
Dysregulation of the Amniotic PPARγ Pathway by Phthalates: Modulation of the Anti-Inflammatory Activity of PPARγ in Human Fetal Membranes

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Cell Culture
2.3. Tissue Collection and Primary Cell Culture
2.4. Characterization of PPAR Isoform by RT-PCR on FM Explants and Cells
2.5. Treatments
2.5.1. MEHP Treatments
2.5.2. Inflammatory Treatment with LPS
2.6. Cytotoxicity
2.7. Cell Viability
2.8. Quantitative RT-PCR
2.9. Western Blot Analysis
2.10. PPARγ Gene Reporter Luciferase Assay
2.11. Cytokine Release Assay
2.12. Statistical Analysis
3. Results
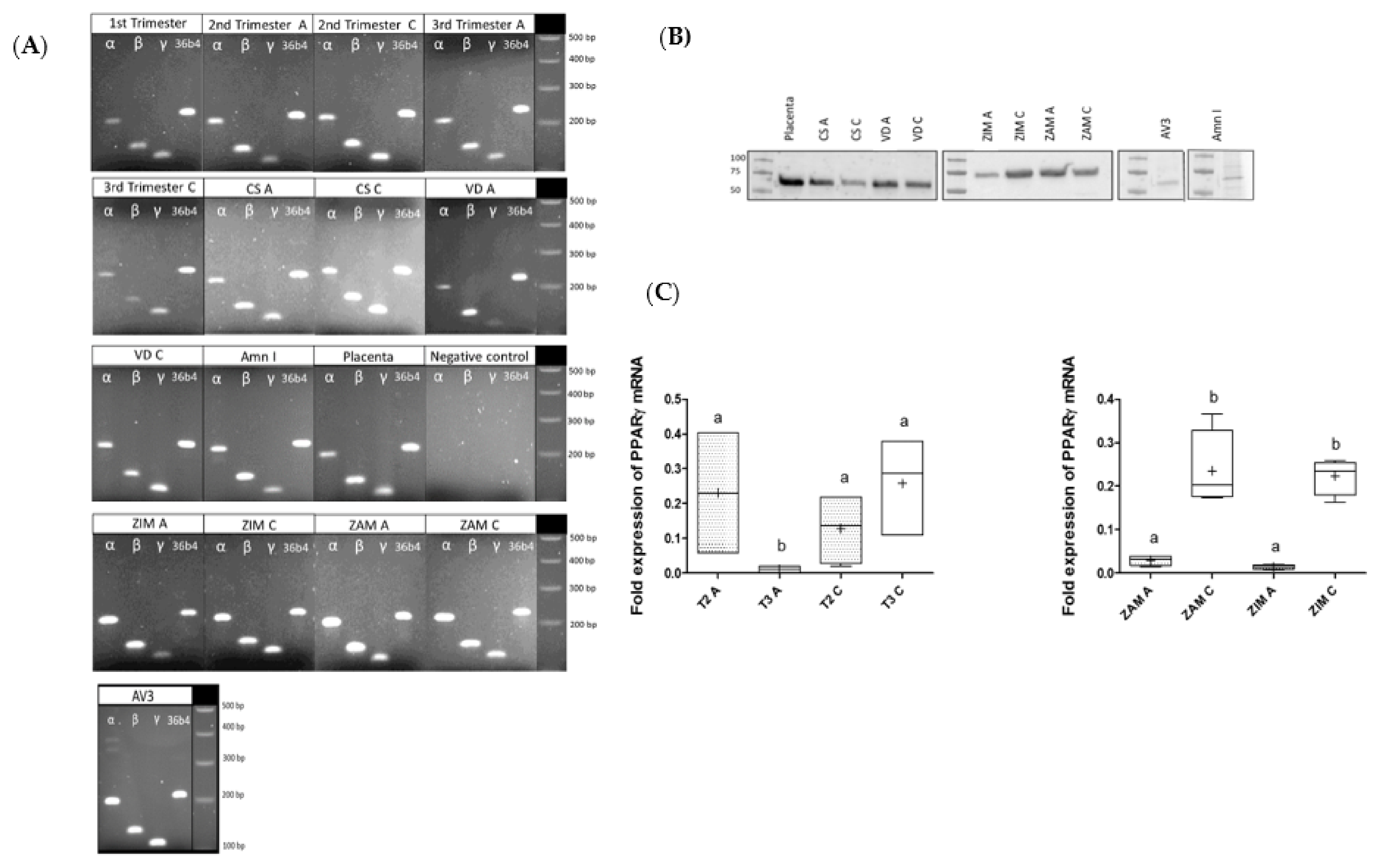
3.1. Characterization of PPAR Isoforms (α, β, γ) Expression (mRNA, Protein) within FMs and Amniotic Cells
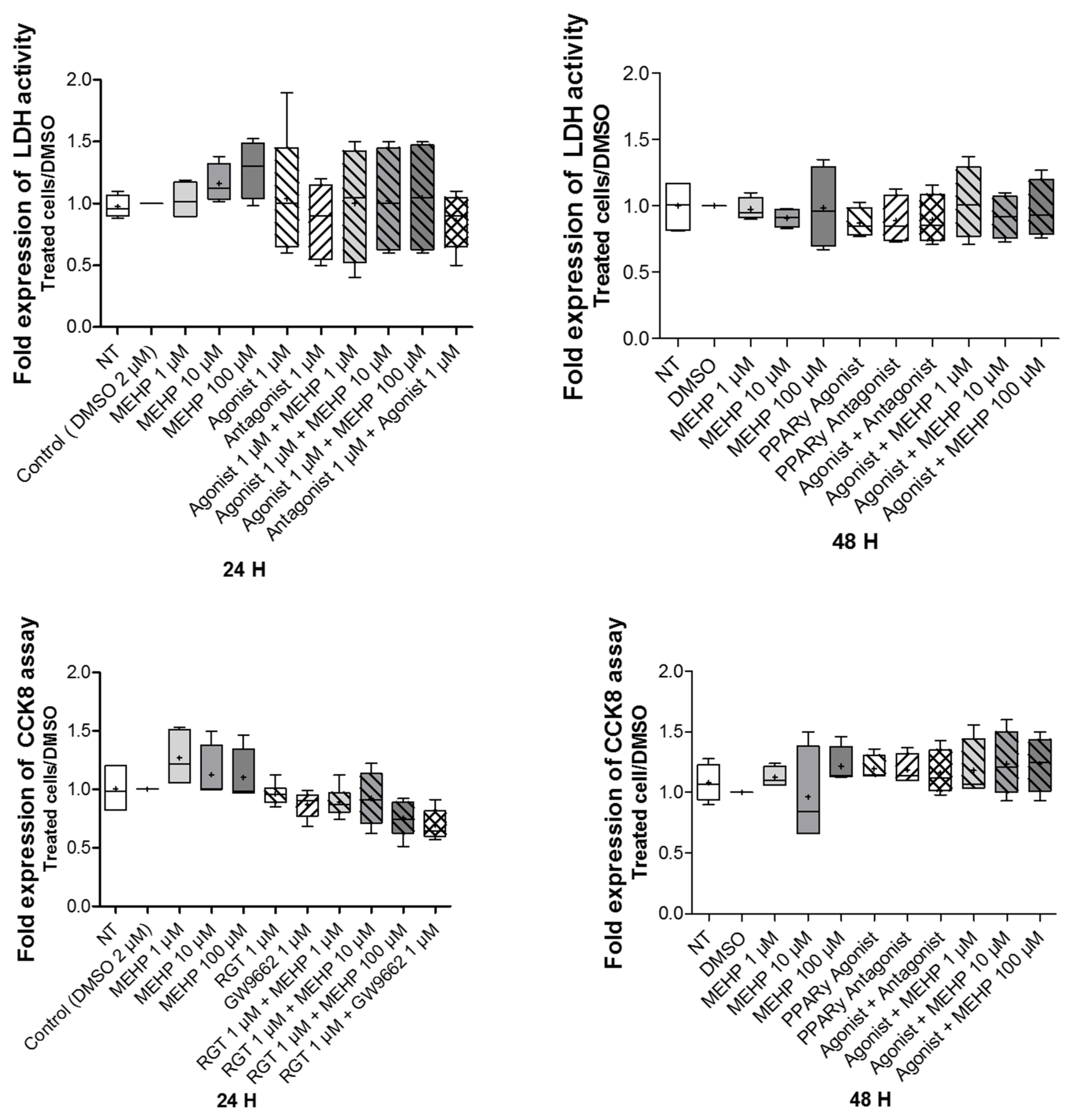
3.2. Effect of MEHP on Cell Viability and Cytotoxicity
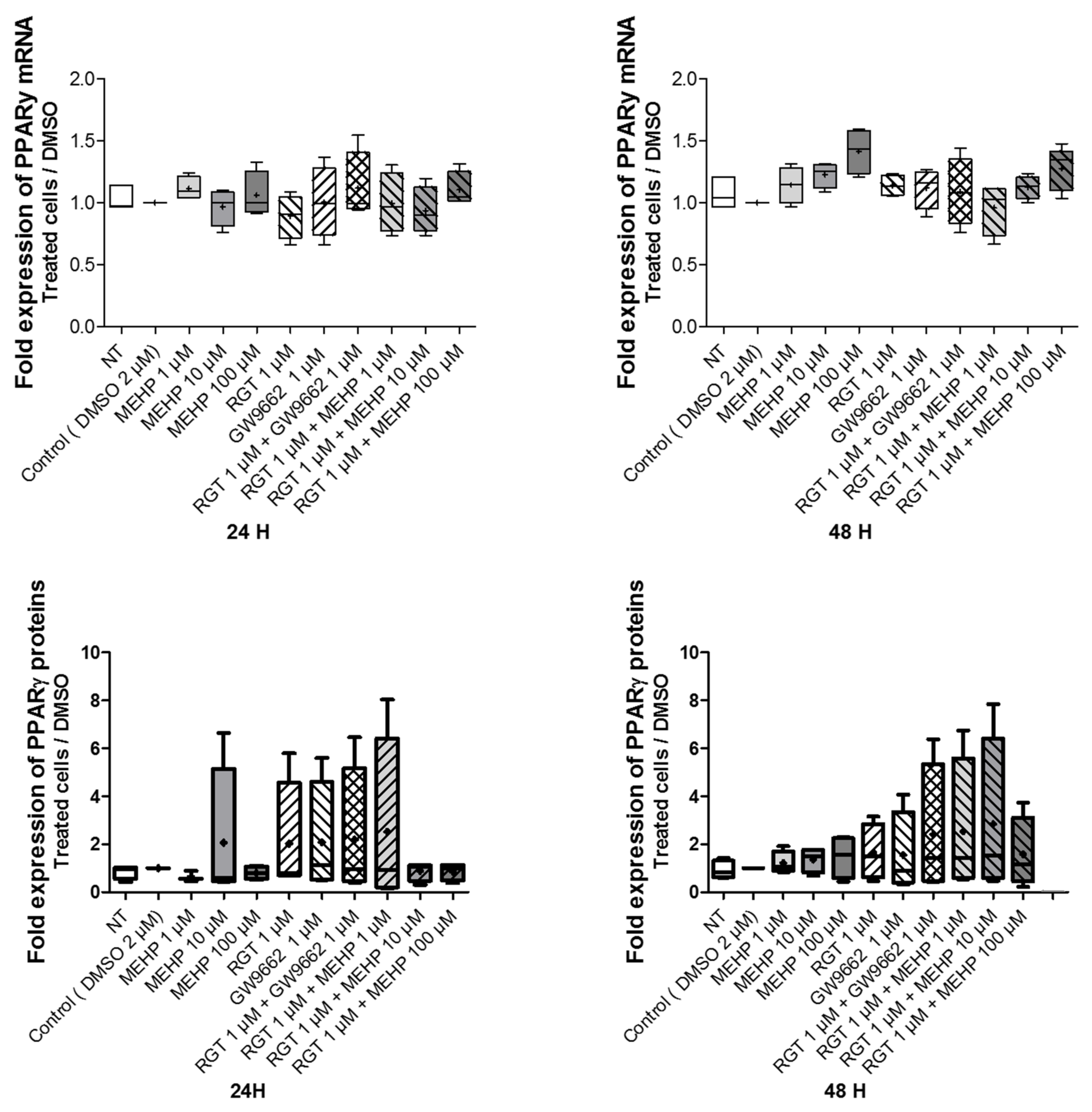
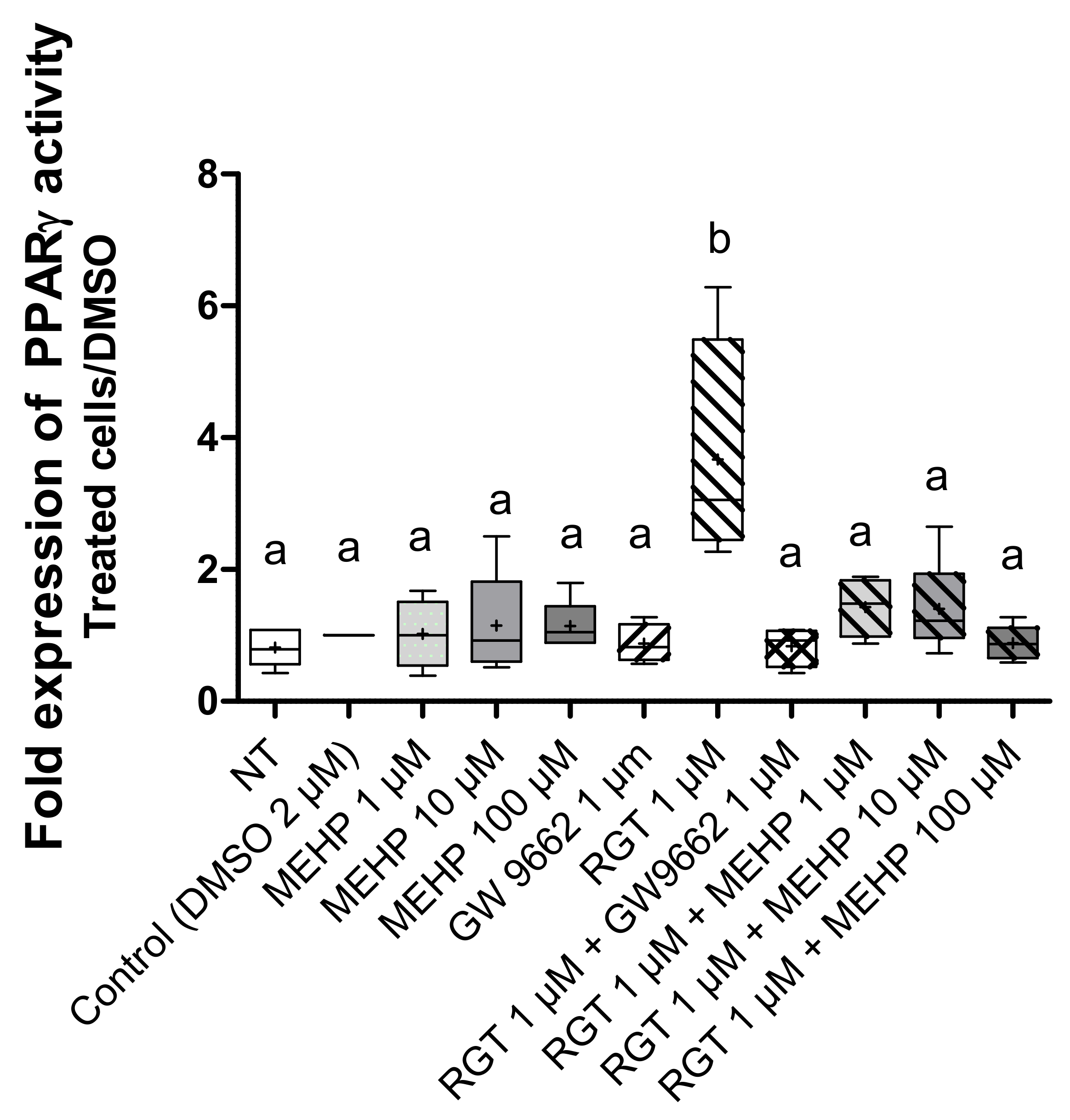
3.3. Effects of MEHP on PPARγ Expression and Transcriptional Activity in AV3 Cells
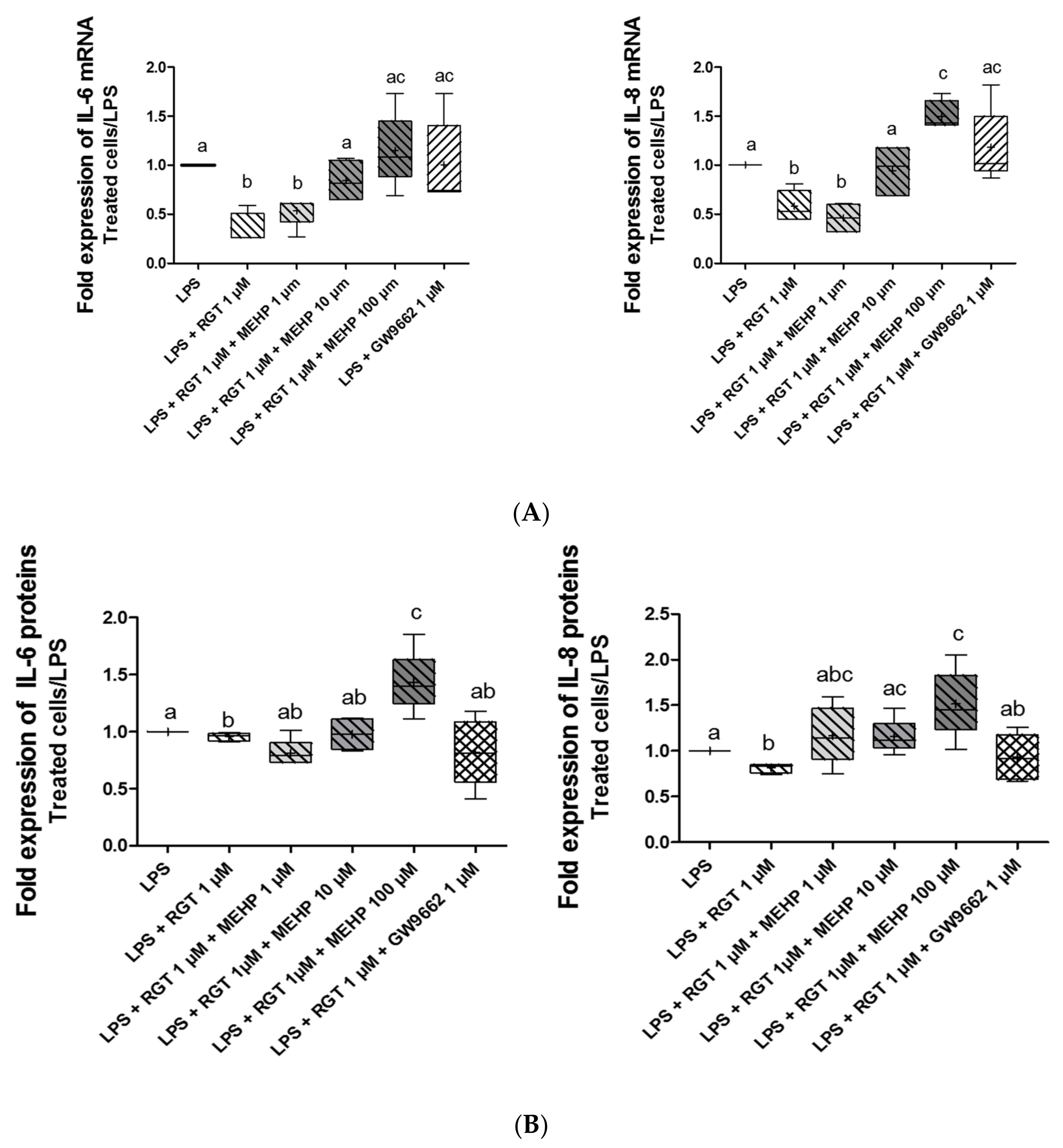
3.4. Effect of MEHP on Anti-Inflammatory Properties of PPARγ
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arikat, S.; Novince, R.W.; Mercer, B.M.; Kumar, D.; Fox, J.M.; Mansour, J.M.; Moore, J.J. Separation of Amnion from Choriodecidua Is an Integral Event to the Rupture of Normal Term Fetal Membranes and Constitutes a Significant Component of the Work Required. Am. J. Obstet. Gynecol. 2006, 194, 211–217. [Google Scholar] [CrossRef]
- Méhats, C.; Schmitz, T.; Marcellin, L.; Breuiller-Fouché, M. Biochemistry of fetal membranes rupture. Gynecol. Obstet. Fertil. 2011, 39, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Menon, R.; Richardson, L.S.; Lappas, M. Fetal Membrane Architecture, Aging and Inflammation in Pregnancy and Parturition. Placenta 2019, 79, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Moore, R.M.; Mercer, B.M.; Mansour, J.M.; Redline, R.W.; Moore, J.J. The Physiology of Fetal Membrane Weakening and Rupture: Insights Gained from the Determination of Physical Properties Revisited. Placenta 2016, 42, 59–73. [Google Scholar] [CrossRef]
- Moore, R.M.; Mansour, J.M.; Redline, R.W.; Mercer, B.M.; Moore, J.J. The Physiology of Fetal Membrane Rupture: Insight Gained from the Determination of Physical Properties. Placenta 2006, 27, 1037–1051. [Google Scholar] [CrossRef]
- Shobokshi, A.; Shaarawy, M. Maternal Serum and Amniotic Fluid Cytokines in Patients with Preterm Premature Rupture of Membranes with and without Intrauterine Infection. Int. J. Gynecol. Obstet. 2002, 79, 209–215. [Google Scholar] [CrossRef]
- Romero, R.; Miranda, J.; Chaemsaithong, P.; Chaiworapongsa, T.; Kusanovic, J.P.; Dong, Z.; Ahmed, A.I.; Shaman, M.; Lannaman, K.; Yoon, B.H.; et al. Sterile and Microbial-Associated Intra-Amniotic Inflammation in Preterm Prelabor Rupture of Membranes. J. Matern.-Fetal Neonatal Med. 2015, 28, 1394–1409. [Google Scholar] [CrossRef]
- Shoaito, H.; Petit, J.; Chissey, A.; Auzeil, N.F.M.; Guibourdenche, J.; Gil, S.; Laprévote, O.; Fournier, T.; Degrelle, S.A. The Role of Peroxisome Proliferator–Activated Receptor Gamma (PPARγ) in Mono(2-Ethylhexyl) Phthalate (MEHP)-Mediated Cytotrophoblast Differentiation. Environ. Health Perspect. 2019, 127, 27003. [Google Scholar] [CrossRef]
- Ciavarella, C.; Motta, I.; Valente, S.; Pasquinelli, G. Pharmacological (or Synthetic) and Nutritional Agonists of PPAR-γ as Candidates for Cytokine Storm Modulation in COVID-19 Disease. Molecules 2020, 25, E2076. [Google Scholar] [CrossRef]
- Lappas, M.; Permezel, M.; Georgiou, H.M.; Rice, G.E. Regulation of Proinflammatory Cytokines in Human Gestational Tissues by Peroxisome Proliferator-Activated Receptor-γ: Effect of 15-Deoxy-Δ12,14-PGJ2 and Troglitazone. J. Clin. Endocrinol. Metab. 2002, 87, 4667–4672. [Google Scholar] [CrossRef] [Green Version]
- Saillenfait, A.-M.; Laudet-Hesbert, A. Phtalates. EMC-Toxicol.-Pathol. 2005, 2, 1–13. [Google Scholar] [CrossRef]
- Kalo, D.; Vitorino Carvalho, A.; Archilla, C.; Duranthon, V.; Moroldo, M.; Levin, Y.; Kupervaser, M.; Smith, Y.; Roth, Z. Mono(2-Ethylhexyl) Phthalate (MEHP) Induces Transcriptomic Alterations in Oocytes and Their Derived Blastocysts. Toxicology 2019, 421, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, K.K.; McElrath, T.F.; Meeker, J.D. Environmental Phthalate Exposure and Preterm Birth. JAMA Pediatr. 2014, 168, 61–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domínguez-Romero, E.; Scheringer, M. A Review of Phthalate Pharmacokinetics in Human and Rat: What Factors Drive Phthalate Distribution and Partitioning? Drug Metab. Rev. 2019, 51, 314–329. [Google Scholar] [CrossRef] [Green Version]
- Qian, Y.; Shao, H.; Ying, X.; Huang, W.; Hua, Y. The Endocrine Disruption of Prenatal Phthalate Exposure in Mother and Offspring. Front. Public Health 2020, 8, 366. [Google Scholar] [CrossRef]
- Marie, C.; Vendittelli, F.; Sauvant-Rochat, M.-P. Obstetrical Outcomes and Biomarkers to Assess Exposure to Phthalates: A Review. Environ. Int. 2015, 83, 116–136. [Google Scholar] [CrossRef]
- Whyatt, R.M.; Adibi, J.J.; Calafat, A.M.; Camann, D.E.; Rauh, V.; Bhat, H.K.; Perera, F.P.; Andrews, H.; Just, A.C.; Hoepner, L.; et al. Prenatal Di(2-Ethylhexyl)Phthalate Exposure and Length of Gestation among an Inner-City Cohort. Pediatrics 2009, 124, e1213–e1220. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Zhang, Y.-W.; Huang, K.; Yan, S.-Q.; Mao, L.-J.; Ge, X.; Xu, Y.-Q.; Xu, Y.-Y.; Sheng, J.; Jin, Z.-X.; et al. Urinary Concentrations of Phthalate Metabolites in Early Pregnancy Associated with Clinical Pregnancy Loss in Chinese Women. Sci. Rep. 2017, 7, 6800. [Google Scholar] [CrossRef]
- Yang, C.; Song, G.; Lim, W. A Mechanism for the Effect of Endocrine Disrupting Chemicals on Placentation. Chemosphere 2019, 231, 326–336. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.-C.; Kuo, P.-L.; Chou, Y.-Y.; Lin, S.-J.; Lee, C.-C. Association between Prenatal Exposure to Phthalates and the Health of Newborns. Environ. Int. 2009, 35, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.S.; Nørgaard-Pedersen, B.; Toft, G.; Hougaard, D.M.; Bonde, J.P.; Cohen, A.; Thulstrup, A.M.; Ivell, R.; Anand-Ivell, R.; Lindh, C.H.; et al. Phthalates and Perfluorooctanesulfonic Acid in Human Amniotic Fluid: Temporal Trends and Timing of Amniocentesis in Pregnancy. Environ. Health Perspect. 2012, 120, 897–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suteau, V.; Briet, C.; Lebeault, M.; Gourdin, L.; Henrion, D.; Rodien, P.; Munier, M. Human Amniotic Fluid-Based Exposure Levels of Phthalates and Bisphenol A Mixture Reduce INSL3/RXFP2 Signaling. Environ. Int. 2020, 138, 105585. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Cook, T.J.; Knipp, G.T. Effects of Di-(2-Ethylhexyl)-Phthalate (DEHP) and Its Metabolites on Fatty Acid Homeostasis Regulating Proteins in Rat Placental HRP-1 Trophoblast Cells. Toxicol. Sci. 2005, 84, 287–300. [Google Scholar] [CrossRef]
- Shang, J.; Mosure, S.A.; Zheng, J.; Brust, R.; Bass, J.; Nichols, A.; Solt, L.A.; Griffin, P.R.; Kojetin, D.J. A Molecular Switch Regulating Transcriptional Repression and Activation of PPARγ. Nat. Commun. 2020, 11, 956. [Google Scholar] [CrossRef]
- McKinnon, B.; Bersinger, N.A.; Mueller, M.D. Peroxisome Proliferating Activating Receptor Gamma-Independent Attenuation of Interleukin 6 and Interleukin 8 Secretion from Primary Endometrial Stromal Cells by Thiazolidinediones. Fertil. Steril. 2012, 97, 657–664. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y. Crosstalk Between Peroxisome Proliferator-Activated Receptor Gamma and the Canonical WNT/β-Catenin Pathway in Chronic Inflammation and Oxidative Stress During Carcinogenesis. Front. Immunol. 2018, 9, 745. [Google Scholar] [CrossRef] [Green Version]
- Tetz, L.M.; Cheng, A.A.; Korte, C.S.; Giese, R.W.; Wang, P.; Harris, C.; Meeker, J.D.; Loch-Caruso, R. Mono-2-Ethylhexyl Phthalate Induces Oxidative Stress Responses in Human Placental Cells in Vitro. Toxicol. Appl. Pharmacol. 2013, 268, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Potter, S.; Hansen, W.R.; Keelan, J.A.; Mitchell, M.D. Characterization of the Amnion-Derived AV3 Cell Line for Use as a Model for Investigation of Prostaglandin-Cytokine Interactions in Human Amnion. Prostaglandins Leukot. Essent. Fat. Acids PLEFA 1999, 61, 373–379. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence 5′-3′ (F: Forward, r: Reverse) | Product Size (bp) | Annealing Temperature (°C) |
|---|---|---|---|
| hsPPARα | F: GATCTAGAGAGCCCGTTATC | 208 | 61 |
| R: GGACCACAGGATAAGTCAC | |||
| hsPPARβ | F: AGTGCCTGGCACTGGGCATG | 222 | 61 |
| R: TCAGGTAGGCATTGTAGATGTGC | |||
| hsPPARγ | F: AGTGGGGATGTCTCATAATGCC | 117 | 61 |
| R: GCAGAGTTTCCTCTGTGATA | |||
| hs36b4 | F: AGGCTTTAGGTATCACCACT | 219 | 61 |
| R:GCAGAGTTTCCTCTGTGATA |
| Gene | Sequence 5′-3′ (F: Forward, r: Reverse) | Product Size (bp) | Annealing Temperature (°C) |
|---|---|---|---|
| hsIL6 | F: AATGAGGAGACTTGCCTGGTG | 143 | 61 |
| R: AGGAACTGGATCAGGACTTTTG | |||
| hsIL8 | F: TGATTTCTGCAGCTCTGTGTG | 154 | 61 |
| R: TCTGTGTTGGCGCAGTGTGG | |||
| hs36B4 | F: AGGCTTTAGGTATCACCACT | 219 | 61 |
| R: GCAGAGTTTCCTCTGTGATA | |||
| hsRSP17 | F: TGCGAGGAGATCGCCATTATC | 169 | 61 |
| R: AAGGCTGAGACCTCAGGAAC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antoine, A.; De Sousa Do Outeiro, C.; Charnay, C.; Belville, C.; Henrioux, F.; Gallot, D.; Blanchon, L.; Minet-Quinard, R.; Sapin, V. Dysregulation of the Amniotic PPARγ Pathway by Phthalates: Modulation of the Anti-Inflammatory Activity of PPARγ in Human Fetal Membranes. Life 2022, 12, 544. https://doi.org/10.3390/life12040544
Antoine A, De Sousa Do Outeiro C, Charnay C, Belville C, Henrioux F, Gallot D, Blanchon L, Minet-Quinard R, Sapin V. Dysregulation of the Amniotic PPARγ Pathway by Phthalates: Modulation of the Anti-Inflammatory Activity of PPARγ in Human Fetal Membranes. Life. 2022; 12(4):544. https://doi.org/10.3390/life12040544
Chicago/Turabian StyleAntoine, Audrey, Coraline De Sousa Do Outeiro, Coline Charnay, Corinne Belville, Fanny Henrioux, Denis Gallot, Loïc Blanchon, Régine Minet-Quinard, and Vincent Sapin. 2022. "Dysregulation of the Amniotic PPARγ Pathway by Phthalates: Modulation of the Anti-Inflammatory Activity of PPARγ in Human Fetal Membranes" Life 12, no. 4: 544. https://doi.org/10.3390/life12040544