
Cancer Cachexia and Antitumor Immunity: Common Mediators and Potential Targets for New Therapies

 , , ,
, , ,
Abstract
:1. Background
2. Common Mediators of Cachexia and the Cancer-Immunity Cycle
2.1. Tumor Necrosis Factor Alpha (TNF-α)
2.2. TNF-Related Weak Inducer of Apoptosis (TWEAK)
2.3. IL-1α and IL-1β
2.4. IL-6
2.5. IL-8
3. Transforming Growth Factor Beta (TGF-β) Family
3.1. Activin
3.2. TGF-β
3.3. Growth Differentiation Factor 15 (GDF15)
4. MDSCS, Cancer Associated Fibroblasts and CCS
4.1. Myeloid Derived Suppressor Cells (MDSCs)
4.2. Cancer Associated Fibroblasts (CAFs)
5. P-Selectin and Cachexia
6. CCS, Autophagy, and Immune Response
7. Cancer Cachexia and Response to Immunotherapy
8. Conclusions
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Primers. 2018, 18, 17105. [Google Scholar] [CrossRef] [PubMed]
- Shachar, S.S.; Williams, G.R.; Muss, H.B.; Nishijima, T.F. Prognostic value of sarcopenia in adults with solid tumours: A meta-analysis and systematic review. Eur. J. Cancer 2016, 57, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.C.H.; Glass, D.J.; Guttridge, D.C. Cancer cachexia: Mediators, signaling, and metabolic pathways. Cell Metab. 2012, 16, 153–166. [Google Scholar] [CrossRef] [PubMed]
- De Matos-Neto, E.M.; Lima, J.D.C.C.; de Pereira, W.O.; Figuerêdo, R.G.; Riccardi, D.M.D.R.; Radloff, K.; Rodrigo, X.; das Neves, R.; Camargo, G.; Linda, F.; et al. Systemic Inflammation in Cachexia—Is Tumor Cytokine Expression Profile the Culprit? Front. Immunol. 2015, 6, 629. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Turner, D.C.; Kondic, A.G.; Anderson, K.M.; Robinson, A.G.; Garon, E.B.; Riess, J.W.; Jain, L.; Mayawala, K.; Kang, J.; Ebbinghaus, S.W.; et al. Pembrolizumab Exposure-Response Assessments Challenged by Association of Cancer Cachexia and Catabolic Clearance. Clin. Cancer Res. 2018, 24, 5841–5849. [Google Scholar] [CrossRef]
- Naik, G.S.; Waikar, S.S.; Johnson, A.E.W.; Buchbinder, E.I.; Haq, R.; Hodi, F.S.; Schoenfeld, J.D.; Ott, P.A. Complex inter-relationship of body mass index, gender and serum creatinine on survival: Exploring the obesity paradox in melanoma patients treated with checkpoint inhibition. J. Immunother. Cancer 2019, 7, 89. [Google Scholar] [CrossRef]
- Rounis, K.; Makrakis, D.; Tsigkas, A.P.; Georgiou, A.; Galanakis, N.; Papadaki, C.; Monastirioti, A.; Vamvakas, L.; Kalbakis, K.; Vardakis, N.; et al. Cancer cachexia syndrome and clinical outcome in patients with metastatic non-small cell lung cancer treated with PD-1/PD-L1 inhibitors: Results from a prospective, observational study. Transl. Lung Cancer Res. 2021, 10, 3538–3549. [Google Scholar] [CrossRef]
- Tracey, K.J.; Lowry, S.F.; Cerami, A. Cachectin: A hormone that triggers acute shock and chronic cachexia. J. Infect. Dis. 1988, 157, 413–420. [Google Scholar] [CrossRef]
- Siddiqui, J.A.; Pothuraju, R.; Jain, M.; Batra, S.K.; Nasser, M.W. Advances in cancer cachexia: Intersection between affected organs, mediators, and pharmacological interventions. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188359. [Google Scholar] [CrossRef]
- Peyta, L.; Jarnouen, K.; Pinault, M.; Coulouarn, C.; Guimaraes, C.; Goupille, C.; de Barros, J.P.P.; Chevalier, S.; Dumas, J.F.; Maillot, F.; et al. Regulation of hepatic cardiolipin metabolism by TNFα: Implication in cancer cachexia. Biochim. Biophys. Acta 2015, 1851, 1490–1500. [Google Scholar] [CrossRef] [PubMed]
- Braun, T.; Zhu, X.; Szumowski, M.; Scott, G.D.; Grossberg, A.; Levasseur, P.R.; Graham, K.; Khan, S.; Damaraju, S.; Colmers, W.F.; et al. Central nervous system inflammation induces muscle atrophy via activation of the hypothalamic-pituitary-adrenal axis. J. Exp. Med. 2011, 208, 2449–2463. [Google Scholar] [CrossRef] [PubMed]
- Sherry, B.A.; Gelin, J.; Fong, Y.; Marano, M.; Wei, H.; Cerami, A.; Lowry, S.F.; Lundholm, K.G.; Moldawer, L.L. Anticachectin/tumor necrosis factor-alpha antibodies attenuate development of cachexia in tumor models. FASEB J. 1989, 3, 1956–1962. [Google Scholar] [CrossRef] [PubMed]
- Tisdale, M.J. Biology of cachexia. J. Natl. Cancer Inst. 1997, 89, 1763–1773. [Google Scholar] [CrossRef]
- Karayiannakis, A.J.; Syrigos, K.N.; Polychronidis, A.; Pitiakoudis, M.; Bounovas, A.; Simopoulos, K. Serum levels of tumor necrosis factor-alpha and nutritional status in pancreatic cancer patients. Anticancer. Res. 2001, 21, 1355–1358. [Google Scholar]
- Maltoni, M.; Fabbri, L.; Nanni, O.; Scarpi, E.; Pezzi, L.; Flamini, E.; Riccobon, A.; Derni, S.; Pallotti, G.; Amadori, D. Serum levels of tumour necrosis factor alpha and other cytokines do not correlate with weight loss and anorexia in cancer patients. Support Care Cancer 1997, 5, 130–135. [Google Scholar] [CrossRef]
- Jatoi, A.; Dakhil, S.R.; Nguyen, P.L.; Sloan, J.A.; Kugler, J.W.; Rowland, K.M., Jr.; Soori, G.S.; Wender, D.B.; Fitch, T.R.; Novotny, P.J.; et al. A placebo-controlled double blind trial of etanercept for the cancer anorexia/weight loss syndrome: Results from N00C1 from the North Central Cancer Treatment Group. Cancer 2007, 110, 1396–1403. [Google Scholar] [CrossRef]
- Wiedenmann, B.; Malfertheiner, P.; Friess, H.; Ritch, P.; Arseneau, J.; Mantovani, G.; Caprioni, F.; Van Cutsem, E.; Richel, D.; Dewitte, M.; et al. A multicenter, phase II study of infliximab plus gemcitabine in pancreatic cancer cachexia. J. Support Oncol. 2008, 6, 18–25. [Google Scholar]
- Bertrand, F.; Montfort, A.; Marcheteau, E.; Imbert, C.; Gilhodes, J.; Filleron, T.; Rochaix, P.; Andrieu-Abadie, N.; Levade, T.; Meyer, N.; et al. TNFα blockade overcomes resistance to anti-PD-1 in experimental melanoma. Nat. Commun. 2017, 8, 2256. [Google Scholar] [CrossRef]
- Weber, J.S.; Antonia, S.J.; Topalian, S.L.; Schadendorf, D.; Larkin, J.M.; Sznol, M.; Liu, H.Y.; Waxman, I.; Robert, C. Safety Profile of Nivolumab Monotherapy: A Pooled Analysis of Patients with Advanced Melanoma. J. Clin. Oncol. 2017, 35, 785–792. [Google Scholar] [CrossRef]
- Padrão, A.I.; Moreira-Gonçalves, D.; Oliveira, P.A.; Teixeira, C.; Faustino-Rocha, A.I.; Helguero, L.; Vitorino, R.; Santos, L.L.; Amado, F.; Duarte, J.A.; et al. Endurance training prevents TWEAK but not myostatin-mediated cardiac remodelling in cancer cachexia. Arch. Biochem. Biophys. 2015, 567, 13–21. [Google Scholar] [CrossRef]
- Johnston, A.J.; Murphy, K.T.; Jenkinson, L.; Laine, D.; Emmrich, K.; Faou, P.; Weston, R.; Jayatilleke, K.M.; Schloegel, J.; Talbo, G.; et al. Targeting of Fn14 Prevents Cancer-Induced Cachexia and Prolongs Survival. Cell 2015, 162, 1365–1378. [Google Scholar] [CrossRef] [PubMed]
- Maecker, H.; Varfolomeev, E.; Kischkel, F.; Lawrence, D.; LeBlanc, H.; Lee, W.; Hurst, S.; Danilenko, D.; Li, J.; Filvaroff, E.; et al. TWEAK attenuates the transition from innate to adaptive immunity. Cell 2005, 123, 931–944. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Fox, M.I.; Belmar, N.A.; Sho, M.; Chao, D.T.; Choi, N.; Fang, Y.; Zhao, V.; Keller, S.F.; Starling, G.C.; et al. Enavatuzumab, a Humanized Anti-TWEAK Receptor Monoclonal Antibody, Exerts Antitumor Activity through Attracting and Activating Innate Immune Effector Cells. J. Immunol. Res. 2017, 2017, 5737159. [Google Scholar] [CrossRef] [PubMed]
- Lassen, U.N.; Meulendijks, D.; Siu, L.L.; Karanikas, V.; Mau-Sorensen, M.; Schellens, J.H.; Jonker, D.J.; Hansen, A.R.; Simcox, M.E.; Schostack, K.J.; et al. A phase I monotherapy study of RG7212, a first-in-class monoclonal antibody targeting TWEAK signaling in patients with advanced cancers. Clin. Cancer Res. 2015, 21, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Lam, E.T.; Eckhardt, S.G.; Messersmith, W.; Jimeno, A.; O’Bryant, C.L.; Ramanathan, R.K.; Weiss, G.J.; Chadha, M.; Oey, A.; Ding, H.T.; et al. Phase I Study of Enavatuzumab, a First-in-Class Humanized Monoclonal Antibody Targeting the TWEAK Receptor, in Patients with Advanced Solid Tumors. Mol. Cancer Ther. 2018, 17, 215–221. [Google Scholar] [CrossRef]
- McDonald, J.J.; McMillan, D.C.; Laird, B.J.A. Targeting IL-1α in cancer cachexia: A narrative review. Curr. Opin. Support Palliat. Care 2018, 12, 453–459. [Google Scholar] [CrossRef]
- Costelli, P.; Llovera, M.; Carbó, N.; García-Martínez, C.; López-Sorianoq, F.J.; Argilés, J.M. Interleukin-1 receptor antagonist (IL-1ra) is unable to reverse cachexia in rats bearing an ascites hepatoma (Yoshida AH-130). Cancer Lett. 1995, 95, 33–38. [Google Scholar] [CrossRef]
- Douvdevani, A.; Huleihel, M.; Zöller, M.; Segal, S.; Apte, R.N. Reduced tumorigenicity of fibrosarcomas which constitutively generate IL-1 alpha either spontaneously or following IL-1 alpha gene transfer. Int. J. Cancer 1992, 51, 822–830. [Google Scholar] [CrossRef]
- Voronov, E.; Weinstein, Y.; Benharroch, D.; Cagnano, E.; Ofir, R.; Dobkin, M.; White, R.M.; Zoller, M.; Barak, V.; Segal, S.; et al. Antitumor and immunotherapeutic effects of activated invasive T lymphoma cells that display short-term interleukin 1alpha expression. Cancer Res. 1999, 59, 1029–1035. [Google Scholar]
- Tjomsland, V.; Spångeus, A.; Välilä, J.; Sandström, P.; Borch, K.; Druid, H.; Falkmer, S.; Falkmer, U.; Messmer, D.; Larsson, M. Interleukin 1α sustains the expression of inflammatory factors in human pancreatic cancer microenvironment by targeting cancer-associated fibroblasts. Neoplasia 2011, 13, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Kuan, E.L.; Ziegler, S.F. A tumor-myeloid cell axis, mediated via the cytokines IL-1α and TSLP, promotes the progression of breast cancer. Nat. Immunol. 2018, 19, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Hickish, T.; Andre, T.; Wyrwicz, L.; Saunders, M.; Sarosiek, T.; Kocsis, J.; Nemecek, R.; Rogowski, W.; Lesniewski-Kmak, K.; Petruzelka, L.; et al. MABp1 as a novel antibody treatment for advanced colorectal cancer: A randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2017, 18, 192–201. [Google Scholar] [CrossRef]
- Scheede-Bergdahl, C.; Watt, H.L.; Trutschnigg, B.; Kilgour, R.D.; Haggarty, A.; Lucar, E.; Vigano, A. Is IL-6 the best pro-inflammatory biomarker of clinical outcomes of cancer cachexia? Clin. Nutr. 2012, 31, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Elkabets, M.; Ribeiro, V.S.G.; Dinarello, C.A.; Ostrand-Rosenberg, S.; Di Santo, J.; Apte, R.N.; Vosshenrich, C.A.J. IL-1β regulates a novel myeloid-derived suppressor cell subset that impairs NK cell development and function. Eur. J. Immunol. 2010, 40, 3347–3357. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.; Lee, O.-Y.; Park, Y.; Seo, M.W.; Lee, D.-S. IL-1β induces IL-6 production and increases invasiveness and estrogen-independent growth in a TG2-dependent manner in human breast cancer cells. BMC Cancer 2016, 16, 724. [Google Scholar] [CrossRef]
- Voigt, C.; May, P.; Gottschlich, A.; Markota, A.; Wenk, D.; Gerlach, I.; Voigt, S.; Stathopoulos, G.T.; Arendt, K.A.M.; Heise, C.; et al. Cancer cells induce interleukin-22 production from memory CD4+ T cells via interleukin-1 to promote tumor growth. Proc. Natl. Acad. Sci. USA 2017, 114, 12994–12999. [Google Scholar] [CrossRef]
- Kaplanov, I.; Carmi, Y.; Kornetsky, R.; Shemesh, A.; Shurin, G.V.; Shurin, M.R.; Dinarello, C.A.; Voronov, E.; Apte, R.N. Blocking IL-1β reverses the immunosuppression in mouse breast cancer and synergizes with anti-PD-1 for tumor abrogation. Proc. Natl. Acad. Sci. USA 2019, 116, 1361–1369. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J.; Ridker, P.; Lorenzatti, A.; Krum, H.; Varigos, J.; et al. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1833–1842. [Google Scholar] [CrossRef]
- Moses, A.G.W.; Maingay, J.; Sangster, K.; Fearon, K.C.H.; Ross, J.A. Pro-inflammatory cytokine release by peripheral blood mononuclear cells from patients with advanced pancreatic cancer: Relationship to acute phase response and survival. Oncol. Rep. 2009, 21, 1091–1095. [Google Scholar] [PubMed]
- Baltgalvis, K.A.; Berger, F.G.; Pena, M.M.O.; Davis, J.M.; Muga, S.J.; Carson, J.A. Interleukin-6 and cachexia in ApcMin/+ mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R393–R401. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, H.; Kamon, H.; Sawa, S.I.; Park, S.J.; Katunuma, N.; Ishihara, K.; Murakami, M.; Hirano, T. IL-6-STAT3 controls intracellular MHC class II alphabeta dimer level through cathepsin S activity in dendritic cells. Immunity 2005, 23, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Diehl, S.; Anguita, J.; Hoffmeyer, A.; Zapton, T.; Ihle, J.N.; Fikrig, E.; Rincón, M. Inhibition of Th1 differentiation by IL-6 is mediated by SOCS1. Immunity 2000, 13, 805–815. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Fujieda, K.; Hirayama, M.; Ikeda, T.; Yuno, A.; Matsumura, K.; Fukuma, D.; Araki, K.; Mizuta, H.; Nakayama, H.; et al. Soluble IL6R Expressed by Myeloid Cells Reduces Tumor-Specific Th1 Differentiation and Drives Tumor Progression. Cancer Res. 2017, 77, 2279–2291. [Google Scholar] [CrossRef]
- Haynes, L.; Eaton, S.M.; Burns, E.M.; Randall, T.D.; Swain, S.L. CD4 T cell memory derived from young naive cells functions well into old age, but memory generated from aged naive cells functions poorly. Proc. Natl. Acad. Sci. USA 2003, 100, 15053–15058. [Google Scholar] [CrossRef]
- Zhou, J.; Qu, Z.; Sun, F.; Han, L.; Li, L.; Yan, S.; Stabile, L.P.; Chen, L.-F.; Siegfried, J.M.; Xiao, G. Myeloid STAT3 Promotes Lung Tumorigenesis by Transforming Tumor Immunosurveillance into Tumor-Promoting Inflammation. Cancer Immunol. Res. 2017, 5, 257–268. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Flint, T.R.; Janowitz, T.; Connell, C.M.; Roberts, E.; Denton, A.; Coll, A.P.; Jodrell, D.I.; Fearon, D.T. Tumor-Induced IL-6 Reprograms Host Metabolism to Suppress Anti-tumor Immunity. Cell Metab. 2016, 24, 672–684. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Fujieda, K.; Miyashita, A.; Fukushima, S.; Ikeda, T.; Kubo, Y.; Senju, S.; Ihn, H.; Nishimura, Y.; Oshiumi, H. Combined Blockade of IL6 and PD-1/PD-L1 Signaling Abrogates Mutual Regulation of Their Immunosuppressive Effects in the Tumor Microenvironment. Cancer Res. 2018, 78, 5011–5022. [Google Scholar] [CrossRef]
- Li, J.; Xu, J.; Yan, X.; Jin, K.; Li, W.; Zhang, R. Targeting Interleukin-6 (IL-6) Sensitizes Anti-PD-L1 Treatment in a Colorectal Cancer Preclinical Model. Med. Sci. Monit. 2018, 24, 5501–5508. [Google Scholar] [CrossRef] [PubMed]
- Damuzzo, V.; Solito, S.; Pinton, L.; Carrozzo, E.; Valpione, S.; Pigozzo, J.; Giancristofaro, R.A.; Chiarion-Sileni, V.; Mandruzzato, S. Clinical implication of tumor-associated and immunological parameters in melanoma patients treated with ipilimumab. Oncoimmunology 2016, 5, e1249559. [Google Scholar] [CrossRef] [PubMed]
- Dixit, N.; Simon, S.I. Chemokines, selectins and intracellular calcium flux: Temporal and spatial cues for leukocyte arrest. Front. Immunol. 2012, 3, 188. [Google Scholar] [CrossRef] [PubMed]
- Gioulbasanis, I.; Patrikidou, A.; Kitikidou, K.; Papadimitriou, K.; Vlachostergios, P.J.; Tsatsanis, C.; Margioris, A.N.; Papandreou, C.N.; Mavroudis, D.; Georgoulias, V. Baseline plasma levels of interleukin-8 in stage IV non-small-cell lung cancer patients: Relationship with nutritional status and prognosis. Nutr. Cancer. 2012, 64, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.-C.; Wang, C.-J.; Chao, Y.-J.; Chen, H.-Y.; Wang, H.-C.; Tung, H.-L.; Lin, J.-T.; Shan, Y.-S. Elevated Serum Interleukin-8 Level Correlates with Cancer-Related Cachexia and Sarcopenia: An Indicator for Pancreatic Cancer Outcomes. J. Clin. Med. 2018, 7, 502. [Google Scholar] [CrossRef]
- Mishalian, I.; Bayuh, R.; Eruslanov, E.; Michaeli, J.; Levy, L.; Zolotarov, L.; Singhal, S.; Albelda, S.M.; Granot, Z.; Fridlender, Z.G. Neutrophils recruit regulatory T-cells into tumors via secretion of CCL17—A new mechanism of impaired antitumor immunity. Int. J. Cancer 2014, 135, 1178–1186. [Google Scholar] [CrossRef]
- Jin, L.; Tao, H.; Karachi, A.; Long, Y.; Hou, A.Y.; Na, M.; Dyson, K.A.; Grippin, A.J.; Deleyrolle, L.P.; Zhang, W.; et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat. Commun. 2019, 10, 4016. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Perez-Gracia, J.L.; Schalper, K.A.; Fusco, J.P.; Gonzalez, A.; Rodriguez-Ruiz, M.E.; Oñate, C.; Perez, G.; Alfaro, C.; Martín-Algarra, S.; et al. Changes in serum interleukin-8 (IL-8) levels reflect and predict response to anti-PD-1 treatment in melanoma and non-small-cell lung cancer patients. Ann. Oncol. 2017, 28, 1988–1995. [Google Scholar] [CrossRef]
- Collins, J.M.; Heery, C.R.; Donahue, R.N.; Palena, C.; Madan, R.A.; Strauss, J.; Gatti-Mays, M.E.; Schlom, J.; Gulley, J.L.; Bilusic, M. Phase I trial of BMS-986253, an anti-IL-8 monoclonal antibody, in patients with metastatic or unresectable solid tumors. J. Clin. Oncol. 2018, 36, 3091. [Google Scholar] [CrossRef]
- Loumaye, A.; de Barsy, M.; Nachit, M.; Lause, P.; Frateur, L.; van Maanen, A.; Trefois, P.; Gruson, D.; Thissen, J.P. Role of Activin A and myostatin in human cancer cachexia. J. Clin. Endocrinol. Metab. 2015, 100, 2030–2038. [Google Scholar] [CrossRef]
- Ding, H.; Zhang, G.; Sin, K.W.T.; Liu, Z.; Lin, R.-K.; Li, M.; Li, Y.-P. Activin A induces skeletal muscle catabolism via p38β mitogen-activated protein kinase. J. Cachexia Sarcopenia Muscle 2017, 8, 202–212. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, J.L.; Lu, J.; Song, Y.; Kwak, K.S.; Jiao, Q.; Rosenfeld, R.; Chen, Q.; Boone, T.; Simonet, W.S.; et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010, 142, 531–543. [Google Scholar] [CrossRef]
- Loumaye, A.; De Barsy, M.; Nachit, M.; Lause, P.; Van Maanen, A.; Trefois, P.; Gruson, D.; Thissen, J.-P. Circulating Activin A predicts survival in cancer patients. J. Cachexia Sarcopenia Muscle 2017, 8, 768–777. [Google Scholar] [CrossRef]
- Semitekolou, M.; Alissafi, T.; Aggelakopoulou, M.; Kourepini, E.; Kariyawasam, H.H.; Kay, A.B.; Robinson, D.S.; Lloyd, C.; Panoutsakopoulou, V.; Xanthou, G. Activin-A induces regulatory T cells that suppress T helper cell immune responses and protect from allergic airway disease. J. Exp. Med. 2009, 206, 1769–1785. [Google Scholar] [CrossRef]
- Ogawa, K.; Funaba, M.; Chen, Y.; Tsujimoto, M. Activin A functions as a Th2 cytokine in the promotion of the alternative activation of macrophages. J. Immunol. 2006, 177, 6787–6794. [Google Scholar] [CrossRef]
- Rautela, J.; Dagley, L.F.; De Oliveira, C.C.; Schuster, I.S.; Hediyeh-Zadeh, S.; Delconte, R.B.; Cursons, J.; Hennessy, R.; Hutchinson, D.S.; Harrison, C.; et al. Therapeutic blockade of Activin-A improves NK cell function and antitumor immunity. Sci. Signal. 2019, 12, eaat7527. Available online: https://stke.sciencemag.org/content/12/596/eaat7527 (accessed on 4 May 2020). [CrossRef]
- Tao, J.J.; Cangemi, N.A.; Makker, V.; Cadoo, K.A.; Liu, J.F.; Rasco, D.W.; Navarro, W.H.; Haqq, C.M.; Hyman, D.M. First-in-Human Phase I Study of the Activin A Inhibitor, STM 434, in Patients with Granulosa Cell Ovarian Cancer and Other Advanced Solid Tumors. Clin. Cancer Res. 2019, 25, 5458–5465. [Google Scholar] [CrossRef]
- Zugmaier, G.; Paik, S.; Wilding, G.; Knabbe, C.; Bano, M.; Lupu, R.; Deschauer, B.; Simpson, S.; Dickson, R.B.; Lippman, M. Transforming Growth Factor β1 Induces Cachexia and Systemic Fibrosis without an Antitumor Effect in Nude Mice. Cancer Res. 1991, 51, 3590–3594. [Google Scholar]
- Waning, D.L.; Mohammad, K.S.; Reiken, S.; Xie, W.; Andersson, D.; John, S.K.; Chiechi, A.; Wright, L.; Umanskaya, A.; Niewolna, M.; et al. Excess TGF-β mediates muscle weakness associated with bone metastases in mice. Nat. Med. 2015, 21, 1262–1271. [Google Scholar] [CrossRef]
- Lima, J.D.; Simoes, E.; de Castro, G.; Morais, M.R.; de Matos-Neto, E.M.; Alves, M.J.; Pinto, N.I.; Figueredo, R.G.; Zorn, T.M.; Felipe-Silva, A.S.; et al. Tumour-derived transforming growth factor-β signalling contributes to fibrosis in patients with cancer cachexia. J. Cachexia Sarcopenia Muscle 2019, 10, 1045–1059. [Google Scholar] [CrossRef]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Polanczyk, M.J.; Walker, E.; Haley, D.; Guerrouahen, B.S.; Akporiaye, E.T. Blockade of TGF-β signaling to enhance the antitumor response is accompanied by dysregulation of the functional activity of CD4+CD25+Foxp3+ and CD4+CD25-Foxp3+ T cells. J. Transl. Med. 2019, 17, 219. [Google Scholar] [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef]
- Lerner, L.; Tao, J.; Liu, Q.; Nicoletti, R.; Feng, B.; Krieger, B.; Mazsa, E.; Siddiquee, Z.; Wang, R.; Huang, L.; et al. MAP3K11/GDF15 axis is a critical driver of cancer cachexia. J. Cachexia Sarcopenia Muscle 2016, 7, 467–482. [Google Scholar] [CrossRef]
- Johnen, H.; Lin, S.; Kuffner, T.; Brown, D.A.; Tsai, V.W.; Bauskin, A.R.; Wu, L.; Pankhurst, G.; Jiang, L.; Junankar, S.; et al. Tumor-induced anorexia and weight loss are mediated by the TGF-beta superfamily cytokine MIC-1. Nat. Med. 2007, 13, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Li, W.; Song, Y.; Wang, L.; Zhang, K.; Yang, J.; Zhang, W.; Su, H.; Zhang, Y. Growth differentiation factor-15 suppresses maturation and function of dendritic cells and inhibits tumor-specific immune response. PLoS ONE 2013, 8, e78618. [Google Scholar] [CrossRef]
- Roth, P.; Junker, M.; Tritschler, I.; Mittelbronn, M.; Dombrowski, Y.; Breit, S.N.; Tabatabai, G.; Wick, W.; Weller, M.; Wischhusen, J. GDF-15 contributes to proliferation and immune escape of malignant gliomas. Clin. Cancer Res. 2010, 16, 3851–3859. [Google Scholar] [CrossRef]
- Ratnam, N.M.; Peterson, J.M.; Talbert, E.E.; Ladner, K.J.; Rajasekera, P.V.; Schmidt, C.R.; Dillhoff, M.E.; Swanson, B.J.; Haverick, E.; Kladney, R.D.; et al. NF-κB regulates GDF-15 to suppress macrophage surveillance during early tumor development. J. Clin. Investig. 2017, 127, 3796–3809. [Google Scholar] [CrossRef]
- Suriben, R.; Chen, M.; Higbee, J.; Oeffinger, J.; Ventura, R.; Li, B.; Mondal, K.; Gao, Z.; Ayupova, D.; Taskar, P.; et al. Antibody-mediated inhibition of GDF15–GFRAL activity reverses cancer cachexia in mice. Nat. Med. 2020, 26, 1264–1270. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef]
- Ohki, S.; Shibata, M.; Gonda, K.; Machida, T.; Shimura, T.; Nakamura, I.; Ohtake, T.; Koyama, Y.; Suzuki, S.; Ohto, H.; et al. Circulating myeloid-derived suppressor cells are increased and correlate to immune suppression, inflammation and hypoproteinemia in patients with cancer. Oncol. Rep. 2012, 28, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Cuenca, A.G.; Cuenca, A.L.; Winfield, R.D.; Joiner, D.N.; Gentile, L.; Delano, M.J.; Kelly-Scumpia, K.M.; Scumpia, P.O.; Matheny, M.K.; Scarpace, P.J.; et al. Novel role for tumor-induced expansion of myeloid-derived cells in cancer cachexia. J. Immunol. 2014, 192, 6111–6119. [Google Scholar] [CrossRef] [PubMed]
- Strauss, L.; Mahmoud, M.A.A.; Weaver, J.D.; Tijaro-Ovalle, N.M.; Christofides, A.; Wang, Q.; Pal, R.; Yuan, M.; Asara, J.; Patsoukis, N.; et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci. Immunol. 2020, 5, eaay1863. [Google Scholar] [CrossRef] [PubMed]
- Paunescu, V.; Bojin, F.M.; Tatu, C.A.; Gavriliuc, O.I.; Rosca, A.; Gruia, A.T.; Tanasie, G.; Bunu, C.; Crisnic, D.; Gherghiceanu, M.; et al. Tumour-associated fibroblasts and mesenchymal stem cells: More similarities than differences. J. Cell. Mol. Med. 2011, 15, 635–646. [Google Scholar] [CrossRef]
- Roberts, E.W.; Deonarine, A.; Jones, J.O.; Denton, A.E.; Feig, C.; Lyons, S.K.; Espeli, M.; Kraman, M.; McKenna, B.; Wells, R.J.; et al. Depletion of stromal cells expressing fibroblast activation protein-α from skeletal muscle and bone marrow results in cachexia and anemia. J. Exp. Med. 2013, 210, 1137–1151. [Google Scholar] [CrossRef]
- Kir, S.; Spiegelman, B.M. Cachexia & Brown Fat: A Burning Issue in Cancer. Trends Cancer 2016, 2, 461–463. [Google Scholar]
- Ziani, L.; Chouaib, S.; Thiery, J. Alteration of the Antitumor Immune Response by Cancer-Associated Fibroblasts. Front. Immunol. 2018, 9, 414. [Google Scholar] [CrossRef]
- Tan, B.H.L.; Fladvad, T.; Braun, T.; Vigano, A.; Strasser, F.; Deans, D.A.C.; Skipworth, R.J.E.; Solheim, T.S.; Damaraju, S.; Ross, J.A.; et al. P-selectin genotype is associated with the development of cancer cachexia. EMBO Mol. Med. 2012, 4, 462–471. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3443952/ (accessed on 4 May 2020). [CrossRef]
- Johns, N.; Stretch, C.; Tan, B.H.; Solheim, T.S.; Sørhaug, S.; Stephens, N.A.; Gioulbasanis, I.; Skipworth, R.J.; Deans, D.C.; Vigano, A.; et al. New genetic signatures associated with cancer cachexia as defined by low skeletal muscle index and weight loss. J. Cachexia Sarcopenia Muscle 2017, 8, 122–130. [Google Scholar] [CrossRef]
- Ley, K.; Kansas, G.S. Selectins in T-cell recruitment to non-lymphoid tissues and sites of inflammation. Nat. Rev. Immunol. 2004, 4, 325–336. [Google Scholar] [CrossRef]
- Borsig, L.; Wong, R.; Hynes, R.O.; Varki, N.M.; Varki, A. Synergistic effects of L- and P-selectin in facilitating tumor metastasis can involve non-mucin ligands and implicate leukocytes as enhancers of metastasis. Proc. Natl. Acad. Sci. USA 2002, 99, 2193–2198. [Google Scholar] [CrossRef]
- Tinoco, R.; Carrette, F.; Barraza, M.L.; Otero, D.C.; Magaña, J.; Bosenberg, M.W.; Swain, S.L.; Bradley, L.M. PSGL-1 Is an Immune Checkpoint Regulator that Promotes T Cell Exhaustion. Immunity 2016, 44, 1190–1203. [Google Scholar] [CrossRef] [PubMed]
- Takeshige, K.; Baba, M.; Tsuboi, S.; Noda, T.; Ohsumi, Y. Autophagy in yeast demonstrated with proteinase-deficient mutants and conditions for its induction. J. Cell. Biol. 1992, 119, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Aversa, Z.; Pin, F.; Lucia, S.; Penna, F.; Verzaro, R.; Fazi, M.; Colasante, G.; Tirone, A.; Fanelli, F.R.; Ramaccini, C.; et al. Autophagy is induced in the skeletal muscle of cachectic cancer patients. Sci. Rep. 2016, 6, 30340. [Google Scholar] [CrossRef]
- Pigna, E.; Berardi, E.; Aulino, P.; Rizzuto, E.; Zampieri, S.; Carraro, U.; Kern, H.; Merigliano, S.; Gruppo, M.; Mericskay, M.; et al. Aerobic Exercise and Pharmacological Treatments Counteract Cachexia by Modulating Autophagy in Colon Cancer. Sci. Rep. 2016, 31, 26991. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, K.; Andersen, S.; Degen, S.; Tadini, V.; Grosjean, J.; Hatakeyama, S.; Tesfahun, A.N.; Moestue, S.; Kim, J.; Nonstad, U.; et al. Cancer cachexia associates with a systemic autophagy-inducing activity mimicked by cancer cell-derived IL-6 trans-signaling. Sci. Rep. 2017, 7, 2046. [Google Scholar] [CrossRef]
- Rosenfeldt, M.T.; Ryan, K.M. The multiple roles of autophagy in cancer. Carcinogenesis 2011, 32, 955–963. [Google Scholar] [CrossRef]
- Randow, F.; Münz, C. Autophagy in the regulation of pathogen replication and adaptive immunity. Trends Immunol. 2012, 33, 475–487. [Google Scholar] [CrossRef]
- Hahn, T.; Akporiaye, E.T. α-TEA as a stimulator of tumor autophagy and enhancer of antigen cross-presentation. Autophagy 2013, 9, 429–431. [Google Scholar] [CrossRef]
- Pua, H.H.; Guo, J.; Komatsu, M.; He, Y.-W. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J. Immunol. 2009, 182, 4046–4055. [Google Scholar] [CrossRef]
- Wei, J.; Long, L.; Yang, K.; Guy, C.; Shrestha, S.; Chen, Z.; Wu, C.; Vogel, P.; Neale, G.; Green, D.R.; et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat. Immunol. 2016, 17, 277–285. [Google Scholar] [CrossRef]
- Liu, K.; Zhao, E.; Ilyas, G.; Lalazar, G.; Lin, Y.; Haseeb, M.; E Tanaka, K.; Czaja, M.J. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy 2015, 11, 271–284. [Google Scholar] [CrossRef]
- Parker, K.H.; Horn, L.A.; Ostrand-Rosenberg, S. High-mobility group box protein 1 promotes the survival of myeloid-derived suppressor cells by inducing autophagy. J. Leukoc. Biol. 2016, 100, 463–470. [Google Scholar] [CrossRef]
- Kichenadasse, G.; Miners, J.O.; Mangoni, A.A.; Rowland, A.; Hopkins, A.M.; Sorich, M.J. Association Between Body Mass Index and Overall Survival with Immune Checkpoint Inhibitor Therapy for Advanced Non-Small Cell Lung Cancer. JAMA Oncol. 2019, 6, 512–518. [Google Scholar] [CrossRef]
- Martini, D.J.; Kline, M.R.; Liu, Y.; Shabto, J.M.; Williams, M.A.; Khan, A.I.; Lewis, C.; Collins, H.; Akce, M.; Kissick, H.T.; et al. Adiposity may predict survival in patients with advanced stage cancer treated with immunotherapy in phase 1 clinical trials. Cancer 2020, 126, 575–582. [Google Scholar] [CrossRef]
- Shiroyama, T.; Nagatomo, I.; Koyama, S.; Hirata, H.; Nishida, S.; Miyake, K.; Fukushima, K.; Shirai, Y.; Mitsui, Y.; Takata, S.; et al. Impact of sarcopenia in patients with advanced non–small cell lung cancer treated with PD-1 inhibitors: A preliminary retrospective study. Sci. Rep. 2019, 9, 2447. [Google Scholar] [CrossRef]
- Roch, B.; Coffy, A.; Jean-Baptiste, S.; Palaysi, E.; Daures, J.-P.; Pujol, J.-L.; Bommart, S. Cachexia—Sarcopenia as a determinant of disease control rate and survival in non-small lung cancer patients receiving immune-checkpoint inhibitors. Lung Cancer 2020, 143, 19–26. [Google Scholar] [CrossRef]
- Chu, M.P.; Li, Y.; Ghosh, S.; Sass, S.; Smylie, M.; Walker, J.; Sawyer, M.B. Body composition is prognostic and predictive of ipilimumab activity in metastatic melanoma. J. Cachexia Sarcopenia Muscle 2020, 11, 748–755. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Clinical Study | Number (n) of Patients | Malignancy Setting | Treatment | Primary Study Point | Results |
|---|---|---|---|---|---|
| Turner et al. * [6] | n = 1453 | Metastatic melanoma and NSCLC | Pembrolizumab | Relationship between Pembrolizumab pharmacokinetics and overall survival | Higher Pembrolizumab clearance (CL0) was an adverse prognostic factor for OS and it paralleled disease parameters associated with CCS (multivariate-adjusted CL0 HR = 1.64; 95% CI, 1.06–2.52 for melanoma and HR = 1.88; 95% CI, 1.22–2.89 for NSCLC). |
| Naik et al. * [7] | n = 139 | Metastatic melanoma | Pembrolizumab or Nivolumab or Nivolumab plus Ipilimumab | Association of baseline BMI (at the beginning of immunotherapy) with treatment outcomes | BMI values > 25 kg/m2 and <35 kg/m2 were a favorable prognostic factor for OS (adjusted-HR: 0.26; 95% CI:0.1–0.71; p-value = 0.008) and PFS (adjusted-HR: 0.43; 95% CI: 0.19–0.95; p-value: 0.038) compared to BMI values 18.5–< 25 kg/m2 |
| Kichenadasse et al. * [104] | n = 2110 | Metastatic NSCLC | Atezolizumab | Association of baseline BMI (at the beginning of immunotherapy) with treatment outcomes and adverse events | A linear association between increasing values of BMI and overall survival was observed. |
| Martini et al. * [105] | n = 90 | Cancer patients that were treated with immunotherapy in the context of phase I clinical trials in a single center | Immunotherapy based treatments | Association of BMI, subcutaneous fat index (SFI), intermuscular fat index (IFI), and visceral fat index (VFI) with survival outcome. | Patients with an SFI ≥ 73 had a significantly longer OS (hazard ratio, 0.20; 95% CI, 0.09–0.46 [p < 0.001]) and PFS (hazard ratio, 0.38; 95% CI, 0.20–0.72 [p = 0.003]) compared with patients with an SFI < 73 and IFI < 3.4 and those with an SFI < 73 and IFI ≥ 3.4) |
| Shiroyama et al. * [106] | n = 42 | Previously treated metastatic NSCLC patients | Nivolumab, Pembrolizumab | Association of sarcopenia (calculated by measuring the cross-sectional area of the psoas muscle at the caudal end of the 3rd lumbar verterbrae) with treatment outcomes | Sarcopenia negatively affected PFS (median, 2.1 vs. 6.8 months, p = 0.004) and response rates (40.0% vs. 9.1%, p = 0.025) |
| Roch et al. * [107] | n = 142 | Metastatic NSCLC | PD1/PDL1 inhibitors | Effect of cachexia (defined as 5% loss of body within the last 6 months) or the effect of evolving sarcopenia (defined as 5% reduction in skeletal muscle index during treatment) on patient outcomes | Cachexia negatively affected disease control rates (59.9 % vs. 41.1 %, odds ratio: 2.60 (95% CI: 1.03–6.58) and OS HR: 6.26 (95% CI: 2.23–17.57). Evolving sarcopenia was an adverse predictor for shorter PFS, HR: 2.45 (95% CI: 1.09–5.53) and OS, HR: 3.87 (95% CI: 1.60–9.34) |
| Rounis et al. # [8] | n = 83 | Metastatic NSCLC | PD1/PDL1 | Association of cachexia (defined as weight loss 5% during the last 6 months since the initiation of immunotherapy or any degree of weight loss ≥ 2% and a BMI < 20 kg/m2 or reduced muscle mass according to tomovision analysis) with treatment outcomes | The presence of cancer cachexia consisted an independent predictor of increased probability of progression as best response to immunotherapy [OR = 8.11 (95% CI: 2.95–22.40, p < 0.001)] and an independent predictor of inferior survival [HR = 2.52 (95% CI: 1.40–2.55, p = 0.002)] |
| Biological Parameter | Implication on Cachexia Pathogenesis | Adverse Effects on Antitumor Immunity | Positive Effects on Antitumor Immunity | Ongoing Clinical Trials Evaluating the Effect of Inhibition of the Referred Biological Parameter in Combination with Immunotherapy in Cancer Patients |
|---|---|---|---|---|
| TNF-α | Inhibition of myocyte differentiation and stimulation of protein degradation [10] Inducing adipose tissue atrophy [11] Triggering sickness behavior at hypothalamus [12] | Impairment of intratumoral CD8+ T cells accumulation and upregulation of TIM3 [19] | - | Certolizumab or infliximab in combination with ipilimumab and nivolumab for advanced melanoma (NCT03293784) |
| TWEAK | Inducement of muscle atrophy via activation of ubiquitin proteolytic system [21] TWEAK/Fn14 inhibition reversed cachexia in mouse models [22] | Inhibition of STAT-1 and suppression of IFN-γ and IL-12 [23] Tweak−/− mice exhibit increased numbers of NK and Th1 cells [23] Tweak inhibition lead to tumor shrinkage and accumulation of CD45+ cells in the TME [24] | - | - |
| IL-1α | Hypothalamus stimulation that leads to proteolytic, lipolytic signals and causes anorexia and early satiety through increased tryptophan plasma levels [27] | Maintenance of tumor suppressive TME through interactions with CAFs [31] Inducing TLSP expression on tumor-infiltrating myeloid cells [32] | IL1α administration resulted in regression in mouse models of lymphoma and fibrosarcoma via accumulation of intratumoral CD8+ T cells [29,30] | - |
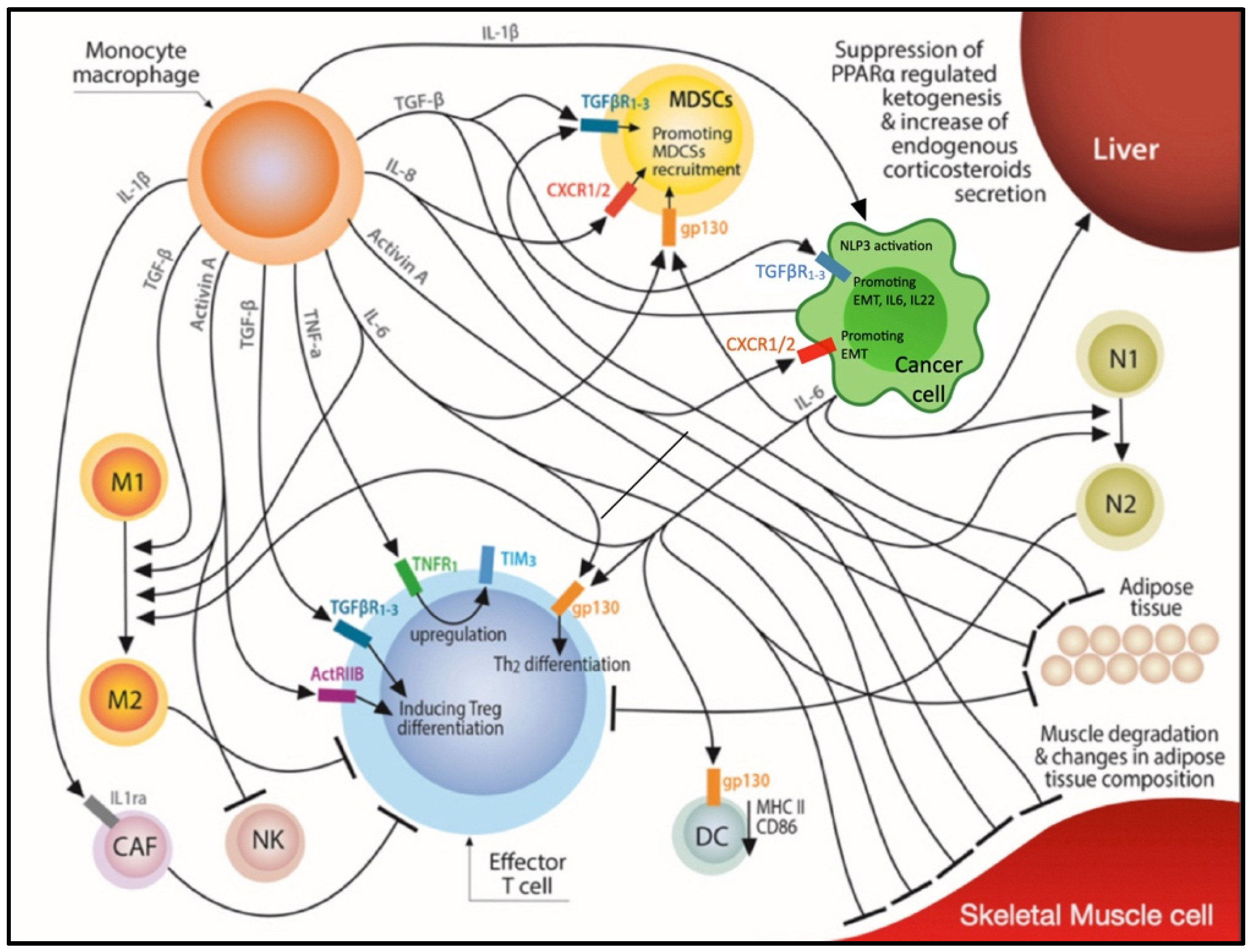
| IL-1β | Increased levels of IL-1β have been associated with cachexia in patients with advanced malignancies [34] | Stimulation of MDSCs [35] Induction of IL-6 and IL-22 expression [36,37] IL1-β deficient mice exhibited improved antitumor immunity compared to wt ones [38] | Canakimumab in combination with pembrolizumab for NSCLC in the metastatic (NCT03631199) or the adjuvant setting (NCT03447769) | |
| IL-6 | Liver stimulation for inducing an acute phase response [1] Upregulation of transcriptional factors that promote myofibrilar breakdown [1] Exogenous administration of IL-6 in ApcMin/+/IL-6−/− mice resulted to the development of a cachexia phenotype [42] Induction of autophagy in skeletal muscle [1] Clinical studies have correlated circulating IL-6 levels with the development of CCS in cancer patients [41] | Reprogramming of hepatic metabolism via suppression of peroxisome proliferator-activated receptor alpha (PPARα) regulated ketogenesis that subsequently induced increased endogenous glucocorticoid secretion leading to impaired antitumor immunity and resistance to immunotherapy in two mouse models of cachexia [49] Suppressing DC function through inhibition of MHC-II and CD80/86 [43] Suppressing T cell function through inhibition of IFN-γ/STAT1 Th1 differentiation [44,45] Suppressing the formation of CD4+ memory cells [46] Macrophage polarization to an M2 phenotype [47] Stimulation of MDSCs [48] | - | Tocilizumab in combination with ipilimumab and nivolumab in patients with unresectable or metastatic melanoma (NCT03999749) Tocilizumab in combination with trastuzumab and pertuzumab in patients with Her2 amplified metastatic breast cancer resistant to trastuzumab (NCT03135171) |
| IL-8 | Elevated circulating levels of IL-8 have been correlated with the development of CCS in cancer patients [54,55] | Recruitment of N2 TANs [56] Recruitment of MDSCs [48] Inhibition of IL-8/CXCR1/2 pathway in experimental models exerts antitumor effects [57] Increased serum levels of IL-8 have been correlated with secondary resistance to immunotherapy and disease progression in patients with metastatic melanoma and NSCLC receiving immunotherapy [58] | - | BMS-986253 in combination with nivolumab for hormone sensitive prostate cancer (NCT03689699), in combination with nivolumab or cabiralizumab in patients with HCC (NCT04050462) and in combination with nivolumab in patients with advanced cancer (NCT03400332) Neoadjuvant Nivolumab combined with CCR2/5-inhibitor or BMS-986253 for NSCLC or HCC (NCT04123379) |
| Activin A | Activin A causes muscle degradation and atrophy through downstream activation of Atrogin 1 and UBR2 and autophagosome formation [61] Pharmacological blockade of Activin A/ActRIIB pathway reversed cancer cachexia and muscle wasting in preclinical models [62] Elevated serum levels of Activin A have been associated with the development of CCS in pancreatic cancer patients [63] | Activin A has been shown to be able to differentiate CD4+ T cells into Tregs in vitro [64] and has the potential to induce polarization of TAMs to an M2 phenotype [65] Activin A/ActRIIB interaction impairs NK cell function via SMAD2/3 signaling and its blockade improved NK cell function and antitumor immunity and slowed melanoma growth in mouse models [66]. | - | - |
| TGF-β | TGF-β release into circulation activates the SMAD3-NOX4-RyR1 pathway leading to muscle dysfunction and development of cachexia in mouse models [69] Elevated serum levels of TGF-β in patients with colorectal cancer were correlated with the development of CCS [70] | TGF-β induces differentiation of CD4+ T cells to Tregs, acts as a chemoattractant for MDSCs in the TME, induces macrophage polarization to an M2 phenotype and promotes EMT [71] TGF-β blockade has shown activity in boosting host’s antitumor immunity mainly via suppressing Treg function [72] Combined blockade of TGF-β and PDL1 had a synergistic effect and lead to a robust antitumor response in mouse models bearing EMT6 tumors [73] | - | SAR439459 in combination with cemiplimab in advanced solid tumors (NCT03192345) MSB0011359C in advanced solid tumors (NCT02699515, NCT02517398), in combination with gemcitabine for pancreatic adenocarcinoma (NCT03451773), in combination with PROSTVAC and CV301 in prostate cancer (NCT03315871), for stage II/III Her2 amplified breast cancer (NCT03620201), in combination with eribulin for metastatic TNBC (NCT03579472), in combination with RT for ER+PR+Her2-breast cancer (NCT03524170) and SBRT for locally recurrent head and neck cancer (NCT04220775) and as monotherapy for MSI-high advanced solid tumors (NCT03436563). |
| GDF15 | GDF-15/GFRAL interaction has been identified as the key trigger for weight loss in animal models of cancer-related cachexia [74] Increased serum levels of GDF15 have been associated with the development of CCS in cancer patients [75] | GDF-15 inhibits dendritic cell maturation in the TME leading to impaired T cell activation [76], Downregulation of GDF-15 using shRNA in a glioblastoma model resulted in increased T cell infiltration in the TME and increased survival [77] Depletion of GDF-15 in orthotopic pancreatic cancer models restored immunosurveillance in the TME resulting in improved tumor control [78]. | - | - |
| MDSCs | Increased numbers of MDSCs in the serum or in the TME have been linked with the development of CCS in multiple experimental models and cancer patients [81,82] | MDSCs suppress antitumor immunity through angiogenesis promotion, production of matrix metalloproteinases, arginine depletion via increased Arg1 activity, ROS production leading to T cell anergy and death, Treg recruitment and expansion and macrophage polarization to an M2 phenotype [80] Specific PD1 ablation in myeloid cells in preclinical tumor models had a more pronounced effect on boosting antitumor immunity compared to specific PD1 ablation on T cells [83] | - | Cabiralizumab in combination with nivolumab for pretreated metastatic pancreatic cancer (NCT03336216) |
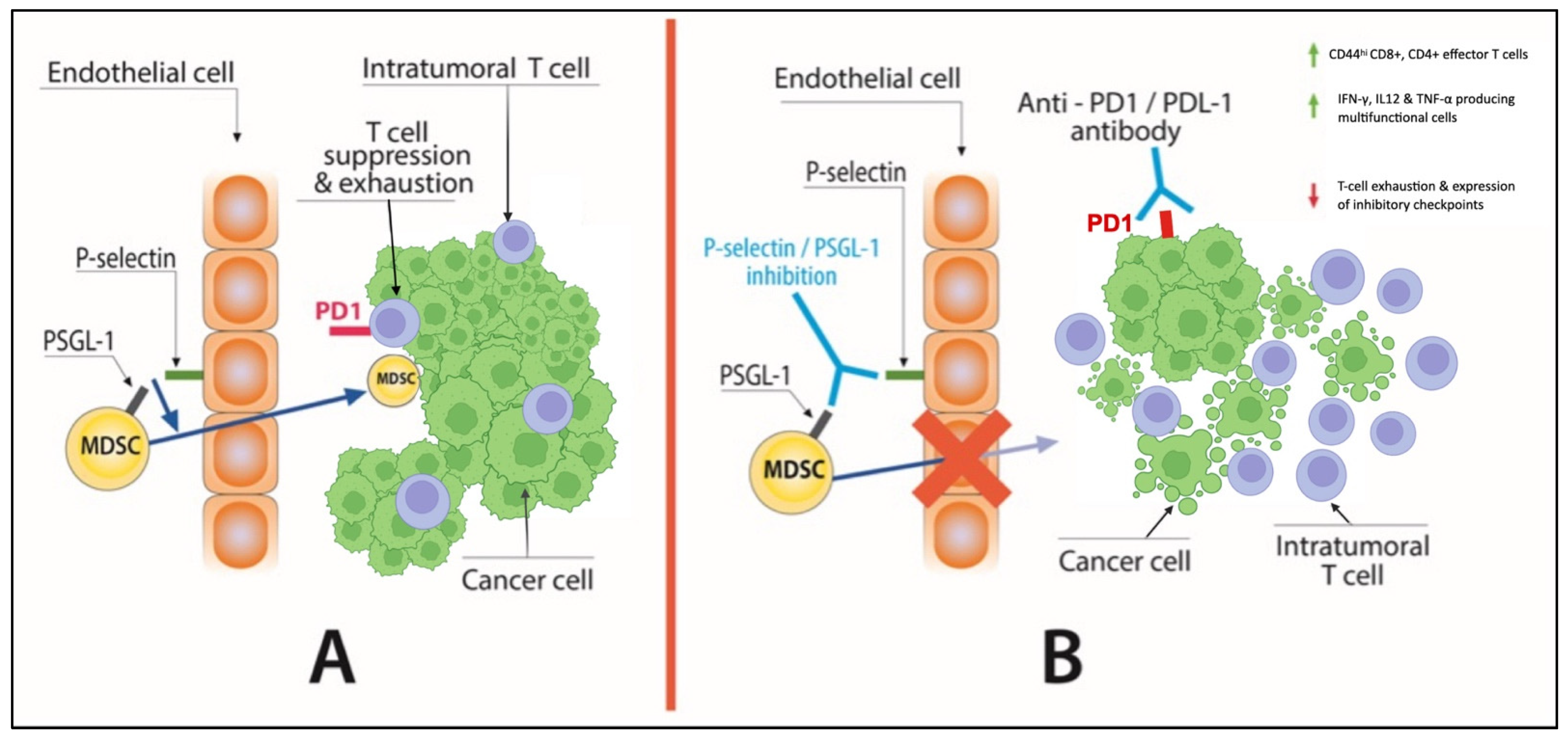
| p-Selectin | A loss-of-function mutation of the gene that encodes for the adhesion molecule P-Selectin (SELP) has been linked with reduced likelihood of developing CCS in the setting of malignancy [88,89] | P-, L- and E-selectin deficient mice have shown the importance of selectins in promoting metastasis and recruiting CD11b+Ly6C+Ly6G+ MDSCs in the TME [91] PSGL-1 ligation on exhausted T cells PSGL-1 due to TCR engagement extinguished ERK and AKT signaling and upregulated PD1 leading to their diminished survival and function [92] Selplg−/− mice demonstrated an improved antitumor immune response and increased intratumoral accumulation of effector CD44hiCD8+ and CD4+ T cells compared had higher frequencies of IFN-γ and IL-2 producing T cells [92]. | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rounis, K.; Makrakis, D.; Gioulbasanis, I.; Ekman, S.; De Petris, L.; Mavroudis, D.; Agelaki, S. Cancer Cachexia and Antitumor Immunity: Common Mediators and Potential Targets for New Therapies. Life 2022, 12, 880. https://doi.org/10.3390/life12060880
Rounis K, Makrakis D, Gioulbasanis I, Ekman S, De Petris L, Mavroudis D, Agelaki S. Cancer Cachexia and Antitumor Immunity: Common Mediators and Potential Targets for New Therapies. Life. 2022; 12(6):880. https://doi.org/10.3390/life12060880
Chicago/Turabian StyleRounis, Konstantinos, Dimitrios Makrakis, Ioannis Gioulbasanis, Simon Ekman, Luigi De Petris, Dimitris Mavroudis, and Sofia Agelaki. 2022. "Cancer Cachexia and Antitumor Immunity: Common Mediators and Potential Targets for New Therapies" Life 12, no. 6: 880. https://doi.org/10.3390/life12060880