
New Potential Immune Biomarkers in the Era of Precision Medicine: Lights and Shadows in Colorectal Cancer

 , ,
, ,
Abstract
:1. Introduction
2. Oncogene Driver Mutations and Therapeutic Implications in CRC
2.1. RAS Mutations
2.2. BRAF Mutations
2.3. ERBB2 Alterations
3. Long-Standing and Emerging Immune Biomarkers in CRC
3.1. Microsatellite Instability/Mismatch Repair Status
3.2. Tumor Mutational Burden
3.3. CD279 (PD-1)/CD274 (PD-L1) Expression
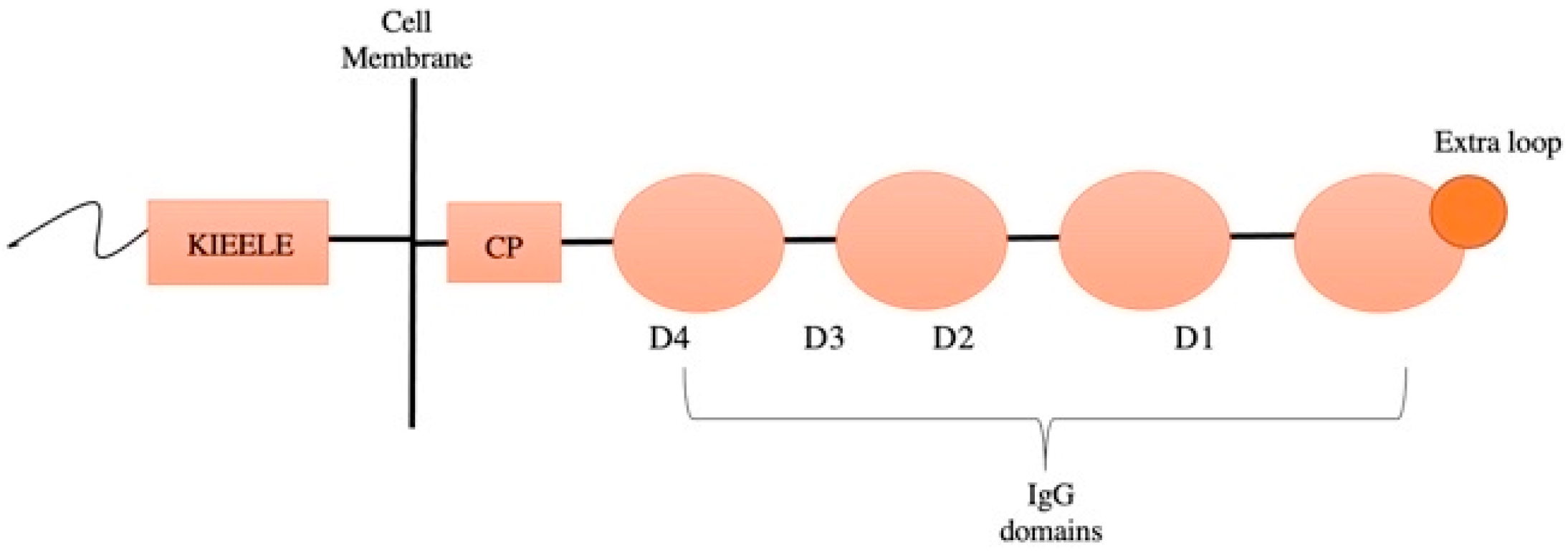
3.4. Lymphocyte-Activation Gene 3 (LAG3)
4. Synergistic Immunotherapy Combinations in CRC
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Goodwin, R.A.; Asmis, T.R. Overview of Systemic Therapy for Colorectal Cancer. Clin. Colon Rectal Surg. 2009, 22, 251–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popat, S.; Hubner, R.; Houlston, R.S. Systematic Review of Microsatellite Instability and Colorectal Cancer Prognosis. J. Clin. Oncol. 2005, 23, 609–618. [Google Scholar] [CrossRef]
- Overman, M.J.; Ernstoff, M.S.; Morse, M.A. Where we stand with immunotherapy in colorectal cancer: Deficient mismatch repair, proficient mismatch repair, and toxicity management. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef] [Green Version]
- Cristescu, R.; Mogg, R.; Ayers, M.; Albright, A.; Murphy, E.; Yearley, J.; Sher, X.; Liu, X.Q.; Lu, H.; Nebozhyn, M.; et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018, 362, eaar3593. [Google Scholar] [CrossRef] [Green Version]
- Addeo, A.; Friedlaender, A.; Banna, G.L.; Weiss, G.J. TMB or not TMB as a biomarker: That is the question. Crit. Rev. Oncol. Hematol. 2021, 163, 103374. [Google Scholar] [CrossRef]
- Prasad, V.; Addeo, A. The FDA approval of pembrolizumab for patients with TMB >10 mut/Mb: Was it a wise decision? No. Ann. Oncol. 2020, 31, 1112–1114. [Google Scholar] [CrossRef] [PubMed]
- Marcus, L.; Fashoyin-Aje, L.A.; Donoghue, M.; Mengdie Yuan, L.R. Pembrolizumab for the treatment of tumor mutational burden-high solid tumors. Clin. Cancer Res. 2021, 27, 4685–4689. [Google Scholar] [CrossRef]
- Wang, F.; Wei, X.L.; Wang, F.H.; Xu, N.; Shen, L.; Dai, G.H.; Yuan, X.L.; Chen, Y.; Yang, S.J.; Shi, J.H.; et al. Safety, efficacy and tumor mutational burden as a biomarker of overall survival benefit in chemo-refractory gastric cancer treated with toripalimab, a PD-1 antibody in phase Ib/II clinical trial NCT02915432. Ann. Oncol. 2019, 30, 1479–1486. [Google Scholar] [CrossRef] [Green Version]
- Ruffo, E.; Wu, R.C.; Bruno, T.C.; Workman, C.J.; Vignalia, D.A.A. Lymphocyte-activation gene 3 (LAG3): The next immune check- point receptor. Semin. Immunol. 2019, 42, 101305. [Google Scholar] [CrossRef]
- Zelba, H.; Bedke, J.; Hennenlotter, J.; Mostböck, S.; Zettl, M.; Zichner, T.; Chandran, A.; Stenzl, A.; Rammensee, H.-G.; Gouttefangeas, C. PD-1 and LAG-3 dominate checkpoint receptor-mediated T-cell inhibition in renal cell carcinoma. Cancer Immunol. Res. 2019, 7, 1891–1899. [Google Scholar] [CrossRef]
- Whitehair, R.; Peres, L.C.; Mills, A.M. Expression of the immune checkpoints LAG-3 and PD-L1 in high-grade serous ovarian carcinoma: Relationship to tumor-associated lymphocytes and germline BRCA status. Int. J. Gynecol. Pathol. 2020, 39, 558–566. [Google Scholar] [CrossRef]
- He, Y.; Yu, H.; Rozeboom, L.; Rivard, C.J.; Ellison, K.; Dziadziuszko, R.; Suda, K.; Ren, S.; Wu, C.; Hou, L.; et al. LAG-3 protein expression in non-small cell lung cancer and its relationship with PD-1/PD-L1 and tumor-infiltrating lymphocytes. J. Thorac. Oncol. 2017, 12, 814–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Liu, Y.D.; Luo, Y.L.; Liu, B.; Huang, Q.; Wang, F.; Zhong, Q. Prognostic value of lymphocyte activation gene-3 (LAG-3) expression in esophageal squamous cell carcinoma. J. Cancer 2018, 9, 4287–4293. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, J.; Gnjatic, S.; Mhawech-Fauceglia, P.; Beck, A.; Miller, A.; Tsuji, T.; Eppolito, C.; Qian, F.; Lele, S.; Shrikant, P.; et al. Tumor-infiltrating NY-ESO-1- specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 7875–7880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burugu, S.; Gao, D.; Leung, S.; Chia, S.K.; Nielsen, T.O. LAG-3+ tumor infiltrating lymphocytes in breast cancer: Clinical correlates and association with PD-1/PD-L1+ tumors. Ann. Oncol. 2017, 28, 2977–2984. [Google Scholar] [CrossRef]
- Hald, S.M.; Rakaee, M.; Martinez, I.; Richardsen, E.; Al-Saad, S.; Paulsen, E.; Blix, E.S.; Kilvaer, T.; Andersen, S.; Busund, L.T.; et al. LAG-3 in Non-Small-cell Lung Cancer: Expression in Primary Tumors and Metastatic Lymph Nodes Is Associated with Improved Survival. Clin. Lung Cancer 2018, 19, 249–259. [Google Scholar] [CrossRef]
- Han, C.B.; Li, F.; Ma, J.T.; Zou, H.W. Concordant KRAS mutations in primary and metastatic colorectal cancer tissue specimens: A meta-analysis and systematic review. Cancer Investig. 2012, 30, 741–747. [Google Scholar] [CrossRef]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab–FOLFOX4 Treatment and RAS Mutations in Colorectal Cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- Van Cutsem, E.; Lenz, H.J.; Köhne, C.H.; Heinemann, V.; Tejpar, S.; Melezínek, I.; Beier, F.; Stroh, C.; Rougier, P.; van Krieken, J.H.; et al. Fluorouracil, Leucovorin, and Irinotecan Plus Cetuximab treatment and RAS mutations in colorectal cancer. J. Clin. Oncol. 2015, 33, 692–700. [Google Scholar] [CrossRef] [Green Version]
- Bokemeyer, C.; Kohne, C.H.; Ciardiello, F.; Lenz, H.-J.; Heinemann, V.; Klinkhardt, U.; Beier, F.; Duecker, K.; van Krieken, J.H.; Tejpar, S. FOLFOX4 plus cetuximab treatment and RAS mutations in colorectal cancer. Eur. J. Cancer 2015, 51, 1243–1252. [Google Scholar] [CrossRef]
- Yokota, T. Are KRAS/BRAF mutations potent prognostic and/or predictive biomarkers in colorectal cancers? Anticancer Agents Med. Chem. 2012, 12, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Schirripa, M.; Cremolini, C.; Loupakis, F.; Morvillo, M.; Bergamo, F.; Zoratto, F.; Salvatore, L.; Antoniotti, C.; Marmorino, F.; Sensi, E.; et al. Role of NRAS mutations as prognostic and predictive markers in metastatic colorectal cancer. Int. J. Cancer 2015, 136, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Loupakis, F.; Cremolini, C.; Masi, G.; Lonardi, S.; Zagonel, V.; Salvatore, L.; Cortesi, E.; Tomasello, G.; Ronzoni, M.; Spadi, R.; et al. Initial Therapy with FOLFOXIRI and Bevacizumab for Metastatic Colorectal Cancer. N. Engl. J. Med. 2014, 371, 1609–1618. [Google Scholar] [CrossRef] [Green Version]
- Verdaguer, H.; Tabernero, J.; Macarulla, T. Ramucirumab in metastatic colorectal cancer: Evidence to date and place in therapy. Ther. Adv. Med. Oncol. 2016, 8, 230–242. [Google Scholar] [CrossRef] [Green Version]
- Syed, Y.Y.; McKeage, K. Aflibercept: A Review in Metastatic Colorectal Cancer. Drugs 2015, 75, 1435–1445. [Google Scholar] [CrossRef]
- Grothey, A.; Van Cutsem, E.; Sobrero, A.; Siena, S.; Falcone, A.; Ychou, M.; Humblet, Y.; Bouché, O.; Mineur, L.; Barone, C.; et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet 2013, 381, 303–312. [Google Scholar] [CrossRef]
- Henry, J.T.; Coker, O.; Chowdhury, S.; Shen, J.P.; Morris, V.K.; Dasari, A.; Raghav, K.; Nusrat, M.; Kee, B.; Parseghian, C.; et al. Comprehensive clinical and molecular characterization of KRAS G12C-mutant colorectal cancer. JCO Precis Oncol. 2021, 5, 613–621. [Google Scholar] [CrossRef]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [Green Version]
- Hong, D.S.; Fakih, M.G.; Strickler, J.H.; Desai, J.; Durm, G.A.; Shapiro, G.I.; Falchook, G.S.; Price, T.J.; Sacher, A.; Denlinger, C.S.; et al. KRAS G12C Inhibition with Sotorasib in advanced solid tumors. N. Engl. J. Med. 2020, 383, 1207–1217. [Google Scholar] [CrossRef]
- Fakih, M.G.; Kopetz, S.; Kuboki, Y.; Kim, T.W.; Munster, P.N.; Krauss, J.C.; Falchook, G.S.; Han, S.; Heinemann, V.; Muro, K.; et al. Sotorasib for previously treated colorectal cancers with KRASG12C Mutation (CodeBreaK100): A prespecified analysis of a single-arm, phase 2 trial. Lancet Oncol. 2022, 23, 115–124. [Google Scholar] [CrossRef]
- Johnson, M.L.; Ou, S.H.I.; Barve, M.; Rybkin, I.I.; Papadopoulos, K.P.; Leal, T.A.; Velastegui, K.; Christensen, J.G.; Kheoh, T.; Chao, R.C.; et al. KRYSTAL-1: Activity and Safety of Adagrasib (MRTX849) in Patients with Colorectal Cancer (CRC) and Other Solid Tumors Harboring a KRAS G12C Mutation. Eur. J. Cancer 2020, 138, S2. [Google Scholar] [CrossRef]
- Amodio, V.; Yaeger, R.; Arcella, P.; Cancelliere, C.; Lamba, S.; Lorenzato, A.; Arena, S.; Montone, M.; Mussolin, B.; Bian, Y.; et al. EGFR blockade reverts resistance to KRAS G12C inhibition in colorectal cancer. Cancer Discov. 2020, 10, 1129–1139. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.; Yaeger, R.D.; Johnson, M.L.; Spira, A.; Klempner, S.J.; Barve, M.A.; Christensen, J.G.; Chi, A.; Der-Torossian, H.; Velastegui, K.; et al. LBA6 KRYSTAL-1: Adagrasib (MRTX849) as Monotherapy or Combined with Cetuximab (Cetux) in Patients (Pts) with Colorectal Cancer (CRC) Harboring a KRASG12C Mutation. Ann. Oncol. 2021, 32, S1294. [Google Scholar] [CrossRef]
- Fakih, M.; Falchook, G.S.; Hong, D.S.; Yaeger, R.D.; Chan, E.; Mather, O.; Cardona, P.; Dai, T.; Strickler, J. CodeBreaK 101 Subprotocol H: Phase Ib Study Evaluating Combination of Sotorasib (Soto), a KRASG12C Inhibitor, and Panitumumab (PMab), an EGFR Inhibitor, in Advanced KRAS p.G12C-Mutated Colorectal Cancer (CRC). Ann. Oncol. 2021, 32, S551. [Google Scholar] [CrossRef]
- Tabernero, J.; Bendell, J.; Corcoran, R.; Kopetz, S.; Lee, J.; Davis, M.; Christensen, J.; Chi, A.; Kheoh, T.; Yaeger, R.; et al. KRYSTAL-10: A randomized phase 3 study of adagrasib (MRTX849) in combination with cetuximab vs chemotherapy in patients with previously treated advanced colorectal cancer with KRASG12C mutation. Ann. Oncol. 2021, 32, S121. [Google Scholar] [CrossRef]
- Pietrantonio, F.; Petrelli, F.; Coinu, A.; di Bartolomeo, M.; Borgonovo, K.; Maggi, C.; Cabiddu, M.; Iacovelli, R.; Bossi, I.; Lonat, V.; et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: A meta-analysis. Eur. J. Cancer 2015, 51, 587–594. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.; Dias, M.M.; Wiese, M.D.; Kichenadasse, G.; McKinnon, R.A.; Karapetis, C.S.; Sorich, M.J. Meta-analysis of BRAF mutation as a predictive biomarker of benefit from anti-EGFR monoclonal antibody therapy for RAS wild-type metastatic colorectal cancer. Br. J. Cancer 2015, 112, 1888–1894. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, R.B.; Ebi, H.; Turke, A.B.; Coffee, E.M.; Nishino, M.; Cogdill, A.P.; Brown, R.D.; della Pelle, P.; Dias-Santagata, D.; Hung, K.E.; et al. EGFR-mediated reactivation of MAPK signaling contributes to insensitivity of BRAF-mutant colorectal cancers to RAF inhibition with vem.murafenib. Cancer Discov. 2012, 2, 227–235. [Google Scholar] [CrossRef] [Green Version]
- Prahallad, A.; Sun, C.; Huang, S.; di Nicolantonio, F.; Salazar, R.; Zecchin, D.; Beijersbergen, R.L.; Bardelli, A.; Bernards, R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 2012, 483, 100–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atreya, C.E.; Van Cutsem, E.; Bendell, J.C.; Andre, T.; Schellens, J.H.; Gordon, M.S.; McRee, A.J.; O’Dwyer, P.J.; Muro, K.; Tabernero, J.; et al. Updated efficacy of the MEK inhibitor trametinib (T), BRAF inhibitor dab- rafenib (D), and anti-EGFR antibody panitumumab (P) in patients (pts) with BRAF V600E mutated (BRAFm) metastatic colorectal cancer (mCRC). J. Clin. Oncol. 2015, 33, 103. [Google Scholar] [CrossRef]
- Van Geel, R.M.J.M.; Tabernero, J.; Elez, E.; Bendell, J.C.; Spreafico, A.; Schuler, M.; Yoshino, T.; Delord, J.; Yamada, Y.; Lolkema, M.P.; et al. A phase Ib dose-escalation study of encorafenib and cetuximab with or without alpelisib in metastatic BRAF-mutant colorectal cancer. Cancer Discov. 2017, 7, 610–619. [Google Scholar] [CrossRef] [Green Version]
- Tabernero, J.; Grothey, A.; Van Cutsem, E.; Yaeger, R.; Wasan, H.; Yoshino, T.; Desai, J.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib plus Cetuximab as a new standard of care for previously treated BRAF V600E–mutant metastatic colorectal cancer: Updated survival results and subgroup analyses from the BEACON Study. J. Clin. Oncol. 2021, 39, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Taieb, J.; Yaeger, R.; Yoshino, T.; Maiello, E.; Elez, E.; Dekervel, J.; Ross, P.; Ruiz Casado, A.; Graham, J.; et al. ANCHOR CRC: Results from a single-arm, phase 2 study of encorafenib, binimetinib plus cetuximab in previously untreated BRAF V600E mutant metastatic colorectal cancer. Ann. Oncol. 2021, 32, S222. [Google Scholar] [CrossRef]
- André, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Carci-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in microsatellite instability-high advanced colorectal cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef] [PubMed]
- Morris, V.K.; Perseghian, C.M.; Eschano, M.; Johnson, B.; Raghav, K.P.S.; Dasari, A.; Huey, R.; Overman, M.J.; Willis, J.; Lee, M.S.; et al. Phase I/II trial of encorafenib, cetuximab, and nivolumab in patients with microsatellite stable, BRAFV600E metastatic colorectal cancer. J. Clin. Oncol. 2022, 40, 12. [Google Scholar] [CrossRef]
- Taieb, J.; Lapeyre-Prost, A.; Laurent Puig, P.; Zaanan, A. Exploring the best treatment options for BRAF-mutant metastatic colon cancer. Br. J. Cancer 2019, 121, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Mauri, G.; Bonazzina, E.; Amatu, A.; Tosi, F.; Bencardino, K.; Gori, V.; Massihnia, D.; Cipani, T.; Spina, F.; Ghezzi, S.; et al. The Evolutionary landscape of treatment for BRAFV600E mutant metastatic colorectal cancer. Cancers 2021, 13, 137. [Google Scholar] [CrossRef]
- Valtorta, E.; Martino, C.; Sartore-Bianchi, A.; Penaullt-Llorca, F.; Viale, G.; Risio, M.; Rugge, M.; Grigioni, W.; Bencardino, K.; Lonardi, S.; et al. Assessment of a HER2 scoring system for colorectal cancer: Results from a validation study. Mod. Pathol. 2015, 28, 1481–1491. [Google Scholar] [CrossRef] [Green Version]
- Bregni, G.; Sciallero, S.; Sobrero, A. HER2 amplification and anti-EGFR sensitivity in advanced colorectal cancer. JAMA Oncol. 2019, 5, 605. [Google Scholar] [CrossRef] [PubMed]
- Pietrantonio, F.; Vernieri, C.; Siravegna, G.; Mennitto, A.; Berenato, R.; Perrone, F.; Gloghini, A.; Tamborini, E.; Lonardi, S.; Morano, F.; et al. Heterogeneity of acquired resistance to anti-EGFR monoclonal antibodies in patients with metastatic colorectal cancer. Clin. Cancer Res. 2017, 23, 2414–2422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghav, K.P.S.; Overman, M.J.; Yu, R.; Meric-Bernstam, F.; Menter, D.; Kee, B.K.; Muranyi, A.; Singh, S.; Routbort, M.; Chen, K.; et al. HER2 amplification as a negative predictive biomarker for anti-epidermal growth factor receptor antibody therapy in metastatic colorectal cancer. J. Clin. Oncol. 2016, 34, 3517. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-targeted therapy with trastuzumab and lapatinib in treatment-refractory, KRAS codon 12/13 wild-type, HER2-positive metastatic colorectal cancer (HERACLES): A proof-of-concept, multicentre, open-label, 62. phase 2 trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Hurwitz, H.; Raghav, K.P.S.; McWilliams, R.R.; Fakih, M.; VanderWalde, A.; Swanton, C.; Kurzrock, R.; Burris, H.; Sweeney, C.; et al. Pertuzumab plus trastuzumab for HER2-Amplified Metastatic Colorectal Cancer (MyPathway): An updated report from a multicentre, open-label, Phase 2a, Multiple Basket Study. Lancet Oncol. 2019, 20, 518–530. [Google Scholar] [CrossRef]
- Siena, S.; Di Bartolomeo, M.; Raghav, K.; Masuishi, T.; Loupakis, F.; Kawakami, H.; Yamaguchi, K.; Nishina, T.; Fakih, M.; Elez, E.; et al. Trastuzumab deruxtecan (DS-8201) in Patients with HER2-expressing metastatic colorectal cancer (DESTINY-CRC01): A multicentre, open-Label, Phase 2 Trial. Lancet Oncol. 2021, 22, 779–789. [Google Scholar] [CrossRef]
- Strickler, J.H.; Ng, K.; Cercek, A.; Fountzilas, C.; Sanchez, F.A.; Hubbard, J.M.; Wu, C.; Siena, S.; Tabernero, J.; Van Cutsem, E.; et al. MOUNTAINEER: Open-Label, Phase II Study of Tucatinib Combined with Trastuzumab for HER2-Positive Metastatic Colorectal Cancer (SGNTUC-017, Trial in Progress). J. Clin. Oncol. 2021, 39, TPS153. [Google Scholar] [CrossRef]
- Ma, J.; Setton, J.; Lee, N.Y.; Riaz, N.; Powell, S.N. The therapeutic significance of mutational signatures from DNA repair deficiency in cancer. Nat. Commun. 2018, 9, 3292. [Google Scholar] [CrossRef]
- Boland, C.R.; Goel, A. Microsatellite Instability in Colorectal Cancer. Gastroenterology 2010, 138, 2073–2087. [Google Scholar] [CrossRef]
- Dolcetti, R.; Viel, A.; Doglioni, C.; Russo, A.; Guidoboni, M.; Capozzi, E.; Vecchiato, N.; Macrì, E.; Fornasarig, M.; Boiocchi, M.; et al. High prevalence of activated intraepithelial cytotoxic T lymphocytes and increased neoplastic cell apoptosis in colorectal carcinomas with microsatellite instability. Am. J. Pathol. 1999, 154, 1805–1813. [Google Scholar] [CrossRef] [Green Version]
- Smyrk, T.C.; Watson, P.; Kaul, K.; Lynch, H.T. Tumor-infiltrating lymphocytes are a marker for microsatellite instability in colorectal carcinoma. Cancer 2001, 91, 2417–2422. [Google Scholar] [CrossRef]
- Roth, A.D.; Delorenzi, M.; Tejpar, S. Integrated analysis of molecular and clinical prognostic factors in stage II/III colon cancer. J. Natl. Cancer Inst. 2012, 104, 1635–1646. [Google Scholar] [CrossRef] [Green Version]
- Mohan, H.M.; Ryan, E.; Balasubramanian, I.; Kennelly, R.; Geraghty, R.; Sclafani, F.; Fennelly, D.; McDermott, R.; Ryan, E.J.; O’Donoghue, D.; et al. Microsatellite instability is associated with reduced disease specific survival in stage III colon cancer. Eur. J. Surg. Oncol. 2016, 42, 1680–1686. [Google Scholar] [CrossRef]
- Marcus, L.; Lemery, S.J.; Keegan, P.; Pazdur, R. FDA approval summary: Pembrolizumab for the treatment of microsatellite instability-high solid tumors. Clin. Cancer Res. 2019, 25, 3753–3758. [Google Scholar] [CrossRef] [Green Version]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Skora, A.E.; Azad, N.S.; Laheru, D.A.; Donehower, R.C.; Luber, B.; et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, D.; Kavan, P.; Kim, T.; Burge, M.; van Cutsem, E.; Hara, H.; Boland, P.; van Laethem, J.; Geva, R.; Taniguchi, H.; et al. Safety and antitumor activity of pembrolizumab in patients with advanced microsatellite instability–high (MSI-H) colorectal cancer: KEYNOT.TE-164. Ann. Oncol. 2018, 29, v107. [Google Scholar] [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Andrè, T.; Lonardi, S.; Wong, M.; Lenz, H.; Gelsomino, F.; Aglietta, M.; Morse, M.; van Cutsem, E.; McDermott, R.S.; Hill, A.G.; et al. Nivolumab + ipilimumab combination in patients with DNA mismatch repair-defi-cient/microsatellite instability-high (dMMR/MSI-H) metastatic colorectal cancer (mCRC): First report of the full cohort from CheckMate-142. J. Clin. Oncol. 2018, 36, 553. [Google Scholar] [CrossRef]
- Andrè, T.; Berton, D.; Curigliano, G.; Ellard, S.; Pérez, J.M.T.; Arkenau, H.; Abdeddaim, C.; Moreno, V.; Guo, W.; Im, E.; et al. Safety and Efficacy of Anti–PD-1 Antibody Dostarlimab in patients (Pts) with Mismatch Repair-Deficient (DMMR) solid cancers: Results from GARNET study. J. Clin. Oncol. 2021, 39. [Google Scholar] [CrossRef]
- Cercek, A.; Lumish, M.; Sinopoli, J.; Weiss, J.; Shia, J.; Lamendola-Essel, M.; el Dika, I.H.; Segal, N.; Shcherba, M.; Sugarman, R.; et al. PD-1 Blockade in Mismatch Repair–Deficient, Locally Advanced Rectal Cancer. N. Engl. J. Med. 2022, 386, 2363–2376. [Google Scholar] [CrossRef]
- Aldea, M.; Andre, F.; Marabelle, A.; Dogan, S.; Barlesi, F.; Soria, J.C. Overcoming resistance to tumor-targeted and immune-targeted therapies. Cancer Discov. 2021, 11, 874–899. [Google Scholar] [CrossRef]
- Doebele, R.C. Acquired resistance is oncogene and drug agnostic. Cancer Cell. 2019, 36, 347–349. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Duanmu, J.; Fu, X.; Li, T.; Jiang, Q. Analyzing and validating the prognostic value and mechanism of colon cancer immune microenvironment. J. Translational. Med. 2020, 18, 324. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kronbichler, A.; Eisenhut, M.; Hong, S.H.; van der Vliet, H.J.; Kang, J.; Shin, J.I.; Gamerith, G. Tumor Mutational Burden and Efficacy of Immune Checkpoint Inhibitors: A Systematic Review and Meta-Analysis. Cancers 2019, 11, 1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fancello, L.; Gandini, S.; Pelicci, P.G.; Mazzarella, L. Tumor mutational burden quantification from targeted gene panels: Major advancements and challenges. J. Immunother. Cancer 2019, 7, 183. [Google Scholar] [CrossRef] [Green Version]
- Yarchoan, M.; Albacker, L.A.; Hopkins, A.C.; Montesion, M.; Murugesan, K.; Vithayathil, T.T.; Zaidi, N.; Azad, N.S.; Laheru, D.A.; Frampton, G.M.; et al. PD.D-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight 2019, 4, e126908. [Google Scholar] [CrossRef] [Green Version]
- Schrock, A.B.; Ouyang, C.; Sandhu, J.; Sokol, E.; Jin, D.; Ross, J.S.; Miller, V.A.; Lim, D.; Amanam, I.; Chao, J.; et al. Tumor mutational burden is predictive of response to immune checkpoint inhibitors in MSI-high metastatic colorectal cancer. Ann. Oncol. 2019, 30, 1096–1103. [Google Scholar] [CrossRef]
- Herbst, R.S.; Lopes, G.; Kowalski, D.M.; Nishio, M.; Wu, Y.-L.; de Castro Junior, G.; Baas, P.; Kim, D.-W.; Gubens, M.A.; Cristescu, R.; et al. Association between Tissue TMB (TTMB) and Clinical Outcomes with Pembrolizumab Monotherapy (Pembro) in PD-L1-positive advanced NSCLC in the KEYNOTE-010 and -042 Trials. Ann. Oncol. 2019, 30, v916–v917. [Google Scholar] [CrossRef]
- Li, Y.; Ma, Y.; Wu, Z.; Zeng, F.; Song, B.; Zhang, Y.; Li, J.; Lui, S.; Wu, M. Tumor Mutational Burden Predicting the Efficacy of Immune Checkpoint Inhibitors in Colorectal Cancer: A Systematic Review and Meta-Analysis. Front. Immunol. 2021, 12, 751407. [Google Scholar] [CrossRef]
- Subbiah, V.; Solit, D.; Chan, T.; Kurzrock, R. The FDA approval of pembrolizumab for adult and pediatric patients with tumor mutational burden (TMB) ≥10: A decision centered on empowering patients and their physicians. Ann. Oncol. 2020, 31, 1115–1118. [Google Scholar] [CrossRef] [PubMed]
- McNamara, M.G.; Jacob, T.; Lamarca, A.; Hubner, R.A.; Valle, J.W.; Amir, E. Impact of high tumor mutational burden in solid tumors and challenges for biomarker application. Cancer Treat. Rev. 2020, 89, 102084. [Google Scholar] [CrossRef]
- Fabrizio, D.A.; George, T.J.; Dunne, R.F., Jr.; Frampton, G.; Sun, J.; Gowen, K.; Kennedy, M.; Greenbowe, J.; Schrock, A.B.; Hezel, A.F.; et al. Beyond microsatellite testing: Assessment of tumor mutational burden identifies subsets of colorectal cancer who may respond to immune checkpoint inhibition. J. Gastrointest. Oncol. 2018, 9, 610–617. [Google Scholar] [CrossRef]
- Xiao, J.; Li, W.; Huang, Y.; Huang, M.; Li, S.; Zhai, X.; Zhao, J.; Gao, C.; Xie, W.; Qin, H.; et al. A next-generation sequencing-based strategy combining microsatellite instability and tumor mutation burden for comprehensive molecular diagnosis of advanced colorectal cancer. BMC Cancer 2021, 21, 282. [Google Scholar] [CrossRef]
- Antoniotti, C.; Korn, W.M.; Marmorino, F.; Rossini, D.; Lonardi, S.; Masi, G.; Randon, G.; Conca, V.; Boccaccino, A.; Tomasello, G.; et al. Tumour mutational burden, microsatellite instability, and actionable alterations in metastatic colorectal cancer: Next-generation sequencing results of TRIBE2 study. Eur. J. Cancer 2018, 155, 73–84. [Google Scholar] [CrossRef]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magrì, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017, 552, 116–120. [Google Scholar] [CrossRef]
- Crisafulli, G.; Sartore-Bianchi, A.; Lazzari, L.; Pietrantonio, F.; Amatu, A.; Macagno, M.; Barault, L.; Cassingena, A.; Bartolini, A.; Luraghi, P.; et al. Temozolomide treatment alters mismatch repair and boosts mutational burden in tumor and blood of colorectal cancer patients. Cancer Discov. 2022, 12, 1656–1675. [Google Scholar] [CrossRef]
- Bortolomeazzi, M.; Keddar, M.R.; Montorsi, L.; Acha-Sagredo, A.; Benedetti, L.; Temelkovski, D.; Choi, S.; Petrov, N.; Todd, K.; Wai, P.; et al. Immunogenomics of Colorectal Cancer Response to Checkpoint Blockade: Analysis of the KEYNOTE 177 Trial and Validation Cohorts. Gastroenterology 2021, 161, 1179–1193. [Google Scholar] [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [Green Version]
- Flies, D.B.; Chen, L. Modulation of immune response by B7 family molecules in tumor microenvironments. Immunol. Investig. 2006, 35, 395–418. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, M.W.; Bledsoe, J.R.; Morales-Oyarvide, V.; Huynh, T.G.; Mino-Kenudson, M. PD-L1 expression in colorectal cancer is associated with microsatellite instability, BRAF mutation, medullary morphology and cytotoxic tumor-infiltrating lymphocytes. Mod. Pathol. 2016, 29, 1104–1112. [Google Scholar] [CrossRef] [PubMed]
- Inaguma, S.; Lasota, J.; Wang, Z.; Felisiak-Golabek, A.; Ikeda, H.; Miettinen, M. Clinicopathologic profile, immunophenotype, and genotype of CD274 (PD-L1)-positive colorectal carcinomas. Mod. Pathol. 2017, 30, 278–285. [Google Scholar] [CrossRef] [Green Version]
- de Guillebon, E.; Roussille, P.; Frouin, E.; Tougeron, D. Anti program death-1/anti program death-ligand 1 in digestive cancers. World J. Gastrointest. Oncol. 2015, 7, 95–101. [Google Scholar] [CrossRef]
- Wang, H.B.; Yao, H.; Li, C.S.; Liang, L.X.; Zhang, Y.; Chen, Y.X.; Fang, J.; Xu, J. Rise of PD-L1 expression during metastasis of colorectal cancer: Implications for immunotherapy. J. Dig. Dis. 2017, 18, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Bae, S.U.; Jeong, W.K.; Baek, S.K.; Kim, N.K.; Hwang, I. Prognostic impact of programmed cell death ligand 1 expression on long-term oncologic outcomes in colorectal cancer. Oncol. Lett. 2018, 16, 5214–5222. [Google Scholar] [CrossRef]
- Droeser, R.A.; Hirt, C.; Viehl, C.T.; Frey, D.M.; Nebiker, C.; Huber, X.; Zlobec, I.; Eppenberger-Castori, S.; Tzankov, A.; Rosso, R.; et al. Clinical impact of programmed cell death ligand 1 expression in colorectal cancer. Eur. J. Cancer 2013, 49, 2233–2242. [Google Scholar] [CrossRef] [PubMed]
- Enkhbat, T.; Nishi, M.; Takasu, C.; Yoshikawa, K.; Jun, H.; Tokunaga, T.; Kashihara, H.; Ishikawa, D.; Shimada, M. Programmed cell death ligand 1 expression is an independent prognostic factor in colorectal cancer. Anticancer Res. 2018, 38, 3367–3373. [Google Scholar] [CrossRef]
- Eriksen, A.C.; Sørensen, F.B.; Lindebjerg, J.; Hager, H.; Christensen, R.d.; Kjær-Frifeldt, S.; Hansen, T.F. Programmed death ligand-1 expression in stage II colon cancer-experiences from a nationwide populationbased cohort. BMC Cancer 2019, 19, 142. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Wu, D.; Li, L.; Chai, Y.; Huang, J. PD-L1 and survival in solid tumors: A meta-analysis. PLoS ONE 2015, 10, e0131403. [Google Scholar] [CrossRef]
- Shen, Z.; Gu, L.; Mao, D.; Chen, M.; Jin, R. Clinicopathological and prognostic significance of PD-L1 expression in colorectal cancer: A systematic review and meta-analysis. World J. Surg. Oncol. 2019, 17, 4. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Xue, R.; Pan, C. Prognostic and clinicopathological value of PD-L1 in colorectal cancer: A systematic review and meta-analysis. Onco Targets Ther. 2019, 12, 3671–3682. [Google Scholar] [CrossRef]
- Li, Y.; He, M.; Zhou, Y.; Yang, C.; Wei, S.; Bian, X.; Christopher, O.; Xie, L. The Prognostic and clinicopathological roles of PD-L1 expression in colorectal cancer: A systematic review and meta-analysis. Front. Pharmacol. 2019, 10, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valentini, A.M.; Di Pinto, F.; Cariola, F.; Guerra, V.; Giannelli, G.; Caruso, M.L.; Pirrelli, M. PD-L1 expression in colorectal cancer defines three subsets of tumor immune microenvironment. Oncotarget 2018, 9, 8584–8596. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.H.; Cavalcanti, M.S.; Segal, N.H.; Hechtman, J.F.; Weiser, M.R.; Smith, J.J.; Garcia-Aguilar, J.; Sadot, E.; Ntiamoah, P.; Markowitz, A.J.; et al. Patterns and prognostic relevance of PD-1 and PD-L1 expression in colorectal carcinoma. Mod. Pathol. 2016, 29, 1433–1442. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.S.; Kwak, Y.; Ahn, S.; Shin, E.; Oh, H.; Kim, D.; Kang, S.; Choe, G.; Kim, W.H.; Lee, H.S. Prognostic implication of CD274 (PD-L1) protein expression in tumor-infiltrating immune cells for microsatellite unstable and stable colorectal cancer. Cancer Immunol. Immunother. 2017, 66, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Yamada, R.; Yamaguchi, T.; Iijima, T.; Wakaume, R.; Takao, M.; Koizumi, K.; Hishima, T.; Horiguchi, S. Differences in histological features and PD-L1 expression between sporadic microsatellite instability and Lynch-syndrome-associated disease in Japanese patients with colorectal cancer. Int. J. Clin. Oncol. 2017, 23, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Long, L.; Zhang, X.; Chen, F.; Pan, Q.; Phiphatwatchara, P.; Zeng, Y.; Chen, H. The promising immune checkpoint LAG-3: From tumor microenvironment to cancer immunotherapy. Genes Cancer 2018, 9, 176–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Sanmamed, M.F.; Datar, I.; Su, T.T.; Ji, L.; Sun, J.; Chen, L.; Chen, Y.; Zhu, G.; Yin, W.; et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell 2019, 176, 334–347. [Google Scholar] [CrossRef] [Green Version]
- Maruhashi, T.; Sugiura, D.; Okazaki, I.M.; Okazaki, T. LAG-3: From Molecular Functions to Clinical Applications. J. Immunother. Cancer 2020, 8, 1–9. [Google Scholar] [CrossRef]
- Hannier, S.; Tournier, M.; Bismuth, G.; Triebel, F. CD3/TCR Complex-Associated Lymphocyte Activation Gene-3 Molecules Inhibit CD3/TCR Signaling. J. Immunol. 1998, 161, 4058–4065. [Google Scholar]
- Lythgoe, M.P.; Liu, D.S.K.; Annels, N.E.; Krell, J.; Frampton, A.E. Gene of the Month: Lymphocyte-Activation Gene 3 (LAG-3). J. Clin. Pathol. 2021, 74, 543–547. [Google Scholar] [CrossRef]
- Shi, A.P.; Tang, X.Y.; Xiong, Y.L.; Zheng, K.; Liu, Y.; Liu, Y.; Lv, Y.; Jiang, T.; Ma, N.; Zhao, J. Immune Checkpoint LAG3 and Its Ligand FGL1 in Cancer. Front. Immunol. 2022, 12, 785091. [Google Scholar] [CrossRef]
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A. LAG3 (CD223) as a Cancer Immunotherapy Target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef]
- Woo, S.R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Badran, S.S.; Grant, L.; Campo, M.V.; Inthagard, J.; Pennel, K.; Quinn, J.; Konanahalli, P.; Hayman, L.; Horgan, P.G.; McMillan, D.C.; et al. Relationship between immune checkpoint proteins, tumour microenvironment characteristics, and prognosis in primary operable colorectal cancer. J. Pathol. Clin. Res. 2021, 7, 121–134. [Google Scholar]
- Chen, B.; Khodadoust, M.S.; Liu, C.L.; Newman, A.M.; Alizadeh, A.A. Profiling Tumor Infiltrating Immune Cells with CIBERSORT. Methods Mol. Biol. 2018, 1711, 243–259. [Google Scholar] [PubMed]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar]
- Zhou, H.; Liu, Z.; Wang, Y.; Wen, X.; Amador, E.H.; Yuan, L.; Ran, X.; Ran, L.X.Y.; Chen, W.; Wen, Y. Colorectal liver metastasis: Molecular mechanism and interventional therapy. Signal. Transduct. Target. Therapy 2022, 7, 1. [Google Scholar] [CrossRef]
- Grosso, J.F.; Kelleher, C.C.; Harris, T.J.; Maris, C.H.; Hipkiss, E.L.; De Marzo, A.; Anders, R.; Netto, G.; Getnet, D.; Bruno, T.C.; et al. LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J. Clin. Investig. 2007, 117, 3383–3392. [Google Scholar] [CrossRef] [Green Version]
- Hamanishi, J.; Mandai, M.; Iwasaki, M.; Okazaki, T.; Tanaka, Y.; Yamaguchi, K.; Higuchi, T.; Yagi, H.; Takakura, K.; Minato, N.; et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 3360–3365. [Google Scholar] [CrossRef] [Green Version]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Gutiérrez, E.C.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; de Menezes, J.J.; et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N. Engl. J. Med. 2022, 386, 24–34. [Google Scholar] [CrossRef]
- Study of Nivolumab and Relatlimab in Patients with Microsatellite Stable (MSS) Advanced Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03642067?term=NCT03642067&draw=2&rank=1 (accessed on 29 June 2022).
- A Study of Coformulated Favezelimab/Pembrolizumab (MK-4280A) Versus Standard of Care in Subjects with Previously Treated Metastatic PD-L1 Positive Colorectal Cancer (MK-4280A-007). Available online: https://clinicaltrials.gov/ct2/show/NCT05064059?term=NCT05064059&draw=2&rank=1 (accessed on 29 June 2022).
- Nivolumab and Ipilimumab in Patients with dMMR and/or MSI Metastatic Colorectal Cancer Resistant to Anti-PD1 Monotherapy. Available online: https://clinicaltrials.gov/ct2/show/NCT05310643?term=NCT05310643&draw=2&rank=1 (accessed on 29 June 2022).
- Envafolimab as Neoadjuvant Immuntherapy in Resectable Local Advanced dMMR/MSI-H Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT05371197?term=NCT05371197&draw=2&rank=1 (accessed on 29 June 2022).
- Atezolizumab with/without IMM-101 in Patients with MSI-h/MMR-D Stage III Colorectal Cancer Ineligible for Oxaliplatin. Available online: https://clinicaltrials.gov/ct2/show/NCT05118724?term=NCT05118724&draw=2&rank=1 (accessed on 29 June 2022).
- Evaluation of Co-formulated Pembrolizumab/Quavonlimab (MK-1308A) Versus Other Treatments in Participants with Microsatellite Instability-High (MSI-H) or Mismatch Repair Deficient (dMMR) Stage IV Colorectal Cancer (CRC) (MK-1308A-008). Available online: https://clinicaltrials.gov/ct2/show/NCT04895722?term=NCT04895722&draw=2&rank=1 (accessed on 29 June 2022).



{kind=link}
{kind=link}
| LAG-3 on Tumor Cells | LAG-3 on Stromal Immune Cells | |||
|---|---|---|---|---|
| N° | Survival | N° | Survival | |
| Low | 253 | 73% | 196 | 63% |
| High | 160 | 65% | 191 | 78% |
| Trial | Phases | Setting/Line | Drugs | Biomarker | End Points |
|---|---|---|---|---|---|
| NCT03642067 [123] | II | Stage IV ≥2 | Nivolumab Relatimab | MSS PD-L1/Mucin (CPM) score ≥ 15% or <15% | Primary: ORR Secondary: AEs |
| NCT05064059 [124] | III | Stage IV ≥2 | Favezelimab/ pembrolizumab vs. Regorafenib TAS 102 | MSI-H/dMMR | Primary: OS Secondary: PFS, ORR, DoR, AEs, TTD |
| NCT05310643 [125] | II | Stage IV 1 and ≥2 | Nivolumab Ipilimumab | MSI-H/dMMR | Primary: ORR Secondary: AEs, DCR, PFS, OS, ctDNA |
| NCT05371197 [126] | II | Stage III Neoadjuvant | Envafolimab | MSI-H/dMMR | Primary: pCR Secondary: DFS, OS, drug safety and feasibility |
| NCT05118724 [127] | II | Stage III (ineligible for oxaliplatin) Adjuvant | Atezolizumab ± IMM-101 | MSI-H/dMMR | Primary: 3 y DFS Secondary: 1, 2, 5 y DFS and OS |
| NCT04895722 [128] | II | Stage IV 1 and ≥2 | Pembrolizumab Quavonlimab Favezelimab Vibostolimab MK-4830 | MSI-H/dMMR | Primary: ORR Secondary: DoR, PFS, OS, AEs |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Damato, A.; Rotolo, M.; Caputo, F.; Borghi, E.; Iachetta, F.; Pinto, C. New Potential Immune Biomarkers in the Era of Precision Medicine: Lights and Shadows in Colorectal Cancer. Life 2022, 12, 1137. https://doi.org/10.3390/life12081137
Damato A, Rotolo M, Caputo F, Borghi E, Iachetta F, Pinto C. New Potential Immune Biomarkers in the Era of Precision Medicine: Lights and Shadows in Colorectal Cancer. Life. 2022; 12(8):1137. https://doi.org/10.3390/life12081137
Chicago/Turabian StyleDamato, Angela, Martina Rotolo, Francesco Caputo, Eleonora Borghi, Francesco Iachetta, and Carmine Pinto. 2022. "New Potential Immune Biomarkers in the Era of Precision Medicine: Lights and Shadows in Colorectal Cancer" Life 12, no. 8: 1137. https://doi.org/10.3390/life12081137