
The Transition from Cancer “omics” to “epi-omics” through Next- and Third-Generation Sequencing

 , , and
, , and
Abstract
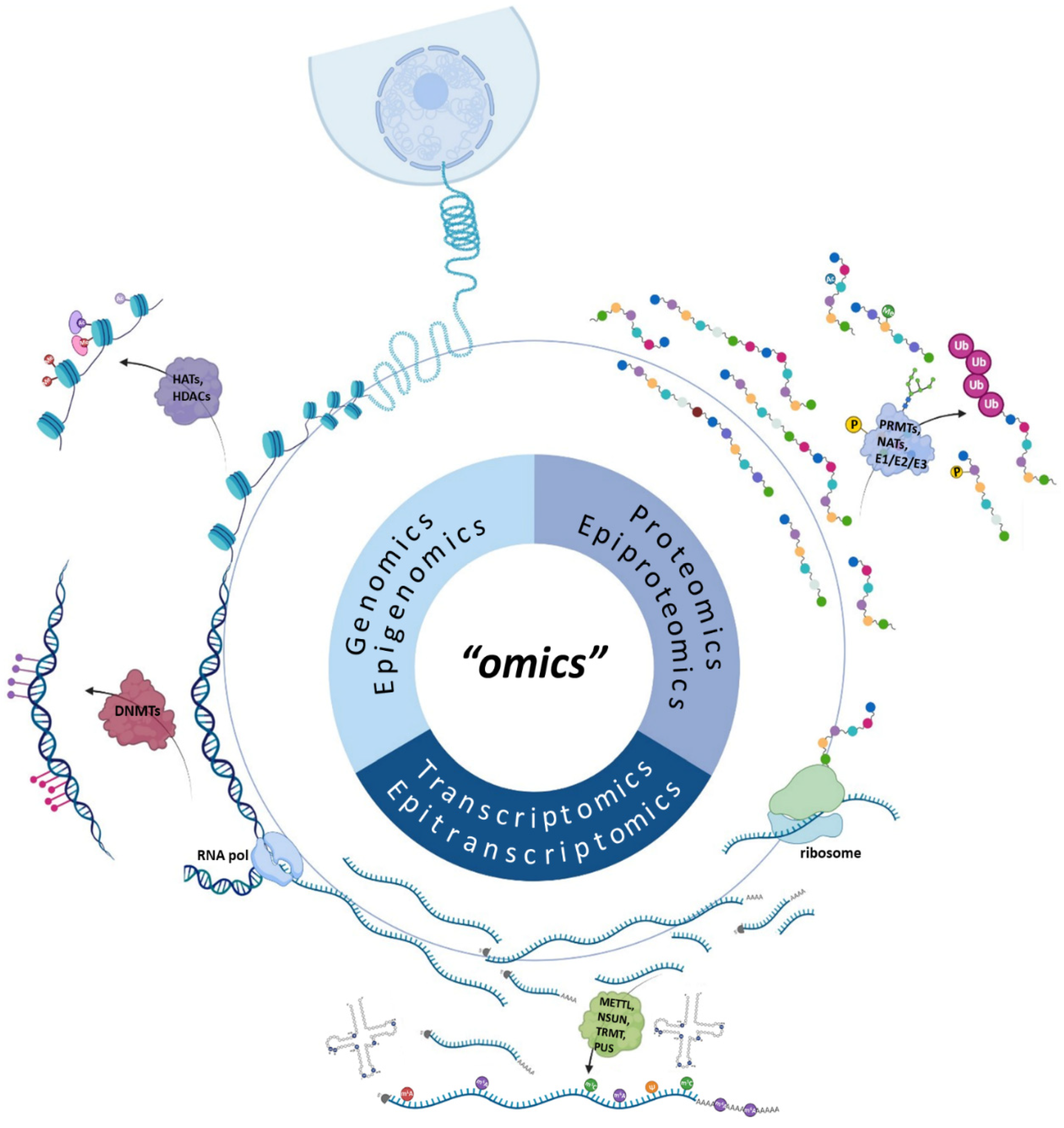
:1. Introduction
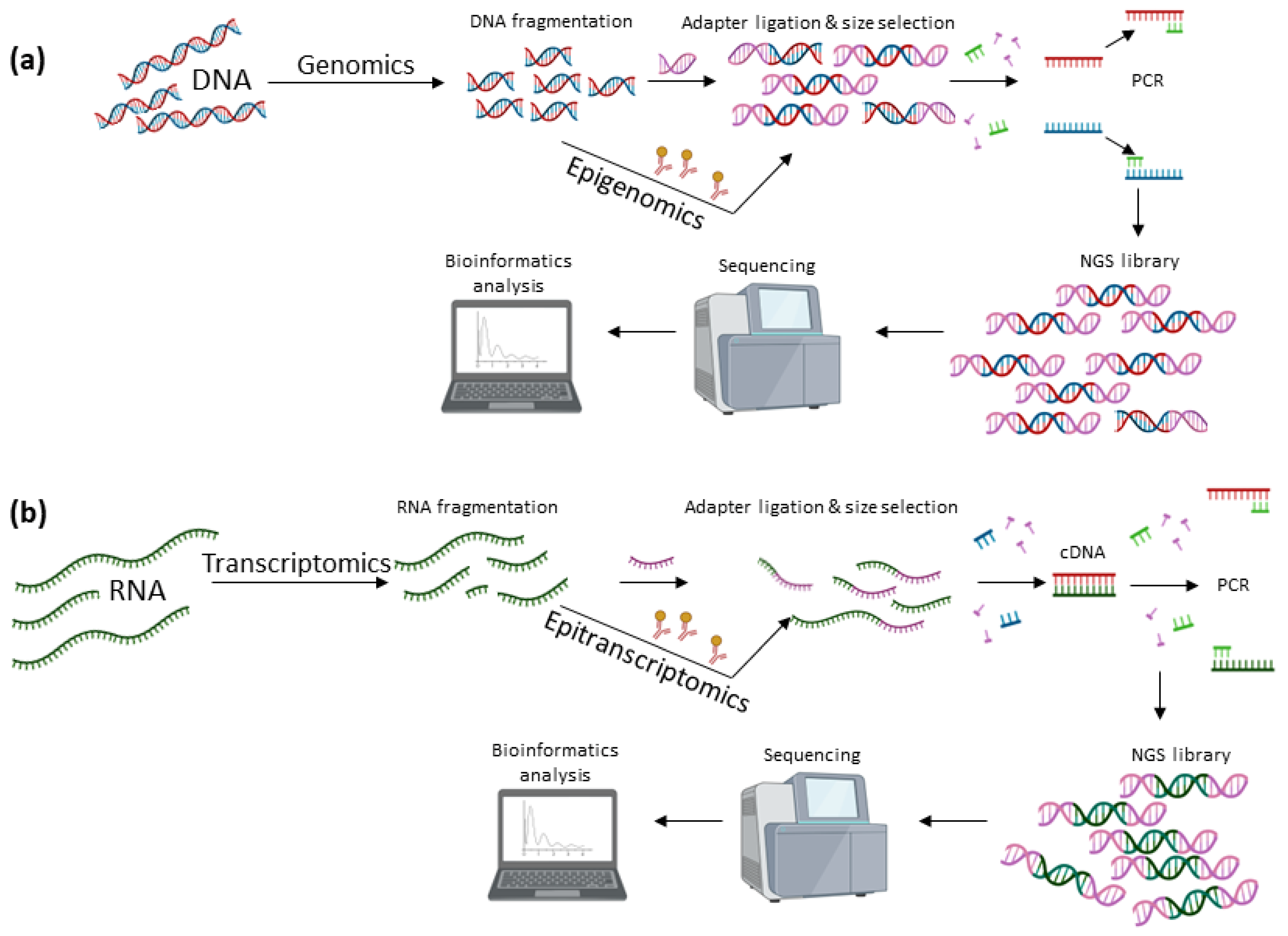
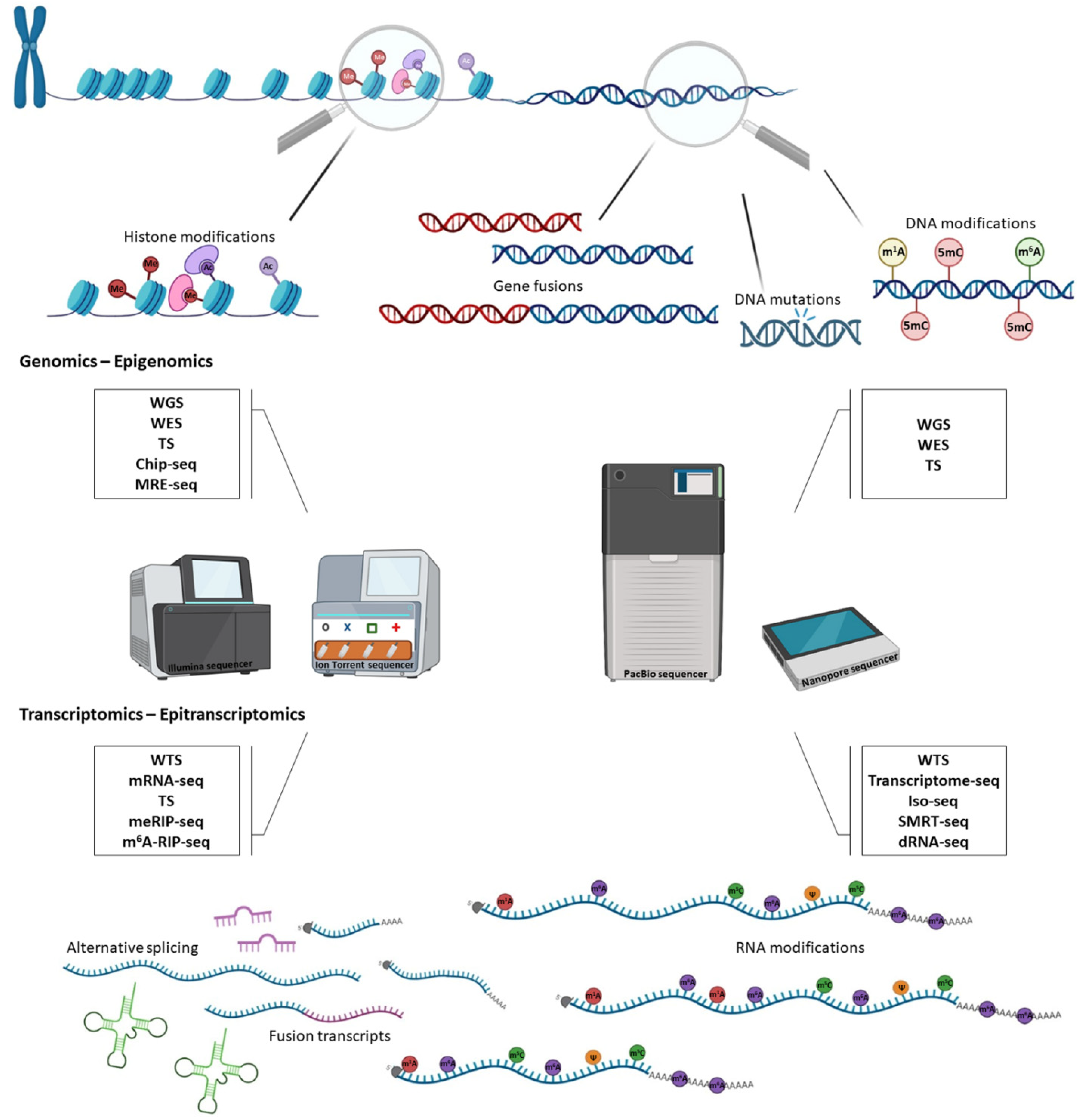
2. The Impact of DNA Sequencing in Cancer Genomics
3. Epigenomics in Cancer Research
4. The Advent of RNA Sequencing for Transcriptome Profiling
5. The Golden Era of Epitranscriptomics in Cancer Research

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transcriptomics/ Epitranscriptomics | Technology | Application | References |
|---|---|---|---|
| Fusion transcripts | NGS | WTS, TS | [157,158,159] |
| TGS | SMRT-seq, Direct RNA-seq | [160] | |
| Alternative splicing | NGS | RNA-seq, targeted RNA sequencing | [161,162] |
| TGS | Direct RNA-seq, WTS | [160,163] | |
| mRNA polydadenylation | NGS | 3′ enriched RNA seq, 3′ mRNA seq, PAT-seq, Poly(A) ClickSeq | [164,165,166,167] |
| TGS | Full length mRNA seq, FLAM-seq, Long read cDNA-seq | [168,169] | |
| ncRNAs/lncRNAs | NGS | scRNA-seq, WTS, WES, AQRNA-seq, ncPRo-seq, miRNA-seq | [170,171,172,173] |
| TGS | Nanopore-induced phase-shift sequencing (NIPSS) | [86] | |
| m6A | NGS | Transcriptome-wide m6A seq, m6A-RIP seq, m6A-REF seq | [174,175] |
| TGS | Direct RNA-seq | [176] | |
| m5C | NGS | RNA-BisSeq, RIP-seq, MeRIP-seq, m5C-RIP seq, AZA-IP seq | [153,154,155,177] |
| TGS | Direct RNA-seq | [178] | |
| Ψ | NGS | Pseudo-seq | [156] |
| TGS | Direct RNA-seq | [179] | |
| m1A | NGS | ARM-seq, m1A-quant seq, m1A-seq | [180,181,182] |
6. The Road Ahead in Proteomics and Epiproteomics
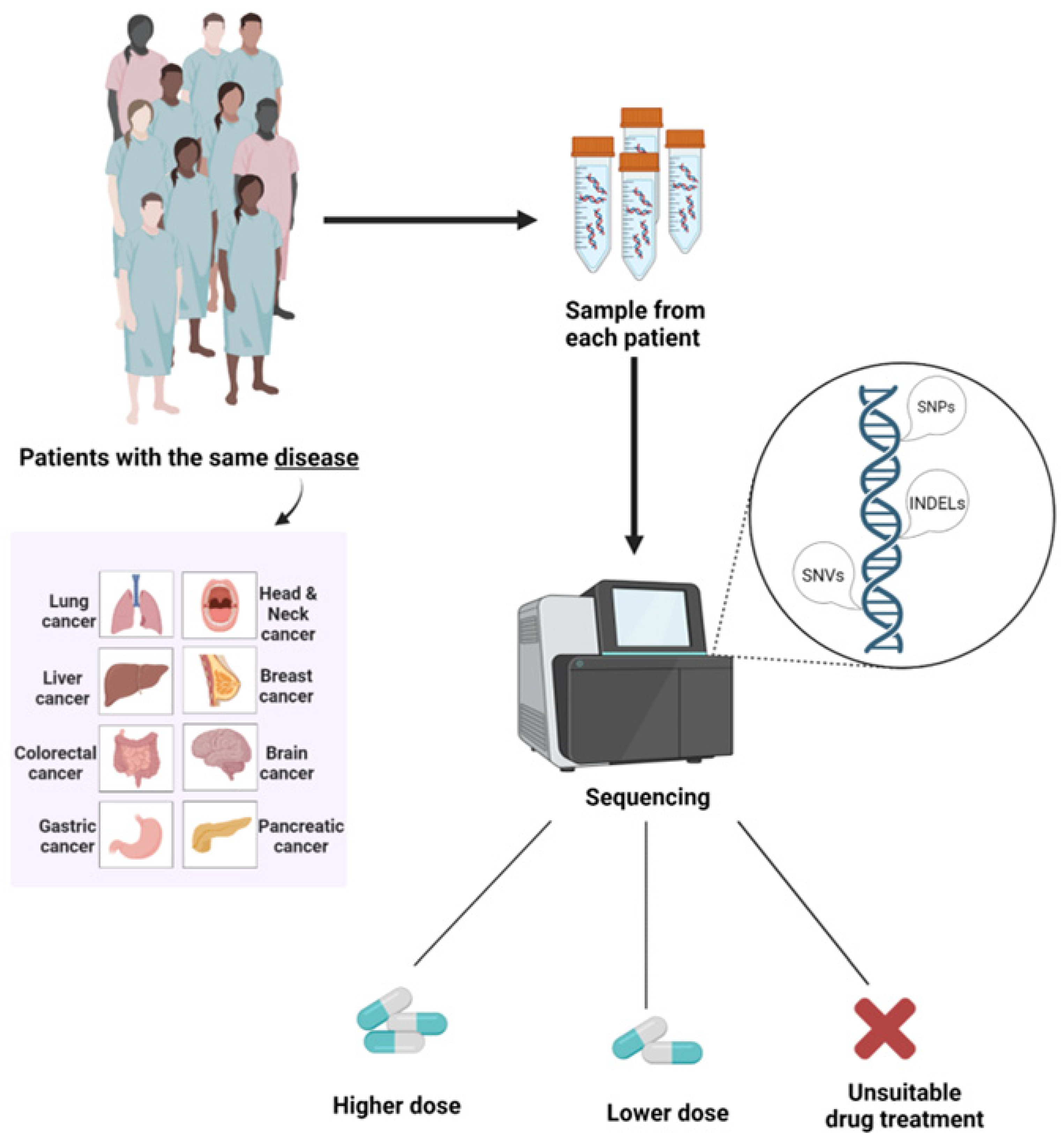
7. Pharmacogenomics in Medical Oncology
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yadav, S.P. The wholeness in suffix -omics, -omes, and the word om. J. Biomol. Tech. 2007, 18, 277. [Google Scholar] [PubMed]
- Olivier, M.; Asmis, R.; Hawkins, G.A.; Howard, T.D.; Cox, L.A. The need for multi-omics biomarker signatures in precision medicine. Int. J. Mol. Sci. 2019, 20, 4781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzoni, C.; Kia, D.A.; Vandrovcova, J.; Hardy, J.; Wood, N.W.; Lewis, P.A.; Ferrari, R. Genome, transcriptome and proteome: The rise of omics data and their integration in biomedical sciences. Brief. Bioinform. 2018, 19, 286–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Liu, D.; Dong, S.; Zeng, L.; Wu, Z.; Zhao, P.; Zhang, L.; Chen, Z.S.; Zou, C. Epitranscriptomics and epiproteomics in cancer drug resistance: Therapeutic implications. Signal Transduct. Target. Ther. 2020, 5, 193. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Epigenomics: Technologies and Applications. Circ. Res. 2018, 122, 1191–1199. [Google Scholar] [CrossRef]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, S.; Hosen, M.I.; Ahmed, M.; Shekhar, H.U. Onco-multi-OMICS approach: A new frontier in cancer research. Biomed. Res. Int. 2018, 2018, 9836256. [Google Scholar] [CrossRef] [Green Version]
- Tomczak, K.; Czerwinska, P.; Wiznerowicz, M. The cancer genome atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- Zhang, J.; Bajari, R.; Andric, D.; Gerthoffert, F.; Lepsa, A.; Nahal-Bose, H.; Stein, L.D.; Ferretti, V. The International Cancer Genome Consortium Data Portal. Nat. Biotechnol. 2019, 37, 367–369. [Google Scholar] [CrossRef]
- Morganti, S.; Tarantino, P.; Ferraro, E.; D’Amico, P.; Viale, G.; Trapani, D.; Duso, B.A.; Curigliano, G. Complexity of genome sequencing and reporting: Next generation sequencing (NGS) technologies and implementation of precision medicine in real life. Crit. Rev. Oncol. Hematol. 2019, 133, 171–182. [Google Scholar] [CrossRef]
- Shendure, J.; Balasubramanian, S.; Church, G.M.; Gilbert, W.; Rogers, J.; Schloss, J.A.; Waterston, R.H. DNA sequencing at 40: Past, present and future. Nature 2017, 550, 345–353. [Google Scholar] [CrossRef]
- Del Giacco, L.; Cattaneo, C. Introduction to genomics. Methods Mol. Biol. 2012, 823, 79–88. [Google Scholar] [CrossRef]
- Pareek, C.S.; Smoczynski, R.; Tretyn, A. Sequencing technologies and genome sequencing. J. Appl. Genet. 2011, 52, 413–435. [Google Scholar] [CrossRef] [Green Version]
- Berger, M.F.; Mardis, E.R. The emerging clinical relevance of genomics in cancer medicine. Nat. Rev. Clin. Oncol. 2018, 15, 353–365. [Google Scholar] [CrossRef]
- Haley, B.; Roudnicky, F. Functional genomics for cancer drug target discovery. Cancer Cell 2020, 38, 31–43. [Google Scholar] [CrossRef]
- Farid, S.G.; Morris-Stiff, G. “OMICS” technologies and their role in foregut primary malignancies. Curr. Probl. Surg. 2015, 52, 409–441. [Google Scholar] [CrossRef]
- Stratton, M.R. Exploring the genomes of cancer cells: Progress and promise. Science 2011, 331, 1553–1558. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wu, G.; Miller, C.P.; Tatevossian, R.G.; Dalton, J.D.; Tang, B.; Orisme, W.; Punchihewa, C.; Parker, M.; Qaddoumi, I.; et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat. Genet. 2013, 45, 602–612. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Benayed, R.; Ho, C.; Mullaney, K.; Sukhadia, P.; Rios, K.; Berry, R.; Rubin, B.P.; Nafa, K.; Wang, L.; et al. Diagnosis of known sarcoma fusions and novel fusion partners by targeted RNA sequencing with identification of a recurrent ACTB-FOSB fusion in pseudomyogenic hemangioendothelioma. Mod. Pathol. 2019, 32, 609–620. [Google Scholar] [CrossRef]
- Stangl, C.; de Blank, S.; Renkens, I.; Westera, L.; Verbeek, T.; Valle-Inclan, J.E.; Gonzalez, R.C.; Henssen, A.G.; van Roosmalen, M.J.; Stam, R.W.; et al. Partner independent fusion gene detection by multiplexed CRISPR-Cas9 enrichment and long read nanopore sequencing. Nat. Commun. 2020, 11, 2861. [Google Scholar] [CrossRef] [PubMed]
- Schulze, K.; Imbeaud, S.; Letouze, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.H.; Li, Y.; Guo, J.; Pei, L.; Rauch, T.A.; Kramer, R.S.; Macmil, S.L.; Wiley, G.B.; Bennett, L.B.; Schnabel, J.L.; et al. Genome-wide DNA methylation maps in follicular lymphoma cells determined by methylation-enriched bisulfite sequencing. PLoS ONE 2010, 5, e13020. [Google Scholar] [CrossRef] [PubMed]
- Gilpatrick, T.; Lee, I.; Graham, J.E.; Raimondeau, E.; Bowen, R.; Heron, A.; Downs, B.; Sukumar, S.; Sedlazeck, F.J.; Timp, W. Targeted nanopore sequencing with Cas9-guided adapter ligation. Nat. Biotechnol. 2020, 38, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Moon, S.J.; Hong, S.; Won, H.H.; Kim, J.H. DBC1 is a key positive regulator of enhancer epigenomic writers KMT2D and p300. Nucleic Acids Res. 2022, 50, 7873–7888. [Google Scholar] [CrossRef]
- Levin, J.Z.; Berger, M.F.; Adiconis, X.; Rogov, P.; Melnikov, A.; Fennell, T.; Nusbaum, C.; Garraway, L.A.; Gnirke, A. Targeted next-generation sequencing of a cancer transcriptome enhances detection of sequence variants and novel fusion transcripts. Genome Biol. 2009, 10, R115. [Google Scholar] [CrossRef] [Green Version]
- Hoogstrate, Y.; Komor, M.A.; Bottcher, R.; van Riet, J.; van de Werken, H.J.G.; van Lieshout, S.; Hoffmann, R.; van den Broek, E.; Bolijn, A.S.; Dits, N.; et al. Fusion transcripts and their genomic breakpoints in polyadenylated and ribosomal RNA-minus RNA sequencing data. Gigascience 2021, 10, giab080. [Google Scholar] [CrossRef]
- Qu, H.; Wang, Z.; Zhang, Y.; Zhao, B.; Jing, S.; Zhang, J.; Ye, C.; Xue, Y.; Yang, L. Long-read nanopore sequencing identifies mismatch repair-deficient related genes with alternative splicing in colorectal cancer. Dis. Markers 2022, 2022, 4433270. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, X.; Lei, W.; Liang, J.; Xu, Y.; Liu, H.; Ma, S. Genome-wide profiling reveals alternative polyadenylation of mRNA in human non-small cell lung cancer. J. Transl. Med. 2019, 17, 257. [Google Scholar] [CrossRef]
- Rabbani, B.; Tekin, M.; Mahdieh, N. The promise of whole-exome sequencing in medical genetics. J. Hum. Genet. 2014, 59, 5–15. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [Green Version]
- Kozarewa, I.; Armisen, J.; Gardner, A.F.; Slatko, B.E.; Hendrickson, C.L. Overview of target enrichment strategies. Curr. Protoc. Mol. Biol. 2015, 112, 7–21. [Google Scholar] [CrossRef]
- Sjoblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; et al. The consensus coding sequences of human breast and colorectal cancers. Science 2006, 314, 268–274. [Google Scholar] [CrossRef]
- Vanlallawma, A.; Lallawmzuali, D.; Pautu, J.L.; Scaria, V.; Sivasubbu, S.; Kumar, N.S. Whole exome sequencing of pediatric leukemia reveals a novel InDel within FLT-3 gene in AML patient from Mizo tribal population, Northeast India. BMC Genom. Data 2022, 23, 23. [Google Scholar] [CrossRef]
- Fewings, E.; Larionov, A.; Redman, J.; Goldgraben, M.A.; Scarth, J.; Richardson, S.; Brewer, C.; Davidson, R.; Ellis, I.; Evans, D.G.; et al. Germline pathogenic variants in PALB2 and other cancer-predisposing genes in families with hereditary diffuse gastric cancer without CDH1 mutation: A whole-exome sequencing study. Lancet Gastroenterol. Hepatol. 2018, 3, 489–498. [Google Scholar] [CrossRef] [Green Version]
- Manier, S.; Park, J.; Capelletti, M.; Bustoros, M.; Freeman, S.S.; Ha, G.; Rhoades, J.; Liu, C.J.; Huynh, D.; Reed, S.C.; et al. Whole-exome sequencing of cell-free DNA and circulating tumor cells in multiple myeloma. Nat. Commun. 2018, 9, 1691. [Google Scholar] [CrossRef] [Green Version]
- Rheinbay, E.; Nielsen, M.M.; Abascal, F.; Wala, J.A.; Shapira, O.; Tiao, G.; Hornshoj, H.; Hess, J.M.; Juul, R.I.; Lin, Z.; et al. Analyses of non-coding somatic drivers in 2658 cancer whole genomes. Nature 2020, 578, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, A.L.; Behr, J.; Singer, J.; Kuipers, J.; Beisel, C.; Schraml, P.; Moch, H.; Beerenwinkel, N. Detailed simulation of cancer exome sequencing data reveals differences and common limitations of variant callers. BMC Bioinform. 2017, 18, 8. [Google Scholar] [CrossRef] [Green Version]
- Royer-Bertrand, B.; Cisarova, K.; Niel-Butschi, F.; Mittaz-Crettol, L.; Fodstad, H.; Superti-Furga, A. CNV detection from exome sequencing data in routine diagnostics of rare genetic disorders: Opportunities and limitations. Genes 2021, 12, 1427. [Google Scholar] [CrossRef]
- Bewicke-Copley, F.; Arjun Kumar, E.; Palladino, G.; Korfi, K.; Wang, J. Applications and analysis of targeted genomic sequencing in cancer studies. Comput. Struct. Biotechnol. J. 2019, 17, 1348–1359. [Google Scholar] [CrossRef]
- Schultzhaus, Z.; Wang, Z.; Stenger, D. CRISPR-based enrichment strategies for targeted sequencing. Biotechnol. Adv. 2021, 46, 107672. [Google Scholar] [CrossRef] [PubMed]
- Nagahashi, M.; Shimada, Y.; Ichikawa, H.; Kameyama, H.; Takabe, K.; Okuda, S.; Wakai, T. Next generation sequencing-based gene panel tests for the management of solid tumors. Cancer Sci. 2019, 110, 6–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennings, L.J.; Arcila, M.E.; Corless, C.; Kamel-Reid, S.; Lubin, I.M.; Pfeifer, J.; Temple-Smolkin, R.L.; Voelkerding, K.V.; Nikiforova, M.N. guidelines for validation of next-generation sequencing-based oncology panels: A joint consensus recommendation of the association for molecular pathology and college of american pathologists. J. Mol. Diagn. 2017, 19, 341–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.S.; Huang, H.D.; Yeh, K.T.; Chang, J.G. Identification of novel mutations in endometrial cancer patients by whole-exome sequencing. Int. J. Oncol. 2017, 50, 1778–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.S.; Huang, H.D.; Yeh, K.T.; Chang, J.G. Genetic alterations in endometrial cancer by targeted next-generation sequencing. Exp. Mol. Pathol. 2016, 100, 8–12. [Google Scholar] [CrossRef]
- Dulak, A.M.; Stojanov, P.; Peng, S.; Lawrence, M.S.; Fox, C.; Stewart, C.; Bandla, S.; Imamura, Y.; Schumacher, S.E.; Shefler, E.; et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat. Genet. 2013, 45, 478–486. [Google Scholar] [CrossRef] [Green Version]
- Heydt, C.; Wolwer, C.B.; Velazquez Camacho, O.; Wagener-Ryczek, S.; Pappesch, R.; Siemanowski, J.; Rehker, J.; Haller, F.; Agaimy, A.; Worm, K.; et al. Detection of gene fusions using targeted next-generation sequencing: A comparative evaluation. BMC Med. Genom. 2021, 14, 62. [Google Scholar] [CrossRef]
- Park, H.J.; Baek, I.; Cheang, G.; Solomon, J.P.; Song, W. Comparison of RNA-based next-generation sequencing assays for the detection of NTRK gene fusions. J. Mol. Diagn. 2021, 23, 1443–1451. [Google Scholar] [CrossRef]
- Miller, D.E.; Sulovari, A.; Wang, T.; Loucks, H.; Hoekzema, K.; Munson, K.M.; Lewis, A.P.; Fuerte, E.P.A.; Paschal, C.R.; Walsh, T.; et al. Targeted long-read sequencing identifies missing disease-causing variation. Am. J. Hum. Genet. 2021, 108, 1436–1449. [Google Scholar] [CrossRef]
- Jeong, M.; Guzman, A.G.; Goodell, M.A. Genome-Wide Analysis of DNA Methylation in hematopoietic cells: DNA methylation analysis by WGBS. Methods Mol. Biol. 2017, 1633, 137–149. [Google Scholar] [CrossRef]
- Nakabayashi, K.; Yamamura, M.; Haseagawa, K.; Hata, K. Reduced representation bisulfite sequencing (RRBS). Methods Mol. Biol. 2022, 2577, 39–51. [Google Scholar] [CrossRef]
- Kirschner, K.; Krueger, F.; Green, A.R.; Chandra, T. Multiplexing for oxidative bisulfite sequencing (oxBS-seq). Methods Mol. Biol. 2018, 1708, 665–678. [Google Scholar] [CrossRef]
- Yu, M.; Han, D.; Hon, G.C.; He, C. Tet-assisted bisulfite sequencing (TAB-seq). Methods Mol. Biol. 2018, 1708, 645–663. [Google Scholar] [CrossRef]
- Song, C.X.; Szulwach, K.E.; Dai, Q.; Fu, Y.; Mao, S.Q.; Lin, L.; Street, C.; Li, Y.; Poidevin, M.; Wu, H.; et al. Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell 2013, 153, 678–691. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Song, C.X.; Szulwach, K.; Wang, Z.; Weidenbacher, P.; Jin, P.; He, C. Chemical modification-assisted bisulfite sequencing (CAB-Seq) for 5-carboxylcytosine detection in DNA. J. Am. Chem. Soc. 2013, 135, 9315–9317. [Google Scholar] [CrossRef] [Green Version]
- Taiwo, O.; Wilson, G.A.; Morris, T.; Seisenberger, S.; Reik, W.; Pearce, D.; Beck, S.; Butcher, L.M. Methylome analysis using MeDIP-seq with low DNA concentrations. Nat. Protoc. 2012, 7, 617–636. [Google Scholar] [CrossRef]
- Lan, X.; Adams, C.; Landers, M.; Dudas, M.; Krissinger, D.; Marnellos, G.; Bonneville, R.; Xu, M.; Wang, J.; Huang, T.H.; et al. High resolution detection and analysis of CpG dinucleotides methylation using MBD-Seq technology. PLoS ONE 2011, 6, e22226. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Zhang, B.; Xing, X.; Wang, T. Combining MeDIP-seq and MRE-seq to investigate genome-wide CpG methylation. Methods 2015, 72, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Flusberg, B.A.; Webster, D.R.; Lee, J.H.; Travers, K.J.; Olivares, E.C.; Clark, T.A.; Korlach, J.; Turner, S.W. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 2010, 7, 461–465. [Google Scholar] [CrossRef] [Green Version]
- Tse, O.Y.O.; Jiang, P.; Cheng, S.H.; Peng, W.; Shang, H.; Wong, J.; Chan, S.L.; Poon, L.C.Y.; Leung, T.Y.; Chan, K.C.A.; et al. Genome-wide detection of cytosine methylation by single molecule real-time sequencing. Proc. Natl. Acad. Sci. USA 2021, 118, e2019768118. [Google Scholar] [CrossRef]
- Pai, S.S.; Ranjan, S.; Mathew, A.R.; Anindya, R.; Meur, G. Analysis of the long-read sequencing data using computational tools confirms the presence of 5-methylcytosine in the Saccharomyces cerevisiae genome. Access Microbiol. 2022, 4, acmi000363. [Google Scholar] [CrossRef]
- Zhang, J.; Xie, S.; Xu, J.; Liu, H.; Wan, S. Cancer biomarkers discovery of methylation modification with direct high-throughput nanopore sequencing. Front. Genet. 2021, 12, 672804. [Google Scholar] [CrossRef]
- Park, P.J. ChIP-seq: Advantages and challenges of a maturing technology. Nat. Rev. Genet. 2009, 10, 669–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hainer, S.J.; Fazzio, T.G. High-resolution chromatin profiling using CUT&RUN. Curr. Protoc. Mol. Biol. 2019, 126, e85. [Google Scholar] [CrossRef] [PubMed]
- Kaya-Okur, H.S.; Wu, S.J.; Codomo, C.A.; Pledger, E.S.; Bryson, T.D.; Henikoff, J.G.; Ahmad, K.; Henikoff, S. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 2019, 10, 1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, L.; Crawford, G.E. DNase-seq: A high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb. Protoc. 2010, 2010, pdb-prot5384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davie, K.; Jacobs, J.; Atkins, M.; Potier, D.; Christiaens, V.; Halder, G.; Aerts, S. Discovery of transcription factors and regulatory regions driving in vivo tumor development by ATAC-seq and FAIRE-seq open chromatin profiling. PLoS Genet. 2015, 11, e1004994. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, J.M.; Sinha, S. Analyzing the global chromatin structure of keratinocytes by MNase-seq. Methods Mol. Biol. 2014, 1195, 49–59. [Google Scholar] [CrossRef]
- Belton, J.M.; McCord, R.P.; Gibcus, J.H.; Naumova, N.; Zhan, Y.; Dekker, J. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods 2012, 58, 268–276. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Fullwood, M.J.; Xu, H.; Mulawadi, F.H.; Velkov, S.; Vega, V.; Ariyaratne, P.N.; Mohamed, Y.B.; Ooi, H.S.; Tennakoon, C.; et al. ChIA-PET tool for comprehensive chromatin interaction analysis with paired-end tag sequencing. Genome Biol. 2010, 11, R22. [Google Scholar] [CrossRef]
- Hrdlickova, R.; Toloue, M.; Tian, B. RNA-Seq methods for transcriptome analysis. Wiley Interdiscip. Rev. RNA 2017, 8, e1364. [Google Scholar] [CrossRef] [Green Version]
- Heidrich, N.; Bauriedl, S.; Schoen, C. Investigating RNA-protein interactions in neisseria meningitidis by RIP-Seq analysis. Methods Mol. Biol. 2019, 1969, 33–49. [Google Scholar] [CrossRef]
- Darnell, R.B. HITS-CLIP: Panoramic views of protein-RNA regulation in living cells. Wiley Interdiscip. Rev. RNA 2010, 1, 266–286. [Google Scholar] [CrossRef] [Green Version]
- Garzia, A.; Morozov, P.; Sajek, M.; Meyer, C.; Tuschl, T. PAR-CLIP for discovering target sites of RNA-binding proteins. Methods Mol. Biol. 2018, 1720, 55–75. [Google Scholar] [CrossRef]
- Helwak, A.; Tollervey, D. Mapping the miRNA interactome by cross-linking ligation and sequencing of hybrids (CLASH). Nat. Protoc. 2014, 9, 711–728. [Google Scholar] [CrossRef] [Green Version]
- Sharma, E.; Sterne-Weiler, T.; O’Hanlon, D.; Blencowe, B.J. Global Mapping of human RNA-RNA interactions. Mol. Cell 2016, 62, 618–626. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.; Quinn, J.; Chang, H.Y. Chromatin isolation by RNA purification (ChIRP). J. Vis. Exp. 2012, 61, e3912. [Google Scholar] [CrossRef]
- Simon, M.D. Capture hybridization analysis of RNA targets (CHART). Curr. Protoc. Mol. Biol. 2013, 101, 21–25. [Google Scholar] [CrossRef]
- Zhou, B.; Li, X.; Luo, D.; Lim, D.H.; Zhou, Y.; Fu, X.D. GRID-seq for comprehensive analysis of global RNA-chromatin interactions. Nat. Protoc. 2019, 14, 2036–2068. [Google Scholar] [CrossRef]
- Jukam, D.; Limouse, C.; Smith, O.K.; Risca, V.I.; Bell, J.C.; Straight, A.F. Chromatin-Associated RNA Sequencing (ChAR-seq). Curr. Protoc. Mol. Biol. 2019, 126, e87. [Google Scholar] [CrossRef]
- Watters, K.E.; Yu, A.M.; Strobel, E.J.; Settle, A.H.; Lucks, J.B. Characterizing RNA structures in vitro and in vivo with selective 2′-hydroxyl acylation analyzed by primer extension sequencing (SHAPE-Seq). Methods 2016, 103, 34–48. [Google Scholar] [CrossRef]
- Siegfried, N.A.; Busan, S.; Rice, G.M.; Nelson, J.A.; Weeks, K.M. RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP). Nat. Methods 2014, 11, 959–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Chang, H.Y.; Artandi, S.E. Analysis of RNA conformation in endogenously assembled RNPs by icSHAPE. STAR Protoc. 2021, 2, 100477. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zheng, D.; Tan, Q.; Wang, M.X.; Gu, L.Q. Nanopore-based detection of circulating microRNAs in lung cancer patients. Nat. Nanotechnol. 2011, 6, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.Q.; Wanunu, M.; Wang, M.X.; McReynolds, L.; Wang, Y. Detection of miRNAs with a nanopore single-molecule counter. Expert Rev. Mol. Diagn. 2012, 12, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yan, S.; Chang, L.; Guo, W.; Wang, Y.; Wang, Y.; Zhang, P.; Chen, H.Y.; Huang, S. Direct microRNA sequencing Using nanopore-induced phase-shift sequencing. iScience 2020, 23, 100916. [Google Scholar] [CrossRef] [Green Version]
- Van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The third revolution in sequencing technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Sereewattanawoot, S.; Suzuki, A. A new era of long-read sequencing for cancer genomics. J. Hum. Genet. 2020, 65, 3–10. [Google Scholar] [CrossRef] [Green Version]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Li, Y.; Zeng, X.; Zhao, Q.; Zhang, Z. Single-cell sequencing technology in tumor research. Clin. Chim. Acta 2021, 518, 101–109. [Google Scholar] [CrossRef]
- Huang, L.; Ma, F.; Chapman, A.; Lu, S.; Xie, X.S. Single-cell whole-genome amplification and sequencing: Methodology and applications. Annu. Rev. Genom. Hum. Genet. 2015, 16, 79–102. [Google Scholar] [CrossRef] [Green Version]
- Yasen, A.; Aini, A.; Wang, H.; Li, W.; Zhang, C.; Ran, B.; Tuxun, T.; Maimaitinijiati, Y.; Shao, Y.; Aji, T.; et al. Progress and applications of single-cell sequencing techniques. Infect. Genet. Evol. 2020, 80, 104198. [Google Scholar] [CrossRef]
- Sanders, A.D.; Falconer, E.; Hills, M.; Spierings, D.C.J.; Lansdorp, P.M. Single-cell template strand sequencing by Strand-seq enables the characterization of individual homologs. Nat. Protoc. 2017, 12, 1151–1176. [Google Scholar] [CrossRef]
- Rivera, C.M.; Ren, B. Mapping human epigenomes. Cell 2013, 155, 39–55. [Google Scholar] [CrossRef] [Green Version]
- Li, Y. Modern epigenetics methods in biological research. Methods 2021, 187, 104–113. [Google Scholar] [CrossRef]
- Nebbioso, A.; Tambaro, F.P.; Dell’Aversana, C.; Altucci, L. Cancer epigenetics: Moving forward. PLoS Genet. 2018, 14, e1007362. [Google Scholar] [CrossRef] [Green Version]
- Bohnsack, K.E.; Hobartner, C.; Bohnsack, M.T. Eukaryotic 5-methylcytosine (m5C) RNA methyltransferases: Mechanisms, cellular functions, and links to disease. Genes 2019, 10, 102. [Google Scholar] [CrossRef] [Green Version]
- Sarda, S.; Hannenhalli, S. Orphan CpG islands as alternative promoters. Transcription 2018, 9, 171–176. [Google Scholar] [CrossRef]
- Joo, J.E.; Dowty, J.G.; Milne, R.L.; Wong, E.M.; Dugue, P.A.; English, D.; Hopper, J.L.; Goldgar, D.E.; Giles, G.G.; Southey, M.C.; et al. Heritable DNA methylation marks associated with susceptibility to breast cancer. Nat. Commun. 2018, 9, 867. [Google Scholar] [CrossRef] [Green Version]
- Usui, G.; Matsusaka, K.; Mano, Y.; Urabe, M.; Funata, S.; Fukayama, M.; Ushiku, T.; Kaneda, A. DNA methylation and genetic aberrations in gastric cancer. Digestion 2021, 102, 25–32. [Google Scholar] [CrossRef]
- Tse, J.W.T.; Jenkins, L.J.; Chionh, F.; Mariadason, J.M. Aberrant DNA methylation in colorectal cancer: What should we target? Trends Cancer 2017, 3, 698–712. [Google Scholar] [CrossRef] [PubMed]
- Zafon, C.; Gil, J.; Perez-Gonzalez, B.; Jorda, M. DNA methylation in thyroid cancer. Endocr. Relat. Cancer 2019, 26, R415–R439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beaulaurier, J.; Schadt, E.E.; Fang, G. Deciphering bacterial epigenomes using modern sequencing technologies. Nat. Rev. Genet. 2019, 20, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Darst, R.P.; Pardo, C.E.; Ai, L.; Brown, K.D.; Kladde, M.P. Bisulfite sequencing of DNA. Curr. Protoc. Mol. Biol. 2010, 91, 7–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Z.; Fang, G.; Korlach, J.; Clark, T.; Luong, K.; Zhang, X.; Wong, W.; Schadt, E. Detecting DNA modifications from SMRT sequencing data by modeling sequence context dependence of polymerase kinetic. PLoS Comput. Biol. 2013, 9, e1002935. [Google Scholar] [CrossRef] [Green Version]
- Wallace, E.V.; Stoddart, D.; Heron, A.J.; Mikhailova, E.; Maglia, G.; Donohoe, T.J.; Bayley, H. Identification of epigenetic DNA modifications with a protein nanopore. Chem. Commun. 2010, 46, 8195–8197. [Google Scholar] [CrossRef]
- Li, Q.N.; Guo, L.; Hou, Y.; Ou, X.H.; Liu, Z.; Sun, Q.Y. The DNA methylation profile of oocytes in mice with hyperinsulinaemia and hyperandrogenism as detected by single-cell level whole genome bisulphite sequencing (SC-WGBS) technology. Reprod. Fertil. Dev. 2018, 30, 1713–1719. [Google Scholar] [CrossRef]
- Audia, J.E.; Campbell, R.M. Histone modifications and cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef]
- Wang, L.; Gao, Y.; Zheng, X.; Liu, C.; Dong, S.; Li, R.; Zhang, G.; Wei, Y.; Qu, H.; Li, Y.; et al. Histone modifications regulate chromatin compartmentalization by contributing to a phase separation mechanism. Mol. Cell 2019, 76, 646–659.e6. [Google Scholar] [CrossRef]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef]
- Grosselin, K.; Durand, A.; Marsolier, J.; Poitou, A.; Marangoni, E.; Nemati, F.; Dahmani, A.; Lameiras, S.; Reyal, F.; Frenoy, O.; et al. High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat. Genet. 2019, 51, 1060–1066. [Google Scholar] [CrossRef]
- Lareau, C.A.; Duarte, F.M.; Chew, J.G.; Kartha, V.K.; Burkett, Z.D.; Kohlway, A.S.; Pokholok, D.; Aryee, M.J.; Steemers, F.J.; Lebofsky, R.; et al. Droplet-based combinatorial indexing for massive-scale single-cell chromatin accessibility. Nat. Biotechnol. 2019, 37, 916–924. [Google Scholar] [CrossRef]
- Ai, S.; Xiong, H.; Li, C.C.; Luo, Y.; Shi, Q.; Liu, Y.; Yu, X.; Li, C.; He, A. Profiling chromatin states using single-cell itChIP-seq. Nat. Cell Biol. 2019, 21, 1164–1172. [Google Scholar] [CrossRef]
- Kumar, S.; Gonzalez, E.A.; Rameshwar, P.; Etchegaray, J.P. Non-Coding RNAs as mediators of epigenetic changes in malignancies. Cancers 2020, 12, 3657. [Google Scholar] [CrossRef]
- Bianchi, M.; Renzini, A.; Adamo, S.; Moresi, V. Coordinated Actions of MicroRNAs with other epigenetic factors regulate skeletal muscle development and adaptation. Int. J. Mol. Sci. 2017, 18, 840. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [Green Version]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Giraldez, M.D.; Spengler, R.M.; Etheridge, A.; Godoy, P.M.; Barczak, A.J.; Srinivasan, S.; De Hoff, P.L.; Tanriverdi, K.; Courtright, A.; Lu, S.; et al. Comprehensive multi-center assessment of small RNA-seq methods for quantitative miRNA profiling. Nat. Biotechnol. 2018, 36, 746–757. [Google Scholar] [CrossRef]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M., Jr.; Jungkamp, A.C.; Munschauer, M.; et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef] [Green Version]
- McGettigan, P.A. Transcriptomics in the RNA-seq era. Curr. Opin. Chem. Biol. 2013, 17, 4–11. [Google Scholar] [CrossRef]
- Zhang, G.; Sun, M.; Wang, J.; Lei, M.; Li, C.; Zhao, D.; Huang, J.; Li, W.; Li, S.; Li, J.; et al. PacBio full-length cDNA sequencing integrated with RNA-seq reads drastically improves the discovery of splicing transcripts in rice. Plant J. 2019, 97, 296–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ura, H.; Togi, S.; Niida, Y. A comparison of mRNA sequencing (RNA-Seq) library preparation methods for transcriptome analysis. BMC Genom. 2022, 23, 303. [Google Scholar] [CrossRef] [PubMed]
- Pyatnitskiy, M.A.; Arzumanian, V.A.; Radko, S.P.; Ptitsyn, K.G.; Vakhrushev, I.V.; Poverennaya, E.V.; Ponomarenko, E.A. Oxford Nanopore MinION Direct RNA-Seq for Systems Biology. Biology 2021, 10, 1131. [Google Scholar] [CrossRef] [PubMed]
- Neckles, C.; Sundara Rajan, S.; Caplen, N.J. Fusion transcripts: Unexploited vulnerabilities in cancer? Wiley Interdiscip. Rev. RNA 2020, 11, e1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Razzaq, S.K.; Vo, A.D.; Gautam, M.; Li, H. Identifying fusion transcripts using next generation sequencing. Wiley Interdiscip. Rev. RNA 2016, 7, 811–823. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Wang, M.; Shi, D.; Zhou, G.; Niu, T.; Hahn, M.G.; O’Neill, M.A.; Kong, Y. DGE-seq analysis of MUR3-related Arabidopsis mutants provides insight into how dysfunctional xyloglucan affects cell elongation. Plant Sci. 2017, 258, 156–169. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Gu, M.; Liu, L.; Liu, Y.; Tian, L. Single-cell RNA sequencing (scRNA-seq) in cardiac tissue: Applications and limitations. Vasc. Health Risk Manag. 2021, 17, 641–657. [Google Scholar] [CrossRef]
- Zhao, J.; Jaffe, A.; Li, H.; Lindenbaum, O.; Sefik, E.; Jackson, R.; Cheng, X.; Flavell, R.A.; Kluger, Y. Detection of differentially abundant cell subpopulations in scRNA-seq data. Proc. Natl. Acad. Sci. USA 2021, 118, e2100293118. [Google Scholar] [CrossRef]
- Goetz, J.J.; Trimarchi, J.M. Transcriptome sequencing of single cells with Smart-Seq. Nat. Biotechnol. 2012, 30, 763–765. [Google Scholar] [CrossRef]
- Singh, M.; Al-Eryani, G.; Carswell, S.; Ferguson, J.M.; Blackburn, J.; Barton, K.; Roden, D.; Luciani, F.; Giang Phan, T.; Junankar, S.; et al. High-throughput targeted long-read single cell sequencing reveals the clonal and transcriptional landscape of lymphocytes. Nat. Commun. 2019, 10, 3120. [Google Scholar] [CrossRef]
- Sarkar, A.; Gasperi, W.; Begley, U.; Nevins, S.; Huber, S.M.; Dedon, P.C.; Begley, T.J. Detecting the epitranscriptome. Wiley Interdiscip. Rev. RNA 2021, 12, e1663. [Google Scholar] [CrossRef]
- Moshitch-Moshkovitz, S.; Dominissini, D.; Rechavi, G. The epitranscriptome toolbox. Cell 2022, 185, 764–776. [Google Scholar] [CrossRef]
- Nachtergaele, S.; He, C. Chemical Modifications in the Life of an mRNA Transcript. Annu. Rev. Genet. 2018, 52, 349–372. [Google Scholar] [CrossRef]
- Nombela, P.; Miguel-Lopez, B.; Blanco, S. The role of m6A, m5C and Psi RNA modifications in cancer: Novel therapeutic opportunities. Mol. Cancer 2021, 20, 18. [Google Scholar] [CrossRef]
- Barbieri, I.; Kouzarides, T. Role of RNA modifications in cancer. Nat. Rev. Cancer 2020, 20, 303–322. [Google Scholar] [CrossRef]
- Lin, S.; Choe, J.; Du, P.; Triboulet, R.; Gregory, R.I. The m6A methyltransferase METTL3 promotes translation in human cancer cells. Mol. Cell 2016, 62, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Eckert, M.A.; Harada, B.T.; Liu, S.M.; Lu, Z.; Yu, K.; Tienda, S.M.; Chryplewicz, A.; Zhu, A.C.; Yang, Y.; et al. m6A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat. Cell Biol. 2018, 20, 1074–1083. [Google Scholar] [CrossRef]
- Liu, T.; Hu, X.; Lin, C.; Shi, X.; He, Y.; Zhang, J.; Cai, K. 5-methylcytosine RNA methylation regulators affect prognosis and tumor microenvironment in lung adenocarcinoma. Ann. Transl. Med. 2022, 10, 259. [Google Scholar] [CrossRef]
- Frye, M.; Watt, F.M. The RNA methyltransferase Misu (NSun2) mediates Myc-induced proliferation and is upregulated in tumors. Curr. Biol. 2006, 16, 971–981. [Google Scholar] [CrossRef] [Green Version]
- Woo, H.H.; Chambers, S.K. Human ALKBH3-induced m1A demethylation increases the CSF-1 mRNA stability in breast and ovarian cancer cells. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 35–46. [Google Scholar] [CrossRef]
- Garus, A.; Autexier, C. Dyskerin: An essential pseudouridine synthase with multifaceted roles in ribosome biogenesis, splicing, and telomere maintenance. RNA 2021, 27, 1441–1458. [Google Scholar] [CrossRef] [PubMed]
- Martinez, N.M.; Su, A.; Burns, M.C.; Nussbacher, J.K.; Schaening, C.; Sathe, S.; Yeo, G.W.; Gilbert, W.V. Pseudouridine synthases modify human pre-mRNA co-transcriptionally and affect pre-mRNA processing. Mol. Cell 2022, 82, 645–659.e9. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Chen, L.; Han, Y.; Zhang, F.; Wang, Y.; Han, Y.; Wang, Y.; Wang, Q.; Guo, X. The Identification of RNA Modification Gene PUS7 as a Potential Biomarker of Ovarian Cancer. Biology 2021, 10, 1130. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Zhuang, Y.; Zhu, C.; Meng, H.; Lu, B.; Xie, B.; Peng, J.; Li, M.; Yi, C. Differential roles of human PUS10 in miRNA processing and tRNA pseudouridylation. Nat. Chem. Biol. 2020, 16, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Jana, S.; Hsieh, A.C.; Gupta, R. Reciprocal amplification of caspase-3 activity by nuclear export of a putative human RNA-modifying protein, PUS10 during TRAIL-induced apoptosis. Cell Death Dis. 2017, 8, e3093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Amariglio, N.; Rechavi, G. Adenosine-to-inosine RNA editing meets cancer. Carcinogenesis 2011, 32, 1569–1577. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Li, Y.; Lin, C.H.; Chan, T.H.; Chow, R.K.; Song, Y.; Liu, M.; Yuan, Y.F.; Fu, L.; Kong, K.L.; et al. Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nat. Med. 2013, 19, 209–216. [Google Scholar] [CrossRef]
- Wetzel, C.; Limbach, P.A. Mass spectrometry of modified RNAs: Recent developments. Analyst 2016, 141, 16–23. [Google Scholar] [CrossRef] [Green Version]
- Giessing, A.M.; Kirpekar, F. Mass spectrometry in the biology of RNA and its modifications. J. Proteom. 2012, 75, 3434–3449. [Google Scholar] [CrossRef]
- Li, X.; Xiong, X.; Yi, C. Epitranscriptome sequencing technologies: Decoding RNA modifications. Nat. Methods 2016, 14, 23–31. [Google Scholar] [CrossRef]
- Meyer, K.D. DART-seq: An antibody-free method for global m6A detection. Nat. Methods 2019, 16, 1275–1280. [Google Scholar] [CrossRef]
- Wang, D.O. Mapping m6A and m1A with mutation signatures. Nat. Methods 2019, 16, 1213–1214. [Google Scholar] [CrossRef]
- Gu, X.; Liang, Z. Transcriptome-Wide Mapping 5-Methylcytosine by m5C RNA Immunoprecipitation Followed by Deep Sequencing in Plant. Methods Mol. Biol. 2019, 1933, 389–394. [Google Scholar] [CrossRef]
- Li, W.; Li, X.; Ma, X.; Xiao, W.; Zhang, J. Mapping the m1A, m5C, m6A and m7G methylation atlas in zebrafish brain under hypoxic conditions by MeRIP-seq. BMC Genom. 2022, 23, 105. [Google Scholar] [CrossRef]
- Khoddami, V.; Cairns, B.R. Transcriptome-wide target profiling of RNA cytosine methyltransferases using the mechanism-based enrichment procedure Aza-IP. Nat. Protoc. 2014, 9, 337–361. [Google Scholar] [CrossRef]
- Carlile, T.M.; Rojas-Duran, M.F.; Gilbert, W.V. Transcriptome-wide identification of pseudouridine modifications using pseudo-seq. Curr. Protoc. Mol. Biol. 2015, 112, 4.25.1–4.25.24. [Google Scholar] [CrossRef] [PubMed]
- Bruno, R.; Fontanini, G. Next generation sequencing for gene fusion analysis in lung cancer: A literature review. Diagnostics 2020, 10, 521. [Google Scholar] [CrossRef]
- Dacic, S.; Villaruz, L.C.; Abberbock, S.; Mahaffey, A.; Incharoen, P.; Nikiforova, M.N. ALK FISH patterns and the detection of ALK fusions by next generation sequencing in lung adenocarcinoma. Oncotarget 2016, 7, 82943–82952. [Google Scholar] [CrossRef] [Green Version]
- Vollbrecht, C.; Lenze, D.; Hummel, M.; Lehmann, A.; Moebs, M.; Frost, N.; Jurmeister, P.; Schweizer, L.; Kellner, U.; Dietel, M.; et al. RNA-based analysis of ALK fusions in non-small cell lung cancer cases showing IHC/FISH discordance. BMC Cancer 2018, 18, 1158. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, H.; Kohnen, M.V.; Prasad, K.; Gu, L.; Reddy, A.S.N. Analysis of transcriptome and epitranscriptome in plants using pacbio Iso-seq and nanopore-based direct RNA SEQUENCING. Front. Genet. 2019, 10, 253. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Gildea, M.A.; Dwyer, Z.W.; Pleiss, J.A. Multiplexed primer extension sequencing: A targeted RNA-seq method that enables high-precision quantitation of mRNA splicing isoforms and rare pre-mRNA splicing intermediates. Methods 2020, 176, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.; Beaudin, A.E.; Olsen, H.E.; Jain, M.; Cole, C.; Palmer, T.; DuBois, R.M.; Forsberg, E.C.; Akeson, M.; Vollmers, C. Nanopore long-read RNAseq reveals widespread transcriptional variation among the surface receptors of individual B cells. Nat. Commun. 2017, 8, 16027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, A.H.; Weng, Z.; Witten, D.M.; Zhu, S.; Foley, J.W.; Lacroute, P.; Smith, C.L.; Tibshirani, R.; van de Rijn, M.; Sidow, A.; et al. 3′-end sequencing for expression quantification (3SEQ) from archival tumor samples. PLoS ONE 2010, 5, e8768. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jia, Q.; Song, Y.; Fu, H.; Wei, G.; Ni, T. Alternative Polyadenylation: Methods, Findings, and Impacts. Genom. Proteom. Bioinform. 2017, 15, 287–300. [Google Scholar] [CrossRef]
- Harrison, P.F.; Powell, D.R.; Clancy, J.L.; Preiss, T.; Boag, P.R.; Traven, A.; Seemann, T.; Beilharz, T.H. PAT-seq: A method to study the integration of 3′-UTR dynamics with gene expression in the eukaryotic transcriptome. RNA 2015, 21, 1502–1510. [Google Scholar] [CrossRef] [Green Version]
- Routh, A.; Ji, P.; Jaworski, E.; Xia, Z.; Li, W.; Wagner, E.J. Poly(A)-ClickSeq: Click-chemistry for next-generation 3′-end sequencing without RNA enrichment or fragmentation. Nucleic Acids Res. 2017, 45, e112. [Google Scholar] [CrossRef] [Green Version]
- Anvar, S.Y.; Allard, G.; Tseng, E.; Sheynkman, G.M.; de Klerk, E.; Vermaat, M.; Yin, R.H.; Johansson, H.E.; Ariyurek, Y.; den Dunnen, J.T.; et al. Full-length mRNA sequencing uncovers a widespread coupling between transcription initiation and mRNA processing. Genome Biol. 2018, 19, 46. [Google Scholar] [CrossRef] [Green Version]
- Legnini, I.; Alles, J.; Karaiskos, N.; Ayoub, S.; Rajewsky, N. FLAM-seq: Full-length mRNA sequencing reveals principles of poly(A) tail length control. Nat. Methods 2019, 16, 879–886. [Google Scholar] [CrossRef]
- Di Bella, S.; La Ferlita, A.; Carapezza, G.; Alaimo, S.; Isacchi, A.; Ferro, A.; Pulvirenti, A.; Bosotti, R. A benchmarking of pipelines for detecting ncRNAs from RNA-Seq data. Brief. Bioinform. 2020, 21, 1987–1998. [Google Scholar] [CrossRef]
- Chen, C.J.; Servant, N.; Toedling, J.; Sarazin, A.; Marchais, A.; Duvernois-Berthet, E.; Cognat, V.; Colot, V.; Voinnet, O.; Heard, E.; et al. ncPRO-seq: A tool for annotation and profiling of ncRNAs in sRNA-seq data. Bioinformatics 2012, 28, 3147–3149. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.F.; Yim, D.; Ma, D.; Huber, S.M.; Davis, N.; Bacusmo, J.M.; Vermeulen, S.; Zhou, J.; Begley, T.J.; DeMott, M.S.; et al. Quantitative mapping of the cellular small RNA landscape with AQRNA-seq. Nat. Biotechnol. 2021, 39, 978–988. [Google Scholar] [CrossRef]
- Motameny, S.; Wolters, S.; Nurnberg, P.; Schumacher, B. Next Generation Sequencing of miRNAs—Strategies, Resources and Methods. Genes 2010, 1, 70–84. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Chen, L.; Tang, Y.; He, Y.; Pan, K.; Yuan, L.; Xie, W.; Chen, S.; Zhao, W.; Yu, D. Transcriptome-wide m6A methylome analysis uncovered the changes of m6A modification in oral pre-malignant cells compared with normal oral epithelial cells. Front. Oncol. 2022, 12, 939449. [Google Scholar] [CrossRef]
- Chen, H.X.; Zhang, Z.; Ma, D.Z.; Chen, L.Q.; Luo, G.Z. Mapping single-nucleotide m6A by m6A-REF-seq. Methods 2022, 203, 392–398. [Google Scholar] [CrossRef]
- Leger, A.; Amaral, P.P.; Pandolfini, L.; Capitanchik, C.; Capraro, F.; Miano, V.; Migliori, V.; Toolan-Kerr, P.; Sideri, T.; Enright, A.J.; et al. RNA modifications detection by comparative Nanopore direct RNA sequencing. Nat. Commun. 2021, 12, 7198. [Google Scholar] [CrossRef]
- Schaefer, M. RNA 5-Methylcytosine Analysis by Bisulfite Sequencing. Methods Enzymol. 2015, 560, 297–329. [Google Scholar] [CrossRef]
- Zhang, S.; Li, R.; Zhang, L.; Chen, S.; Xie, M.; Yang, L.; Xia, Y.; Foyer, C.H.; Zhao, Z.; Lam, H.M. New insights into Arabidopsis transcriptome complexity revealed by direct sequencing of native RNAs. Nucleic Acids Res. 2020, 48, 7700–7711. [Google Scholar] [CrossRef]
- Carlile, T.M.; Martinez, N.M.; Schaening, C.; Su, A.; Bell, T.A.; Zinshteyn, B.; Gilbert, W.V. mRNA structure determines modification by pseudouridine synthase 1. Nat. Chem. Biol. 2019, 15, 966–974. [Google Scholar] [CrossRef]
- Cozen, A.E.; Quartley, E.; Holmes, A.D.; Hrabeta-Robinson, E.; Phizicky, E.M.; Lowe, T.M. ARM-seq: AlkB-facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nat. Methods 2015, 12, 879–884. [Google Scholar] [CrossRef]
- Zhou, H.; Rauch, S.; Dai, Q.; Cui, X.; Zhang, Z.; Nachtergaele, S.; Sepich, C.; He, C.; Dickinson, B.C. Evolution of a reverse transcriptase to map N1-methyladenosine in human messenger RNA. Nat. Methods 2019, 16, 1281–1288. [Google Scholar] [CrossRef] [PubMed]
- Safra, M.; Sas-Chen, A.; Nir, R.; Winkler, R.; Nachshon, A.; Bar-Yaacov, D.; Erlacher, M.; Rossmanith, W.; Stern-Ginossar, N.; Schwartz, S. The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature 2017, 551, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.X.; Zhang, X.S.; Sui, N. Advances in the profiling of N6-methyladenosine (m6A) modifications. Biotechnol. Adv. 2020, 45, 107656. [Google Scholar] [CrossRef] [PubMed]
- Potapov, V.; Fu, X.; Dai, N.; Correa, I.R., Jr.; Tanner, N.A.; Ong, J.L. Base modifications affecting RNA polymerase and reverse transcriptase fidelity. Nucleic Acids Res. 2018, 46, 5753–5763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilfan, I.D.; Tsai, Y.C.; Clark, T.A.; Wegener, J.; Dai, Q.; Yi, C.; Pan, T.; Turner, S.W.; Korlach, J. Analysis of RNA base modification and structural rearrangement by single-molecule real-time detection of reverse transcription. J. Nanobiotechnology 2013, 11, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noakes, M.T.; Brinkerhoff, H.; Laszlo, A.H.; Derrington, I.M.; Langford, K.W.; Mount, J.W.; Bowman, J.L.; Baker, K.S.; Doering, K.M.; Tickman, B.I.; et al. Increasing the accuracy of nanopore DNA sequencing using a time-varying cross membrane voltage. Nat. Biotechnol. 2019, 37, 651–656. [Google Scholar] [CrossRef]
- Johnson, D.T.; Harris, R.A.; French, S.; Blair, P.V.; You, J.; Bemis, K.G.; Wang, M.; Balaban, R.S. Tissue heterogeneity of the mammalian mitochondrial proteome. Am. J. Physiol. Cell Physiol. 2007, 292, C689–C697. [Google Scholar] [CrossRef] [Green Version]
- Shruthi, B.S.; Vinodhkumar, P.; Selvamani. Proteomics: A new perspective for cancer. Adv. Biomed. Res. 2016, 5, 67. [Google Scholar] [CrossRef]
- Collins, B.C.; Aebersold, R. Proteomics goes parallel. Nat. Biotechnol. 2018, 36, 1051–1053. [Google Scholar] [CrossRef]
- Restrepo-Perez, L.; Joo, C.; Dekker, C. Paving the way to single-molecule protein sequencing. Nat. Nanotechnol. 2018, 13, 786–796. [Google Scholar] [CrossRef]
- Mantini, G.; Pham, T.V.; Piersma, S.R.; Jimenez, C.R. Computational analysis of phosphoproteomics data in Multi-Omics cancer studies. Proteomics 2021, 21, e1900312. [Google Scholar] [CrossRef]
- Tang, L. Next-generation peptide sequencing. Nat. Methods 2018, 15, 997. [Google Scholar] [CrossRef]
- Swaminathan, J.; Boulgakov, A.A.; Hernandez, E.T.; Bardo, A.M.; Bachman, J.L.; Marotta, J.; Johnson, A.M.; Anslyn, E.V.; Marcotte, E.M. Highly parallel single-molecule identification of proteins in zeptomole-scale mixtures. Nat. Biotechnol. 2018, 36, 1076–1082. [Google Scholar] [CrossRef]
- Ouldali, H.; Sarthak, K.; Ensslen, T.; Piguet, F.; Manivet, P.; Pelta, J.; Behrends, J.C.; Aksimentiev, A.; Oukhaled, A. Electrical recognition of the twenty proteinogenic amino acids using an aerolysin nanopore. Nat. Biotechnol. 2020, 38, 176–181. [Google Scholar] [CrossRef]
- Wu, W.; Hu, W.; Kavanagh, J.J. Proteomics in cancer research. Int. J. Gynecol. Cancer 2002, 12, 409–423. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.W.; Jo, H.S.; Bae, S.; Seo, Y.; Song, P.; Song, M.; Yoon, J.H. Application of proteomics in cancer: Recent trends and approaches for biomarkers discovery. Front. Med. 2021, 8, 747333. [Google Scholar] [CrossRef]
- Wheeler, H.E.; Maitland, M.L.; Dolan, M.E.; Cox, N.J.; Ratain, M.J. Cancer pharmacogenomics: Strategies and challenges. Nat. Rev. Genet. 2013, 14, 23–34. [Google Scholar] [CrossRef]
- Katsila, T.; Patrinos, G.P. Whole genome sequencing in pharmacogenomics. Front. Pharmacol. 2015, 6, 61. [Google Scholar] [CrossRef] [Green Version]
- Cerea, G.; Ricotta, R.; Schiavetto, I.; Maugeri, M.R.; Sartore-Bianchi, A.; Moroni, M.; Artale, S.; Siena, S. Cetuximab for treatment of metastatic colorectal cancer. Ann. Oncol. 2006, 17 (Suppl. 7), vii66–vii67. [Google Scholar] [CrossRef]
- Mondaca, S.; Lebow, E.S.; Namakydoust, A.; Razavi, P.; Reis-Filho, J.S.; Shen, R.; Offin, M.; Tu, H.Y.; Murciano-Goroff, Y.; Xu, C.; et al. Clinical utility of next-generation sequencing-based ctDNA testing for common and novel ALK fusions. Lung Cancer 2021, 159, 66–73. [Google Scholar] [CrossRef]
- Onidani, K.; Shoji, H.; Kakizaki, T.; Yoshimoto, S.; Okaya, S.; Miura, N.; Sekikawa, S.; Furuta, K.; Lim, C.T.; Shibahara, T.; et al. Monitoring of cancer patients via next-generation sequencing of patient-derived circulating tumor cells and tumor DNA. Cancer Sci. 2019, 110, 2590–2599. [Google Scholar] [CrossRef] [PubMed]




| Abbreviation | Definition |
|---|---|
| 5caC | 5-carboxylcytosine |
| 5fC | 5-formylcytosine |
| 5hmC | 5-hydroxymethylcytosine |
| 5mC | 5-methylcytosine |
| ATAC-seq | Assay for transposase-accessible chromatin using sequencing |
| A-to-I | Adenosine to inosine |
| CAB-seq | Chemical modification-assisted bisulfite sequencing |
| ChAR-seq | Chromatin-associated RNA sequencing |
| CHART | Capture hybridization analysis of RNA targets |
| ChIA-PET | Chromatin interaction analysis with paired-end tag |
| ChIP-seq | Chromatin immunoprecipitation followed by sequencing |
| ChIRP | Chromatin isolation by RNA purification |
| CLASH | Cross-linking ligation and sequencing of hybrids |
| CMC | Cyclohexyl-Methylmorpholino-Carbodiimide |
| CNVs | Copy number variations |
| CpG | Cytosine-phosphate-guanine |
| CTCs | Circulating tumor cells |
| ctDNA | Cell-free tumor DNA |
| CUT&RUN | Cleavage Under Targets and Release Using Nuclease |
| CUT&Tag | Cleavage Under Targets and Tagmentation |
| DGE-Seq | Digital gene expression sequencing |
| DNase-seq | DNase I hypersensitive sites sequencing |
| Drop-ChIP | Droplet-based single-cell ChIP-seq |
| FAIRE-seq | Formaldehyde-assisted isolation of regulatory elements with sequencing |
| fCAB-seq | 5fC chemically assisted bisulfite sequencing |
| GRID-seq | Global RNA interactions with DNA by deep sequencing |
| HITS-CLIP | High-throughput sequencing of RNA isolated by crosslinking immunoprecipitation |
| ICGC | International Cancer Genome Consortium |
| icSHAPE | In vivo click selective 2′-hydroxyl acylation and profiling experiment |
| LIGR-seq | Ligation of interacting RNA followed by high-throughput sequencing |
| m1A | N1-methyladenosine |
| m5C | 5-methylcytosine |
| m6A | N6-methyladenosine |
| MBD-seq | Methyl-CpG-binding domain sequencing |
| MeDIP-seq | Methylated DNA immunoprecipitation sequencing |
| MNase | Micrococcal nuclease |
| MNase-seq | MNase digestion of chromatin followed by sequencing |
| MPS | Massive parallel sequencing |
| MREBS | Methylation-sensitive restriction enzyme bisulfite sequencing |
| MRE-seq | Methylation-sensitive restriction enzyme sequencing |
| NGS | Next-generation sequencing |
| ONT | Oxford Nanopore Technologies |
| OxBS-seq | Oxidative bisulfite sequencing |
| PacBio | Pacific Biosciences |
| PAR-CLIP | Photoactivable-ribonucleoside-enhanced-CLIP |
| PCR | Polymerase chain reaction |
| PTMs | Post-translational modifications |
| PUS | Pseudouridine synthase |
| RIP-seq | RNA immunoprecipitation sequencing |
| RISC | RNA-induced silencing complex |
| RNAi | RNA interference |
| RRBS | Reduced representation bisulfite sequencing |
| scChIP-seq | Single-cell ChIP-seq |
| scDNA-seq | Single-cell DNA sequencing |
| scitChIPseq | Single-cell simultaneous indexing and tagmentation-based ChIP-seq |
| scRNA-seq | Single-cell RNA sequencing |
| scRRBS | Single-cell RRBS |
| SCS | Single-cell sequencing |
| scWGBS | Single-cell WGBS |
| SHAPE-MaP | Selective 2′-hydroxyl acylation analyzed by primer extension and mutational profiling |
| SHAPE-seq | Selective 2′-hydroxyl acylation analyzed by primer extension sequencing |
| siRNAs | Small interfering RNAs |
| SMRT | Single-molecule real-time |
| SVs | Structural variants |
| TAB-seq | TET-assisted bisulfite sequencing |
| TCGA | The Cancer Genome Atlas |
| TGS | Third-generation sequencing |
| TRAIL | TNF-related apoptosis-inducing ligand |
| TS | Targeted sequencing |
| UTRs | Untranslated regions |
| WES | Whole-exome sequencing |
| WGA | Whole-genome amplification |
| WGBS | Whole-genome bisulfite sequencing |
| WGS | Whole-genome sequencing |
| WTS | Whole-transcriptome sequencing |
| Ψ | Pseudouridine |
| Gene | “omics” | Cancer Type | Technology | Application | Reference |
|---|---|---|---|---|---|
| MYB | Genomics/ chromosomal rearrangement | Low grade glioma | NGS | WGS | [19] |
| ACTB-FOSB | Genomics/ gene fusion | Pseudomyogenic hemangioendothelioma | NGS | Targeted RNA-seq | [20] |
| KMT2A-MLTT2/4 | Genomics/ gene fusion | Acute myeloid leukemia | TGS | Nanopore sequencing | [21] |
| CDKN2A | Genomics/ mutation | Hepatocellular carcinoma | NGS | WES | [22] |
| IRF-4 | Epigenomics/ DNA methylation | Lymphoma | NGS | WGBS | [23] |
| KRT19 | Epigenomics/ DNA methylation | Breast cancer | TGS | Nanopore sequencing | [24] |
| DBC1 | Epigenomics/ histone modification | Colorectal cancer | NGS | ChIP-seq RNA-seq | [25] |
| BCR-ABL1 | Transcriptomics/ fusion transcripts | Chronic myelogenous leukaemia | NGS | Targeted RNA-seq | [26,27] |
| MLH1 | Transcriptomics/ alternative splicing | Colorectal cancer | TGS | Long read RNA-seq | [28] |
| CSTF2 | Transcriptomics/ alternative polyadenylation | Non-small cell lung cancer | NGS | IVT-SAPAS | [29] |
| Genomics/Epigenomics | Technology | Application | References |
|---|---|---|---|
| DNA mutations | NGS | WES, WGS, TS | [44,45] |
| Larger SVs, CNVs, gene fusions | NGS | [46,47,48] | |
| TGS | [21,49] | ||
| DNA methylation | NGS | WGBS | [50] |
| RRBS | [51] | ||
| OxBS-seq | [52] | ||
| TAB-seq | [53] | ||
| fCAB-seq | [54] | ||
| CAB-seq | [55] | ||
| MeDIP-seq | [56] | ||
| MBD-seq | [57] | ||
| MRE-seq | [58] | ||
| TGS | SMRT sequencing | [59,60] | |
| Nanopore sequencing | [61,62] | ||
| Histone modifications | NGS | ChIP-seq | [63] |
| CUT&RUN | [64] | ||
| CUT&Tag | [65] | ||
| Chromatin accessibility | NGS | DNase-seq | [66] |
| FAIRE-seq | [67] | ||
| ATAC-seq | [67] | ||
| Nucleosome positioning | NGS | MNase-seq | [68] |
| 3D genome structure | NGS | Hi-C | [69] |
| ChIA-PET | [70] | ||
| ncRNAs | NGS | RNA-seq | [71] |
| RIP-seq | [72] | ||
| HITS-CLIP | [73] | ||
| PAR-CLIP | [74] | ||
| CLASH | [75] | ||
| LIGR-seq | [76] | ||
| ChIRP | [77] | ||
| CHART | [78] | ||
| GRID-seq | [79] | ||
| ChAR-seq | [80] | ||
| SHAPE-seq | [81] | ||
| SHAPE-MaP | [82] | ||
| icSHAPE | [83] | ||
| TGS | Nanopore sequencing | [84,85,86] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Athanasopoulou, K.; Daneva, G.N.; Boti, M.A.; Dimitroulis, G.; Adamopoulos, P.G.; Scorilas, A. The Transition from Cancer “omics” to “epi-omics” through Next- and Third-Generation Sequencing. Life 2022, 12, 2010. https://doi.org/10.3390/life12122010
Athanasopoulou K, Daneva GN, Boti MA, Dimitroulis G, Adamopoulos PG, Scorilas A. The Transition from Cancer “omics” to “epi-omics” through Next- and Third-Generation Sequencing. Life. 2022; 12(12):2010. https://doi.org/10.3390/life12122010
Chicago/Turabian StyleAthanasopoulou, Konstantina, Glykeria N. Daneva, Michaela A. Boti, Georgios Dimitroulis, Panagiotis G. Adamopoulos, and Andreas Scorilas. 2022. "The Transition from Cancer “omics” to “epi-omics” through Next- and Third-Generation Sequencing" Life 12, no. 12: 2010. https://doi.org/10.3390/life12122010