.; Tófoli, C.F.d.; Duran, D.S.; Lourenço, E.M.; Braidotti, J.C.; Almeida, L.P.; Fidalgo, V.H.; et al. Biota and water quality in the Riacho grande reservoir, Billings complex (São Paulo, Brazil). Acta Limnol. Bras. 2006, 18, 267–280. [Google Scholar]
- Leme, E.; Silva, E.P.; Rodrigues, P.S.; Silva, I.R.; Martins, M.F.M.; Bondan, E.F.; Bernardi, M.M.; Kirsten, T.B. Billings reservoir water used for human consumption presents microbiological contaminants and induces both behavior impairments and astrogliosis in zebrafish. Ecotoxicol. Environ. Saf. 2018, 161, 364–373. [Google Scholar] [CrossRef]
- Moschini-Carlos, V.; Bortoli, S.; Pinto, E.; Nishimura, P.Y.; Freitas, L.G.; Pompêo, M.L.M.; Dörr, F. Cyanobacteria and cyanotoxin in the Billings reservoir (São Paulo, SP, Brazil). Limnetica 2009, 28, 273–282. [Google Scholar] [CrossRef]
- de Carvalho, L.R.; Sant’Anna, C.L.; Gemelgo, M.C.P.; de P. Azevedo, M.T. Cyanobacterial occurrence and detection of microcystin by planar chromatography in surface water of Billings and Guarapiranga Reservoirs, SP, Brazil. Braz. J. Bot. 2007, 30, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Paulino, G.V.B.; Felix, C.R.; Silvan, C.G.; Andersen, G.L.; Landell, M.F. Bacterial community and environmental factors associated to rivers runoff and their possible impacts on coral reef conservation. Mar. Pollut. Bull. 2020, 156, 111233. [Google Scholar] [CrossRef]
- Lorenzi, A.S.; Chia, M.A.; Lopes, F.A.C.; Silva, G.G.Z.; Edwards, R.A.; Bittencourt-Oliveira, M.D.C. Cyanobacterial biodiversity of semiarid public drinking water supply reservoirs assessed via next-generation DNA sequencing technology. J. Microbiol. 2019, 57, 450–460. [Google Scholar] [CrossRef]
- Herlemann, D.P.; Labrenz, M.; Jurgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef] [Green Version]
- da Pereira Fonseca, T.A.; Pessoa, R.; Felix, A.C.; Sanabani, S.S. Diversity of Bacterial Communities on Four Frequently Used Surfaces in a Large Brazilian Teaching Hospital. Int. J. Environ. Res. Public Health 2016, 13, 152. [Google Scholar] [CrossRef] [Green Version]
- da Pereira Fonseca, T.A.; Pessoa, R.; Sanabani, S.S. Molecular Analysis of Bacterial Microbiota on Brazilian Currency Note Surfaces. Int. J. Environ. Res. Public Health 2015, 12, 13276–13288. [Google Scholar] [CrossRef] [Green Version]
- Hisbergues, M.; Christiansen, G.; Rouhiainen, L.; Sivonen, K.; Borner, T. PCR-based identification of microcystin-producing genotypes of different cyanobacterial genera. Arch. Microbiol. 2003, 180, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Rantala, A.; Fewer, D.P.; Hisbergues, M.; Rouhiainen, L.; Vaitomaa, J.; Borner, T.; Sivonen, K. Phylogenetic evidence for the early evolution of microcystin synthesis. Proc. Natl. Acad. Sci. USA 2004, 101, 568–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahe, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Myers, E.W.; Miller, W. Optimal alignments in linear space. Comput. Appl. Biosci. CABIOS 1988, 4, 11–17. [Google Scholar] [CrossRef]
- Wheeler, T.J.; Eddy, S.R. nhmmer: DNA homology search with profile HMMs. Bioinformatics 2013, 29, 2487–2489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Chao, A.; Lee, S.-M. Estimating the Number of Classes via Sample Coverage. J. Am. Stat. Assoc. 1992, 87, 210–217. [Google Scholar] [CrossRef]
- Chao, A. Estimating the population size for capture-recapture data with unequal catchability. Biometrics 1987, 43, 783–791. [Google Scholar] [CrossRef]
- Burnham, K.P.; Overton, W.S. Robust Estimation of Population Size When Capture Probabilities Vary Among Animals. Ecology 1979, 60, 927–936. [Google Scholar] [CrossRef]
- Magurran, A.E. Measuring Biological Diversity; Elsevier: Amsterdam, The Netherlands, 2013. [Google Scholar]
- Chao, A.; Shen, T.-J. Nonparametric estimation of Shannon’s index of diversity when there are unseen species in sample. Environ. Ecol. Stat. 2003, 10, 429–443. [Google Scholar] [CrossRef]
- Beals, E.W. Bray-Curtis Ordination: An Effective Strategy for Analysis of Multivariate Ecological Data. In Advances in Ecological Research; MacFadyen, A., Ford, E.D., Eds.; Academic Press: Cambridge, MA, USA, 1984; Volume 14, pp. 1–55. [Google Scholar]
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol. Electron. 2001, 4, 1–9. [Google Scholar]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soares, L.A.; Santos, A.C.A.D.; Duarte, I.C.S.; Romagnoli, E.M.; Calijuri, M.d.C. Distribution of Archaeal and Bacterial communities in a subtropical reservoir. Acta Limnol. Bras. 2015, 27, 411–420. [Google Scholar] [CrossRef] [Green Version]
- Lehours, A.C.; Evans, P.; Bardot, C.; Joblin, K.; Gerard, F. Phylogenetic diversity of archaea and bacteria in the anoxic zone of a meromictic lake (Lake Pavin, France). Appl. Environ. Microbiol. 2007, 73, 2016–2019. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Su, J.; Zhang, M.; Luo, Z.; Li, X.; Chai, B. Bacterial Community Spacing Is Mainly Shaped by Unique Species in the Subalpine Natural Lakes of China. Front. Microbiol. 2021, 12, 669131. [Google Scholar] [CrossRef]
- Dias, R.M.; de Oliveira, A.G.; Baumgartner, M.T.; Angulo-Valencia, M.A.; Agostinho, A.A. Functional erosion and trait loss in fish assemblages from Neotropical reservoirs: The man beyond the environment. Fish Fish. 2021, 22, 377–390. [Google Scholar] [CrossRef]
- Qu, J.; Jia, C.; Liu, Q.; Li, Z.; Liu, P.; Yang, M.; Zhao, M.; Li, W.; Zhu, H.; Zhang, Q. Dynamics of Bacterial Community Diversity and Structure in the Terminal Reservoir of the South-To-North Water Diversion Project in China. Water 2018, 10, 709. [Google Scholar] [CrossRef] [Green Version]
- Toyama, D.; Kishi, L.T.; Santos-Junior, C.D.; Soares-Costa, A.; de Oliveira, T.C.; de Miranda, F.P.; Henrique-Silva, F. Metagenomics Analysis of Microorganisms in Freshwater Lakes of the Amazon Basin. Genome Announc. 2016, 4, e01440-16. [Google Scholar] [CrossRef] [Green Version]
- Santos-Junior, C.D.; Kishi, L.T.; Toyama, D.; Soares-Costa, A.; Oliveira, T.C.; de Miranda, F.P.; Henrique-Silva, F. Metagenome Sequencing of Prokaryotic Microbiota Collected from Rivers in the Upper Amazon Basin. Genome Announc. 2017, 5, e01450-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabral, B.C.A.; Hoffmann, L.; Budowle, B.; Urmenyi, T.P.; Moura-Neto, R.S.; Azevedo, S.; Silva, R. Planktonic microbial profiling in water samples from a Brazilian Amazonian reservoir. MicrobiologyOpen 2018, 7, e00523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gracas, D.A.; Miranda, P.R.; Barauna, R.A.; McCulloch, J.A.; Ghilardi, R., Jr.; Schneider, M.P.; Silva, A. Microbial diversity of an anoxic zone of a hydroelectric power station reservoir in Brazilian Amazonia. Microb. Ecol. 2011, 62, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Albertsen, M.; Karst, S.M.; Ziegler, A.S.; Kirkegaard, R.H.; Nielsen, P.H. Back to Basics—The Influence of DNA Extraction and Primer Choice on Phylogenetic Analysis of Activated Sludge Communities. PLoS ONE 2015, 10, e0132783. [Google Scholar] [CrossRef] [PubMed]
- Simek, K.; Hornak, K.; Jezbera, J.; Nedoma, J.; Vrba, J.; Straskrabova, V.; Macek, M.; Dolan, J.R.; Hahn, M.W. Maximum growth rates and possible life strategies of different bacterioplankton groups in relation to phosphorus availability in a freshwater reservoir. Environ. Microbiol. 2006, 8, 1613–1624. [Google Scholar] [CrossRef]
- Bouvy, M.; Nascimento, S.M.; Molica, R.J.R.; Ferreira, A.; Huszar, V.; Azevedo, S.M.F.O. Limnological features in Tapacurá reservoir (northeast Brazil) during a severe drought. Hydrobiologia 2003, 493, 115–130. [Google Scholar] [CrossRef]
- do Carmo Bittencourt-Oliveira, M. Detection of potential microcystin-producing cyanobacteria in Brazilian reservoirs with a mcyB molecular marker. Harmful Algae 2003, 2, 51–60. [Google Scholar] [CrossRef]
- Bittencourt-Oliveira Mdo, C.; Piccin-Santos, V.; Moura, A.N.; Aragao-Tavares, N.K.; Cordeiro-Araujo, M.K. Cyanobacteria, microcystins and cylindrospermopsin in public drinking supply reservoirs of Brazil. An. Da Acad. Bras. De Cienc. 2014, 86, 297–310. [Google Scholar] [CrossRef] [Green Version]
- Dantas, E.W.; Moura, A.N.; Bittencourt-Oliveira Mdo, C. Cyanobacterial blooms in stratified and destratified eutrophic reservoirs in semi-arid region of Brazil. An. Da Acad. Bras. De Cienc. 2011, 83, 1327–1338. [Google Scholar] [CrossRef] [Green Version]
- Moura, A.N.; Dantas, E.W.; Oliveira, H.S.; Bittencourt-Oliveira, M.C. Vertical and temporal dynamics of cyanobacteria in the Carpina potable water reservoir in northeastern Brazil. Braz. J. Biol. Rev. Brasleira De Biol. 2011, 71, 451–459. [Google Scholar] [CrossRef]
- McMahon, K.D.; Read, E.K. Microbial contributions to phosphorus cycling in eutrophic lakes and wastewater. Annu. Rev. Microbiol. 2013, 67, 199–219. [Google Scholar] [CrossRef] [PubMed]
- Paerl, H.W. Dynamics of Blue-Green Algal (’Microcystis aeruginosa’) Blooms in the Lower Neuse River, North Carolina: Causative Factors and Potential Controls Raleigh; Report No. 229; University of North Carolina Water Resources Research Institute: Raleigh, NC, USA, 1987. [Google Scholar]
- Paerl, H.W.; Huisman, J. Climate change: A catalyst for global expansion of harmful cyanobacterial blooms. Environ. Microbiol. Rep. 2009, 1, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Pope, P.B.; Patel, B.K. Metagenomic analysis of a freshwater toxic cyanobacteria bloom. FEMS Microbiol. Ecol. 2008, 64, 9–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parulekar, N.N.; Kolekar, P.; Jenkins, A.; Kleiven, S.; Utkilen, H.; Johansen, A.; Sawant, S.; Kulkarni-Kale, U.; Kale, M.; Saebo, M. Characterization of bacterial community associated with phytoplankton bloom in a eutrophic lake in South Norway using 16S rRNA gene amplicon sequence analysis. PLoS ONE 2017, 12, e0173408. [Google Scholar] [CrossRef] [PubMed]
- Newton, R.J.; Jones, S.E.; Eiler, A.; McMahon, K.D.; Bertilsson, S. A guide to the natural history of freshwater lake bacteria. Microbiol. Mol. Biol. Rev. MMBR 2011, 75, 14–49. [Google Scholar] [CrossRef] [Green Version]
- Walter, Z.; Natascha, S.; Meinhard, S. Seasonal patterns of the bacterioplankton community composition in a large mesotrophic lake. Aquat. Microb. Ecol. 2003, 31, 211–225. [Google Scholar]
- Patel, V.; Munot, H.; Shouche, Y.S.; Madamwar, D. Response of bacterial community structure to seasonal fluctuation and anthropogenic pollution on coastal water of Alang-Sosiya ship breaking yard, Bhavnagar, India. Bioresour. Technol. 2014, 161, 362–370. [Google Scholar] [CrossRef]
- Ghai, R.; Rodriguez-Valera, F.; McMahon, K.D.; Toyama, D.; Rinke, R.; Cristina Souza de Oliveira, T.; Wagner Garcia, J.; Pellon de Miranda, F.; Henrique-Silva, F. Metagenomics of the water column in the pristine upper course of the Amazon river. PLoS ONE 2011, 6, e23785. [Google Scholar] [CrossRef]
- Paerl, H.W.; Otten, T.G. Harmful cyanobacterial blooms: Causes, consequences, and controls. Microb. Ecol. 2013, 65, 995–1010. [Google Scholar] [CrossRef]
- Ghai, R.; Mizuno, C.M.; Picazo, A.; Camacho, A.; Rodriguez-Valera, F. Key roles for freshwater Actinobacteria revealed by deep metagenomic sequencing. Mol. Ecol. 2014, 23, 6073–6090. [Google Scholar] [CrossRef]
- Goodfellow, M.; Williams, S.T. Ecology of actinomycetes. Annu. Rev. Microbiol. 1983, 37, 189–216. [Google Scholar] [CrossRef] [PubMed]
- Zwirglmaier, K.; Keiz, K.; Engel, M.; Geist, J.; Raeder, U. Seasonal and spatial patterns of microbial diversity along a trophic gradient in the interconnected lakes of the Osterseen Lake District, Bavaria. Front. Microbiol. 2015, 6, 1168. [Google Scholar] [CrossRef] [PubMed]
- Warnecke, F.; Sommaruga, R.; Sekar, R.; Hofer, J.S.; Pernthaler, J. Abundances, identity, and growth state of actinobacteria in mountain lakes of different UV transparency. Appl. Environ. Microbiol. 2005, 71, 5551–5559. [Google Scholar] [CrossRef] [Green Version]
- Buckling, A.; Wills, M.A.; Colegrave, N. Adaptation limits diversification of experimental bacterial populations. Science 2003, 302, 2107–2109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, R.P.; Monchy, S.; Cardinale, M.; Taghavi, S.; Crossman, L.; Avison, M.B.; Berg, G.; van der Lelie, D.; Dow, J.M. The versatility and adaptation of bacteria from the genus Stenotrophomonas. Nat. Rev. Microbiol. 2009, 7, 514–525. [Google Scholar] [CrossRef]
- Lu, X.; Fan, C.; He, W.; Deng, J.; Yin, H. Sulfur-containing amino acid methionine as the precursor of volatile organic sulfur compounds in algea-induced black bloom. J. Environ. Sci. 2013, 25, 33–43. [Google Scholar] [CrossRef]
- Patel, C.B.; Shanker, R.; Gupta, V.K.; Upadhyay, R.S. Q-PCR Based Culture-Independent Enumeration and Detection of Enterobacter: An Emerging Environmental Human Pathogen in Riverine Systems and Potable Water. Front. Microbiol. 2016, 7, 172. [Google Scholar] [CrossRef] [Green Version]
- Bernardet, J.F.; Vancanneyt, M.; Matte-Tailliez, O.; Grisez, L.; Tailliez, P.; Bizet, C.; Nowakowski, M.; Kerouault, B.; Swings, J. Polyphasic study of Chryseobacterium strains isolated from diseased aquatic animals. Syst. Appl. Microbiol. 2005, 28, 640–660. [Google Scholar] [CrossRef]
- Zamora, L.; Fernandez-Garayzabal, J.F.; Sanchez-Porro, C.; Palacios, M.A.; Moore, E.R.; Dominguez, L.; Ventosa, A.; Vela, A.I. Flavobacterium plurextorum sp. nov. Isolated from Farmed Rainbow Trout (Oncorhynchus mykiss). PLoS ONE 2013, 8, e67741. [Google Scholar] [CrossRef]
- Zamora, L.; Fernandez-Garayzabal, J.F.; Svensson-Stadler, L.A.; Palacios, M.A.; Dominguez, L.; Moore, E.R.; Vela, A.I. Flavobacterium oncorhynchi sp. nov., a new species isolated from rainbow trout (Oncorhynchus mykiss). Syst. Appl. Microbiol. 2012, 35, 86–91. [Google Scholar] [CrossRef]
- Hlavsa, M.C.; Roberts, V.A.; Anderson, A.R.; Hill, V.R.; Kahler, A.M.; Orr, M.; Garrison, L.E.; Hicks, L.A.; Newton, A.; Hilborn, E.D.; et al. Surveillance for waterborne disease outbreaks and other health events associated with recreational water—United States, 2007–2008. MMWR Surveill Summ 2011, 60, 1–32. [Google Scholar] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample ID | DO * mg/L | Temp | pH | Average Water Depth (km) | Turbidity (NTU) | Sulfate mg/L | Phosphorus mg/L | Ammonia mg/L | Nitrate mg/L |
|---|---|---|---|---|---|---|---|---|---|
| B1S | 4 | 20.5 | 7.3 | 9.5 | 3.1 | 0.4 | 0.1 | 2.4 | 0.9 |
| B2S | 7.1 | 21.2 | 8.3 | 5.5 | 3.1 | 0.5 | 0.2 | 2.8 | 1.1 |
| B3S | 5.7 | 21.2 | 7.5 | 3.2 | 2.5 | 0.4 | 0.1 | 2.8 | 2.2 |
| B4S | 5.5 | 22.1 | 7.0 | 2.1 | 1.9 | 0.6 | 0.0 | 2.7 | 7.4 |
| B5S | 7.4 | 21.5 | 8.1 | 6.9 | 2.5 | 0.4 | 0.2 | 2.7 | 0.9 |
| B6S | 8.3 | 21.6 | 8.7 | 12.8 | 4.3 | 0.3 | 0.3 | 2.2 | 1.2 |
| B7S | 7.5 | 21.2 | 8.2 | 12.4 | 6.9 | 0.3 | 0.1 | 2.6 | 0.9 |
| B8S | 7.3 | 21.1 | 7.9 | 10.1 | 15.0 | 0.4 | 0.3 | 2.5 | 1.2 |
| B9S | 7.2 | 21.2 | 7.6 | 9 | 11.5 | 0.7 | 0.3 | 2.5 | 1 |
| B10S | 7.9 | 21.5 | 7.9 | 7.6 | 18.5 | 0.3 | 0.8 | 2.9 | 2.2 |
| B11S | 5.8 | 21.5 | 7.6 | 5.9 | 12.0 | 0.6 | 0.1 | 3 | 1.8 |
| B12S | 4.3 | 21.2 | 7.5 | 8.1 | 14.1 | 0.5 | 0.0 | 2.6 | 1.4 |
| B13S | 6.7 | 20.8 | 7.8 | 10.4 | 10.0 | 0.5 | 0.1 | 2.7 | 1.9 |
| B14S | 9.5 | 20.9 | 8.5 | 3.9 | 12.0 | 0.54 | 0.1 | 3.3 | 1.8 |
| B15S | 4.7 | 20.9 | 7.5 | 13.6 | 6.2 | 0.56 | 0.0 | 2.7 | 1.7 |
| B16S | 9.2 | 21 | 8.6 | 10.1 | 12.9 | 0.4 | 0.1 | 2.8 | 1.3 |
| B17S | 7.9 | 20.6 | 7.7 | 6.9 | 11.8 | 0.6 | 0.1 | 2.7 | 2.3 |
| B18S | 6.5 | 20.5 | 7.5 | 8.9 | 18.0 | 0.4 | 0.2 | 2.7 | 2.65 |
| B19S | 6.3 | 20.5 | 7.6 | 6.1 | 7.8 | 0.5 | 0.1 | 2.1 | 1.3 |
| B20S | 6.5 | 20.9 | 7.3 | 4.5 | 10.5 | 0.3 | 0.1 | 2.6 | 2.9 |
| B21S | 7.7 | 20.9 | 7.6 | 7 | 12.0 | 0.5 | 0.2 | 2.8 | 2.2 |
| B22S | 9.4 | 20.6 | 8.3 | 5.4 | 11.0 | 0.5 | 0.2 | 2.1 | 1.1 |
| B23S | 9 | 21 | 7.3 | 5.8 | 6.5 | 0.4 | 0.2 | 2.4 | 1.8 |
| B24S | 7.9 | 21.3 | 8.0 | 5.5 | 8.1 | 0.5 | 0.0 | 2.9 | 1.02 |
| B25S | 5.8 | 21.8 | 7.6 | 6.9 | 5.1 | 0.5 | 0.1 | 3.4 | 1.98 |
| B26S | 7.6 | 21.6 | 8.0 | 6.8 | 6.5 | 0.4 | 0.1 | 3.2 | 0.9 |
| B27S | 9.2 | 20.8 | 7.4 | 3.2 | 5.3 | 0.3 | 0.1 | 2.4 | 0.8 |
| B28S | 9.3 | 20.6 | 7.9 | 7.1 | 4.8 | 0.4 | 0.1 | 2.8 | 0.9 |
| B29S | 9.3 | 21.6 | 8.9 | 8.7 | 5.3 | 0.5 | 0.1 | 2.9 | 1.2 |
| B30S | 8.1 | 20.9 | 7.8 | 10.2 | 8.3 | 0.4 | 0.2 | 2.3 | 2.8 |
| Sample ID | DO * mg/L | Temp | pH | Average Water Depth (km) | Turbidity (NTU) | Sulfate mg/L | Phosphorus mg/L | Ammonia mg/L | Nitrate mg/L |
|---|---|---|---|---|---|---|---|---|---|
| B1F | 3.7 | 20.6 | 7.4 | 9.5 | 4.1 | 0.4 | 0.1 | 2.5 | 1.1 |
| B2F | 6.4 | 20.8 | 8.3 | 5.5 | 3.0 | 0.5 | 0.2 | 2.3 | 1.2 |
| B3F | 5.1 | 19.8 | 7.2 | 3.2 | 3.9 | 0.4 | 0.2 | 2.8 | 1.5 |
| B4F | 4 | 18.8 | 7 | 2.1 | 1.2 | 0.1 | 0.1 | 2.4 | 1.2 |
| B5F | 7.1 | 21.1 | 8.2 | 6.9 | 3.0 | 0.6 | 0.1 | 2.3 | 0.05 |
| B6F | 7.1 | 21.7 | 8.3 | 12.8 | 4.4 | 0.4 | 0.1 | 3.2 | 0.09 |
| B7F | 5.6 | 21.2 | 8.1 | 12.4 | 6.3 | 0.5 | 0.1 | 2.8 | 1.2 |
| B8F | 4.6 | 21.1 | 7.9 | 10.1 | 8.1 | 0.2 | 0.1 | 2.2 | 1.1 |
| B9F | 4.5 | 20.9 | 7.2 | 9 | 9.8 | 0.4 | 0.0 | 2.4 | 0.9 |
| B10F | 4.8 | 21.2 | 7.6 | 7.6 | 13.6 | 0.5 | 0.1 | 3.7 | 0.8 |
| B11F | 3.5 | 21.2 | 7.6 | 5.9 | 10.3 | 0.2 | 0.0 | 3.1 | 1.1 |
| B12F | 3.5 | 20.9 | 7.4 | 8.1 | 6.7 | 0.3 | 0.0 | 2.8 | 0.8 |
| B13F | 4.9 | 20.8 | 7.7 | 10.4 | 10.3 | 0.5 | 0.0 | 3.1 | 1.2 |
| B14F | 8.3 | 21 | 8.3 | 3.9 | 31.9 | 0.3 | 0.0 | 2.5 | 0.9 |
| B15F | 4.3 | 20.8 | 7.5 | 13.6 | 6.1 | 0.5 | 0.1 | 2.7 | 1.2 |
| B16F | 8.4 | 20.6 | 8.5 | 10.1 | 11.5 | 0.4 | 1.2 | 2.3 | 0.09 |
| B17F | 6.3 | 20.5 | 7.8 | 6.9 | 16.5 | 0.4 | 1.1 | 2.6 | 1.1 |
| B18F | 6 | 20.3 | 7.6 | 8.9 | 13.1 | 0.4 | 0.1 | 2.7 | 0.09 |
| B19F | 5.9 | 20.5 | 7.6 | 6.1 | 9.9 | 0.8 | 1.1 | 2.6 | 1 |
| B20F | 5.5 | 20.4 | 7.5 | 4.5 | 9.9 | 0.6 | 2.1 | 2.4 | 2.7 |
| B21F | 6.6 | 20.5 | 7.7 | 7 | 8.4 | 0.3 | 0.9 | 2.8 | 1.1 |
| B22F | 7.8 | 20.5 | 8.4 | 5.4 | 10.2 | 0.6 | 1.2 | 2.7 | 1.6 |
| B23F | 6.9 | 20.9 | 8.1 | 5.8 | 8.2 | 0.5 | 1.3 | 2.3 | 1.8 |
| B24F | 7.1 | 20.7 | 8 | 5.5 | 6.8 | 0.4 | 0.9 | 3 | 2.1 |
| B25F | 6.6 | 20.9 | 7.9 | 6.9 | 6.2 | 0.5 | 1.2 | 3.5 | 1.9 |
| B26F | 7.6 | 21.3 | 8 | 6.8 | 7.8 | 0.5 | 1.9 | 2.9 | 2.3 |
| B27F | 8.3 | 20.5 | 7.8 | 3.2 | 3.4 | 0.6 | 2.1 | 3.1 | 1.9 |
| B28F | 7.6 | 20.5 | 7.9 | 7.1 | 7.2 | 0.4 | 1.9 | 2.9 | 1.6 |
| B29F | 8.2 | 20.6 | 8.6 | 8.7 | 7.7 | 0.5 | 2.1 | 2.4 | 1.1 |
| B30F | 7.7 | 20.5 | 8 | 10.2 | 7.6 | 0.5 | 0.9 | 2.3 | 1.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marcondes, M.A.; Nascimento, A.; Pessôa, R.; Victor, J.R.; Duarte, A.J.d.S.; Clissa, P.B.; Sanabani, S.S. Characterization of Bacterial Communities from the Surface and Adjacent Bottom Layers of Water in the Billings Reservoir. Life 2022, 12, 1280. https://doi.org/10.3390/life12081280
Marcondes MA, Nascimento A, Pessôa R, Victor JR, Duarte AJdS, Clissa PB, Sanabani SS. Characterization of Bacterial Communities from the Surface and Adjacent Bottom Layers of Water in the Billings Reservoir. Life. 2022; 12(8):1280. https://doi.org/10.3390/life12081280
Chicago/Turabian StyleMarcondes, Marta Angela, Andrezza Nascimento, Rodrigo Pessôa, Jefferson Russo Victor, Alberto José da Silva Duarte, Patricia Bianca Clissa, and Sabri Saeed Sanabani. 2022. "Characterization of Bacterial Communities from the Surface and Adjacent Bottom Layers of Water in the Billings Reservoir" Life 12, no. 8: 1280. https://doi.org/10.3390/life12081280