
NF-kB in Signaling Patterns and Its Temporal Dynamics Encode/Decode Human Diseases


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. NF-κB Signaling
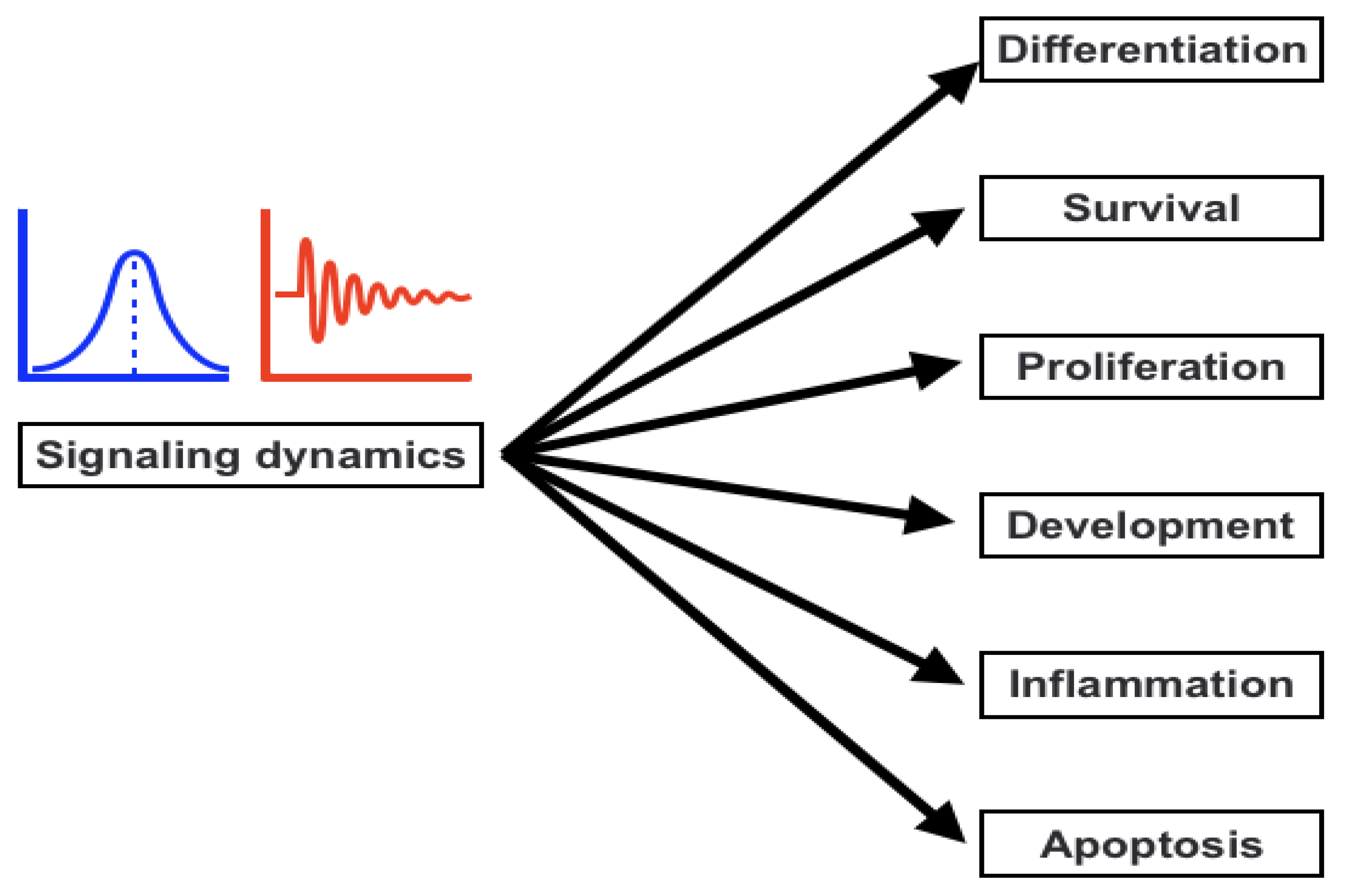
3. Signaling Dynamics and the Association with Signaling Parameters
4. Cell-Fate Decision
5. Encoding and Decoding Cellular Information
6. NF-κB Signaling and Molecular Targets for Therapeutic Purpose of Human Diseases
7. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Purvis, J.E.; Lahav, G. Encoding and Decoding Cellular Information through Signaling Dynamics. Cell 2013, 152, 945–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klil-Drori, A.J.; Azoulay, L.; Pollak, M.N. Cancer, obesity, diabetes, and antidiabetic drugs: Is the fog clearing? Nat. Rev. Clin. Oncol. 2016, 14, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Davis, D.M.; Purvis, J.E. Computational analysis of signaling patterns in single cells. Semin Cell Dev Biol. 2015, 37, 35–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Bernardo, J.; Eppstein, M.J. Evolving modular genetic regulatory networks with a recursive, top-down approach. Syst. Synth. Biol. 2015, 9, 179–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batchelor, E.; Loewer, A.; Mock, C.; Lahav, G. Stimulus-dependent dynamics of p53 in single cells. Mol. Syst. Biol. 2011, 7, 488. [Google Scholar] [CrossRef] [PubMed]
- Regev-Rudzki, N.; Wilson, D.W.; Carvalho, T.G.; Sisquella, X.; Coleman, B.M.; Rug, M.; Bursac, D.; Angrisano, F.; Gee, M.; Hill, A.F.; et al. Cell-Cell Communication between Malaria-Infected Red Blood Cells via Exosome-like Vesicles. Cell 2013, 153, 1120–1133. [Google Scholar] [CrossRef] [Green Version]
- Ruch, R.J. Intercellular communication, homeostasis, and toxicology. Toxicol. Sci. 2002, 68, 265–266. [Google Scholar] [CrossRef] [Green Version]
- Cotari, J.W.; Voisinne, G.; Dar, O.E.; Karabacak, V.; Altan-Bonnet, G. Cell-to-Cell Variability Analysis Dissects the Plasticity of Signaling of Common Chain Cytokines in T Cells. Sci. Signal. 2013, 6, ra17. [Google Scholar] [CrossRef] [Green Version]
- Poltorak, M.; Arndt, B.; Kowtharapu, B.S.; Reddycherla, A.V.; Witte, V.; Lindquist, J.A.; Schraven, B.; Simeoni, L. TCR activation kinetics and feedback regulation in primary human T cells. Cell Commun. Signal. 2013, 11, 4. [Google Scholar] [CrossRef] [Green Version]
- Lim, W.A.; Pawson, T. Phosphotyrosine Signaling: Evolving a New Cellular Communication System. Cell 2010, 142, 661–667. [Google Scholar] [CrossRef]
- Matolcsi, M.; Giordano, N. A Novel Explanation for Observed CaMKII Dynamics in Dendritic Spines with Added EGTA or BAPTA. Biophys. J. 2015, 108, 975–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Kafrawy, S.A.; El-Daly, M.M.; Bajrai, L.H.; Alandijany, T.A.; Faizo, A.A.; Mobashir, M.; Ahmed, S.S.; Ahmed, S.; Alam, S.; Jeet, R.; et al. Genomic profiling and network-level understanding uncover the potential genes and the pathways in hepatocellular carcinoma. Front. Genet. 2022, 13, 880440. [Google Scholar] [CrossRef]
- Orton, R.J.; Sturm, O.E.; Vyshemirsky, V.; Calder, M.; Gilbert, D.R.; Kolch, W. Computational modelling of the receptor-tyrosine-kinase-activated MAPK pathway. Biochem. J. 2005, 392, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Hogenesch, J.B.; Ueda, H.R. Understanding systems-level properties: Timely stories from the study of clocks. Nat. Rev. Genet. 2011, 12, 407–416. [Google Scholar] [CrossRef]
- Stelzl, U.; Worm, U.; Lalowski, M.; Haenig, C.; Brembeck, F.H.; Goehler, H.; Stroedicke, M.; Zenkner, M.; Schoenherr, A.; Koeppen, S.; et al. A Human Protein-Protein Interaction Network: A Resource for Annotating the Proteome. Cell 2005, 122, 957–968. [Google Scholar] [CrossRef] [Green Version]
- Yao, G.; Lee, T.J.; Mori, S.; Nevins, J.R.; You, L. A bistable Rb–E2F switch underlies the restriction point. Nat. Cell Biol. 2008, 10, 476–482. [Google Scholar] [CrossRef]
- Sobie, E.A. Bistability in Biochemical Signaling Models. Sci. Signal. 2011, 4, tr10. [Google Scholar] [CrossRef] [Green Version]
- Kholodenko, B.N. Cell-signalling dynamics in time and space. Nat. Rev. Mol. Cell Biol. 2006, 7, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Schleich, K.; Lavrik, I.N. Mathematical modeling of apoptosis. Cell Commun. Signal. 2013, 11, 44. [Google Scholar] [CrossRef] [Green Version]
- Prashar, Y.; Ritu; Gill, N.S. Emerging role of various signaling pathways in the pathogenesis and therapeutics of atherosclerosis. Rev. Vasc. Med. 2017, 10, 1–12. [Google Scholar] [CrossRef]
- Loewer, A.; Lahav, G. Cellular Conference Call: External Feedback Affects Cell-Fate Decisions. Cell 2006, 124, 1128–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niiro, H.; Clark, E.A. Decision making in the immune system: Regulation of B-cell fate by antigen-receptor signals. Nat. Rev. Immunol. 2002, 2, 945–956. [Google Scholar] [CrossRef] [PubMed]
- White, K.L.; Rider, D.N.; Kalli, K.R.; Knutson, K.L.; Jarvik, G.P.; Goode, E.L. Genomics of the NF-κB signaling pathway: Hypothesized role in ovarian cancer. Cancer Causes Control 2011, 22, 785–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricaño-Ponce, I.; Wijmenga, C. Mapping of Immune-Mediated Disease Genes. Annu. Rev. Genom. Hum. Genet. 2013, 14, 325–353. [Google Scholar] [CrossRef] [PubMed]
- Habashy, N.H.; Kodous, A.S.; Abu-Serie, M.M. Targeting ROS/NF-κB signaling pathway by the seedless black Vitis vinifera polyphenolsin CCl. Sci. Rep. 2021, 11, 16575. [Google Scholar] [CrossRef] [PubMed]
- Abu-Serie, M.M.; Hamouda, A.F.; Habashy, N.H. Acacia senegal gumattenuates systemic toxicityin CCl. Sci. Rep. 2021, 11, 20316. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB Signaling: Navigating Downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Mostafizar, M.; Cortes-Pérez, C.; Snow, W.; Djordjevic, J.; Adlimoghaddam, A.; Albensi, B.C. Challenges with Methods for Detecting and Studying the Transcription Factor Nuclear Factor Kappa B (NF-κB) in the Central Nervous System. Cells 2021, 10, 1335. [Google Scholar] [CrossRef]
- Yan, F.; Liu, L.; Wang, Q. Combinatorial dynamics of protein synthesis time delay and negative feedback loop in NF-κB signalling pathway. IET Syst. Biol. 2020, 14, 284–291. [Google Scholar] [CrossRef]
- Prescott, J.A.; Mitchell, J.P.; Cook, S.J. Inhibitory feedback control of NF-κB signalling in health and disease. Biochem. J. 2021, 478, 2619–2664. [Google Scholar] [CrossRef] [PubMed]
- ARAUJO, R.; LIOTTA, L. A control theoretic paradigm for cell signaling networks: A simple complexity for a sensitive robustness. Curr. Opin. Chem. Biol. 2006, 10, 81–87. [Google Scholar] [CrossRef]
- Restifo, N.P.; Smyth, M.J.; Snyder, A. Acquired resistance to immunotherapy and future challenges. Nat. Rev. Cancer 2016, 16, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Kim, J.; Guan, K.-L. AMPK and mTOR in Cellular Energy Homeostasis and Drug Targets. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 381–400. [Google Scholar] [CrossRef] [PubMed]
- Buszczak, M.; Signer, R.A.J.; Morrison, S.J. Cellular Differences in Protein Synthesis Regulate Tissue Homeostasis. Cell 2014, 159, 242–251. [Google Scholar] [CrossRef] [Green Version]
- Furusawa, C.; Kaneko, K. A Dynamical-Systems View of Stem Cell Biology. Science 2012, 338, 215–217. [Google Scholar] [CrossRef]
- de Souza, N. A systems view of cellular reprogramming. Nat. Meth. 2014, 11, 987. [Google Scholar] [CrossRef]
- Burzyn, D.; Kuswanto, W.; Kolodin, D.; Shadrach, J.L.; Cerletti, M.; Jang, Y.; Sefik, E.; Tan, T.G.; Wagers, A.J.; Benoist, C.; et al. A Special Populationof Regulatory T Cells Potentiates Muscle Repair. Cell 2013, 155, 1282–1295. [Google Scholar] [CrossRef] [Green Version]
- Gutierrez-Arcelus, M.; Rich, S.S.; Raychaudhuri, S. Autoimmune diseases—Connecting risk alleles with molecular traits of the immune system. Nat. Rev. Genet. 2016, 17, 160–174. [Google Scholar] [CrossRef] [Green Version]
- Kurosaki, T.; Shinohara, H.; Baba, Y. B Cell Signaling and Fate Decision. Annu. Rev. Immunol. 2010, 28, 21–55. [Google Scholar] [CrossRef]
- Baxt, L.A.; Garza-Mayers, A.C.; Goldberg, M.B. Bacterial Subversion of Host Innate Immune Pathways. Science 2013, 340, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Shalkami, A.S.; Hassan, M.; Bakr, A.G. Anti-inflammatory, antioxidant and anti-apoptotic activity of diosmin in acetic acid-induced ulcerative colitis. Hum. Exp. Toxicol. 2017, 37, 78–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouwmeester, T.; Bauch, A.; Ruffner, H.; Angrand, P.-O.; Bergamini, G.; Croughton, K.; Cruciat, C.; Eberhard, D.; Gagneur, J.; Ghidelli, S.; et al. A physical and functional map of the human TNF-α/NF-κB signal transduction pathway. Nat. Cell Biol. 2004, 6, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Helmi, N.; Alammari, D.; Mobashir, M. Role of potential COVID-19 immune system associated genes and the potential pathways linkage with type-2 diabetes. Combinatorial Chemistry & High Throughput Screening. 2022, 25, 2452–2462. [Google Scholar]
- Zhou, T.; Hu, Z.; Yang, S.; Sun, L.; Yu, Z.; Wang, G. Review Article Role of Adaptive and Innate Immunity in Type 2 Diabetes Mellitus. J. Diabetes Res. 2018, 2018, 7457269. [Google Scholar] [CrossRef]
- Stephenson, E.; Reynolds, G.; Botting, R.A.; Calero-Nieto, F.J.; Morgan, M.D.; Tuong, Z.K.; Bach, K.; Sungnak, W.; Worlock, K.B.; Yoshida, M.; et al. Single-cell multi-omics analysis of the immune response in COVID-19. Nat. Med. 2021, 27, 904–916. [Google Scholar] [CrossRef]
- Chalkiadaki, A.; Guarente, L. The multifaceted functions of sirtuins in cancer. Nat. Rev. Cancer 2015, 15, 608–624. [Google Scholar] [CrossRef]
- Machado-Oliveira, G.; Ramos, C.; Marques, A.R.A.; Vieira, O.V. Cell Senescence, Multiple Organelle Dysfunction and Atherosclerosis. Cells 2020, 9, 2146. [Google Scholar] [CrossRef]
- Long, E.O.; Sik Kim, H.; Liu, D.; Peterson, M.E.; Rajagopalan, S. Controlling Natural Killer Cell Responses: Integration of Signals for Activation and Inhibition. Annu. Rev. Immunol. 2013, 31, 227–258. [Google Scholar] [CrossRef] [Green Version]
- Kuballa, P.; Nolte, W.M.; Castoreno, A.B.; Xavier, R.J. Autophagy and the Immune System. Annu. Rev. Immunol. 2012, 30, 611–646. [Google Scholar] [CrossRef]
- Bezbradica, J.S.; Medzhitov, R. Integration of cytokine and heterologous receptor signaling pathways. Nat. Immunol. 2009, 10, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Tigno-Aranjuez, J.T.; Bai, X.; Abbott, D.W. A Discrete Ubiquitin-Mediated Network Regulates the Strength of NOD2 Signaling. Mol. Cell. Biol. 2012, 33, 146–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Källstig, E.; McCabe, B.D.; Schneider, B.L. The Links between ALS and NF-κB. Int. J. Mol. Sci. 2021, 22, 3875. [Google Scholar] [CrossRef] [PubMed]
- Bagaev, A.V.; Garaeva, A.Y.; Lebedeva, E.S.; Pichugin, A.V.; Ataullakhanov, R.I.; Ataullakhanov, F.I. Elevated pre-activation basal level of nuclear NF-κB in native macrophages accelerates Lps- induced translocation of cytosolic NF-κB into the cell nucleus. Sci. Rep. 2019, 9, 4563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, L.; Kim, M.K.; Noonan, A.M.; Sagher, E.; Kohlhammer, H.; Wright, G.; Lyle, L.T.; Steeg, P.S.; Anver, M.; Bowtell, D.D.; et al. A dual role for Caspase8 and NF. Nat. Publ. Group 2015, 1, 15053. [Google Scholar]
- Martin, G.S. Cell signaling and cancer. Cancer Cell 2003, 4, 167–174. [Google Scholar] [CrossRef] [Green Version]
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.-M.; Chi, W.-Y.; Liang, J.; Takayanagi, S.; Iglesias, P.A.; Huang, C.-H. Deciphering cell signaling networks with massively multiplexed biosensor barcoding. Cell 2021, 184, 6193–6206. [Google Scholar] [CrossRef]
- Tyson, J.J.; Chen, K.C.; Novák, B. Sniffers, buzzers, toggles and blinkers: Dynamics of regulatory and signaling pathways in the cell. Current Opinion in Cell Biology 2003, 15, 221–231. [Google Scholar] [CrossRef]
- Danko, C.G.; Hah, N.; Luo, X.; Martins, A.L.; Core, L.; Lis, J.T.; Siepel, A.; Kraus, W.L. Signaling Pathways Differentially Affect RNA Polymerase II Initiation, Pausing, and Elongation Rate in Cells. Mol. Cell 2013, 50, 212–222. [Google Scholar] [CrossRef] [Green Version]
- Han, B.; Li, X.; Ai, R.-S.; Deng, S.-Y.; Ye, Z.-Q.; Deng, X.; Ma, W.; Xiao, S.; Wang, J.-Z.; Wang, L.-M.; et al. Atmospheric particulate matter aggravates cns demyelination through involvement of TLR-4/NF-kB signaling and microglial activation. eLife 2022, 11, e72247. [Google Scholar] [CrossRef] [PubMed]
- Heltberg, M.L.; Krishna, S.; Jensen, M.H. On chaotic dynamics in transcription factors and the associated effects in differential gene regulation. Nat. Commun. 2018, 10, 71. [Google Scholar] [CrossRef] [PubMed]
- Dorrington, M.G.; Fraser, I.D.C. NF-κB Signaling in Macrophages: Dynamics, Crosstalk, and Signal Integration. Front. Immunol. 2019, 10, 705. [Google Scholar] [CrossRef] [PubMed]
- Kuznetsova, T.; Prange, K.H.M.; Glass, C.K.; Winther, M.P.J. Transcriptional and epigeneticregulation of macrophagesin atherosclerosis. Nat. Rev. Cardiol. 2019, 17, 216–228. [Google Scholar] [CrossRef]
- Moore, K.E.; Carlson, S.M.; Camp, N.D.; Cheung, P.; James, R.G.; Chua, K.F.; Wolf-Yadlin, A.; Gozani, O. A General Molecular Affinity Strategy for Global Detection and Proteomic Analysis of Lysine Methylation. Mol. Cell 2013, 50, 444–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbri, G.; Rasi, S.; Rossi, D.; Trifonov, V.; Khiabanian, H.; Ma, J.; Grunn, A.; Fangazio, M.; Capello, D.; Monti, S.; et al. Analysis of the chronic lymphocytic leukemia coding genome: Role of NOTCH1mutational activation. J. Exp. Med. 2011, 208, 1389–1401. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Autophagy and Cellular Immune Responses. Immunity 2013, 39, 211–227. [Google Scholar] [CrossRef] [Green Version]
- Khan, N.; Mukhtar, H. Biochemical Pharmacology. Biochem. Pharmacol. 2013, 85, 667–672. [Google Scholar] [CrossRef] [Green Version]
- Bui, J.D.; Schreiber, R.D. Cancer immunosurveillance, immunoediting and inflammation: Independent or interdependent processes? Curr. Opin. Immunol. 2007, 19, 203–208. [Google Scholar] [CrossRef]
- Koosha, S.; Alshawsh, M.A.; Looi, C.Y.; Seyedan, A.; Mohamed, Z. An Association Map on the Effect of Flavonoids on the Signaling Pathways in Colorectal Cancer. Int. J. Med. Sci. 2016, 13, 374–385. [Google Scholar] [CrossRef] [Green Version]
- Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Gonda, T.J.; Ramsay, R.G. Directly targeting transcriptional dysregulation in cancer. Nat. Rev. Cancer 2015, 15, 686–694. [Google Scholar] [CrossRef] [PubMed]
- da Silva, H.B.; Amaral, E.P.; Nolasco, E.L.; de Victo, N.C.; Atique, R.; Jank, C.C.; Anschau, V.; Zerbini, L.F.; Correa, R.G. Dissecting Major Signaling Pathways throughout the Development of Prostate Cancer. Prostate Cancer 2013, 2013, 920612. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Koldobskiy, M.A.; Göndör, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Mobashir, M.; Madhusudhan, T.; Isermann, B.; Beyer, T.; Schraven, B. Negative Interactions and Feedback Regulations Are Required for Transient Cellular Response. Sci. Rep. 2014, 4, 3718. [Google Scholar] [CrossRef] [Green Version]
- Mobashir, M.; Schraven, B.; Beyer, T. Simulated evolution of signal transduction networks. PLoS ONE 2012, 7, e50905. [Google Scholar] [CrossRef]
- Mobashir, M. Mathematical Modeling and Evolution of Signal Transduction Pathways and Networks. Ph.D. Thesis, Otto-von-Guericke-University Magdeburg, Magdeburg, Germany, 2013. [Google Scholar]
- Vafai, S.B.; Mootha, V.K. A Common Pathway for a Rare Disease? Science 2013, 342, 1453–1454. [Google Scholar] [CrossRef]
- Pant, D.K.; Ghosh, A. A systems biology approach for the study of cumulative oncogenes with applications to the MAPK signal transduction pathway. Biophys. Chem. 2006, 119, 49–60. [Google Scholar] [CrossRef]
- Rousseau, F.; Schymkowitz, J. A systems biology perspective on protein structural dynamics and signal transduction. Curr. Opin. Struct. Biol. 2005, 15, 23–30. [Google Scholar] [CrossRef]
- Mosaddeghi, P.; Eslami, M.; Farahmandnejad, M.; Akhavein, M.; Ranjbarfarrokhi, R.; Khorraminejad-Shirazi, M.; Shahabinezhad, F.; Taghipour, M.; Dorvash, M.; Sakhteman, A.; et al. A systems pharmacology approach to identify the autophagy-inducing effects of Traditional Persian medicinal plants. Sci. Rep. 2020, 11, 336. [Google Scholar] [CrossRef]
- Werner, H.M.J.; Mills, G.B.; Ram, P.T. Cancer Systems Biology: A peek into the future of patient care? Nat. Rev. Clin. Oncol. 2014, 11, 167–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, E.; Zou, J.; Zaman, N.; Beitel, L.K.; Trifiro, M.; Paliouras, M. Seminars in Cancer Biology. Semin. Cancer Biol. 2013, 23, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Zaman, N.; Mcgee, S.; Milanese, J.-S.; Masoudi-Nejad, A.; O’Connor-McCourt, M. Seminars in Cancer Biology. Semin. Cancer Biol. 2015, 30, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Finley, S.D.; Chu, L.-H.; Popel, A.S. Computational systems biology approaches to anti-angiogenic cancer therapeutics. Drug Discov. Today 2014, 20, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Qutub, A.A. Systems approaches for synthetic biology: A pathwaytoward mammalian design. Front. Physiol. 2013, 4, 285. [Google Scholar]
- Lin, Y.C.; Huang, D.Y.; Chu, C.L.; Lin, Y.L.; Lin, W.W. The Tyrosine Kinase Syk Differentially Regulates Toll-like Receptor Signaling Downstream of the Adaptor Molecules TRAF6 and TRAF3. Sci. Signal. 2013, 6, ra71. [Google Scholar] [CrossRef] [PubMed]
- Lillemeier, B.F. How membrane structures control T cell signaling. 2012, 3, 291. 3.
- Zhang, Y.; Du, Y.; Le, W.; Wang, K.; Kieffer, N.; Zhang, J. Redox Control of the Survival of Healthy and Diseased Cells. Antioxid. Redox Signal. 2011, 15, 2867–2908. [Google Scholar] [CrossRef] [PubMed]
- Breinig, M.; Klein, F.A.; Huber, W.; Boutros, M. A chemical-genetic interaction map of small molecules using high-throughput imaging in cancer cells. Mol. Syst. Biol. 2015, 11, 846. [Google Scholar] [CrossRef]
- Reinartz, S.; Finkernagel, F.; Adhikary, T.; Rohnalter, V.; Schumann, T.; Schober, Y.; Nockher, W.A.; Nist, A.; Stiewe, T.; Jansen, J.M.; et al. A transcriptome-based global map of signaling pathways in the ovarian cancer microenvironment associated with clinical outcome. Genome Biol. 2016, 17, 108. [Google Scholar] [CrossRef] [Green Version]
- Speer, T.; Rohrer, L.; Blyszczuk, P.; Shroff, R.; Kuschnerus, K.; Kränkel, N.; Kania, G.; Zewinger, S.; Akhmedov, A.; Shi, Y.; et al. Abnormal High-Density Lipoprotein Induces Endothelial Dysfunctionvia Activation of Toll-like Receptor-2. Immunity 2013, 38, 754–768. [Google Scholar] [CrossRef] [Green Version]
- Smyth, J.T.; Hwang, S.-Y.; Tomita, T.; DeHaven, W.I.; Mercer, J.C.; Putney, J.W. Activation and regulation of store-operated calcium entry. J. Cell. Mol. Med. 2010, 14, 2337–2349. [Google Scholar] [CrossRef] [PubMed]
- McClean, M.N.; Mody, A.; Broach, J.R.; Ramanathan, S. Cross-talk and decision making in MAP kinase pathways. Nat. Genet. 2007, 39, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Stuart, R.O.; Wachsman, W.; Berry, C.C.; Wang-Rodriguez, J.; Wasserman, L.; Klacansky, I.; Masys, D.; Arden, K.; Goodison, S.; McClelland, M.; et al. In silico dissection of cell-type-associated patterns of gene expression in prostate cancer. Proc. Natl. Acad. Sci. USA 2004, 101, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Kholodenko, B.; Yaffe, M.B.; Kolch, W. Computational Approaches for Analyzing Information Flow in Biological Networks. Sci. Signal. 2012, 5, re1. [Google Scholar] [CrossRef]
- Kholodenko, B.N. Four-dimensional organization of protein kinase signaling cascades: The roles of diffusion, endocytosis and molecular motors. J. Exp. Biol. 2003, 206, 2073–2082. [Google Scholar] [CrossRef] [Green Version]
- Kholodenko, B.N.; Demin, O.V.; Markevich, N.I.; Kiyatkin, A.; Moehren, G.; Hoek, J.B. Signal processing at the Ras circuit: What shapes Ras activation patterns? Syst. Biol. 2004, 1, 104–113. [Google Scholar]
- Boris, N. Kholodenko Spatially distributed cell signalling. FEBS Lett. 2009, 583, 4006–4012. [Google Scholar]
- Kholodenko, B.N.; Kiyatkin, A.; Bruggeman, F.J.; Sontag, E.; Westerhoff, H.V.; Hoek, J.B. Untangling the wires: A strategy to trace functional interactions in signaling and gene networks. Proc. Natl. Acad. Sci. USA 2002, 99, 12841–12846. [Google Scholar] [CrossRef] [Green Version]
- Aksamitiene, E.; Kiyatkin, A.; Kholodenko, B.N. Cross-talk between mitogenic Ras/MAPK and survival PI3K/Akt pathways: A fine balance. Biochm. Soc. Trans. 2012, 40, 139–146. [Google Scholar] [CrossRef]
- Bluthgen, N.; Bruggeman, F.J.; Legewie, S.; Herzel, H.; Westerhoff, H.V.; Kholodenko, B.N. Effects of sequestration on signal transduction cascades. FEBS J. 2006, 273, 895–906. [Google Scholar] [CrossRef]
- Winstead, C.J.; Weaver, C.T. Dwelling on T Cell Fate Decisions. Cell 2013, 153, 739–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.K.; Matallanas, D.G.; Romano, D.; Kholodenko, B.N.; Kolch, W. Competing to coordinate cell fate decisions: The MST2-Raf-1 signaling device. Cell Cycle 2015, 14, 189–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiterer, V.; Fey, D.; Kolch, W. Pseudophosphatase STYX modulates cell-fate decisions and cell migration by spatiotemporal regulation of ERK1/2. Proc. Natl. Acad. Sci. USA 2013, 110, E2934–E2943. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.; Srikanth, R.; Ahlfors, H.; Lahesmaa, R.; Rao, K.V.S. Capturing cell-fate decisions from the molecular signatures of a receptor-dependent signaling response. Mol. Syst. Biol. 2007, 3, 150. [Google Scholar] [CrossRef] [PubMed]
- Formosa-Jordan, P.; Ibañes, M. Competition in Notch Signaling with Cis Enriches Cell Fate Decisions. PLoS ONE 2014, 9, e95744. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, J. Quantifying Cell Fate Decisions for Differentiation and Reprogramming of a Human Stem Cell Network: Landscape and Biological Paths. PLoS Comput. Biol. 2013, 9, e1003165. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Cao, X.; Biase, F.H.; Yu, P.; Zhong, S. Time-variant clustering model for understanding cell fate decisions. Proc. Natl. Acad. Sci. USA 2014, 111, E4797–E4806. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Durfee, L.A.; Huibregtse, J.M. A Cotranslational Ubiquitination Pathway for Quality Control of Misfolded Proteins. Mol. Cell 2013, 50, 368–378. [Google Scholar] [CrossRef] [Green Version]
- Cui, Q.; Ma, Y.; Jaramillo, M.; Bari, H.; Awan, A.; Yang, S.; Zhang, S.; Liu, L.; Lu, M.; O’Connor-McCourt, M.; et al. A map of human cancer signaling. Mol. Syst. Biol. 2007, 3, 152. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef] [Green Version]
- Khosravi, M.; Poursaleh, A.; Ghasempour, G.; Farhad, S.; Najafi, M. The effects of oxidative stress on the development of atherosclerosis. Biol. Chem. 2019, 400, 711–732. [Google Scholar] [CrossRef] [PubMed]
- Tata, P.R.; Rajagopal, J. Plasticity in the lung: Making and breaking cell identity. Development 2017, 144, 755–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loukovaara, S.; Gucciardo, E.; Repo, P.; Lohi, J.; Salven, P.; Lehti, K. A Case of Abnormal Lymphatic-Like Differentiation and Endothelial Progenitor Cell Activation in Neovascularization Associated with Hemi-Retinal Vein Occlusion. Case Rep. Ophthalmol. 2015, 6, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Michelini, R.H.; Doedens, A.L.; Goldrath, A.W.; Hedrick, S.M. Differentiation of CD8 memory T cells depends on Foxo1. J. Exp. Med. 2013, 210, 1189–1200. [Google Scholar] [CrossRef]
- Chen, B.; Xue, Z.; Yang, G.; Shi, B.; Yang, B.; Yan, Y.; Wang, X.; Han, D.; Huang, Y.; Dong, W. Akt-Signal Integration Is Involved in the Differentiation of Embryonal Carcinoma Cells. PLoS ONE 2013, 8, e64877. [Google Scholar] [CrossRef]
- Rebhahn, J.A.; Deng, N.; Sharma, G.; Livingstone, A.M.; Huang, S.; Mosmann, T.R. An animated landscape representation of CD4 +T-cell differentiation, variability, and plasticity: Insights into the behavior of populations versus cells. Eur. J. Immunol. 2014, 44, 2216–2229. [Google Scholar] [CrossRef]
- Moustakas, A.; Pardali, K.; Gaal, A.; Heldin, C.H. Mechanisms of TGF-β signaling in regulation of cell growth and differentiation. Immunol. Lett. 2002, 82, 85–91. [Google Scholar] [CrossRef]
- Ma, Y.; Adjemian, S.; Mattarollo, S.R.; Yamazaki, T.; Aymeric, L.; Yang, H.; Catani, J.P.P.; Hannani, D.; Duret, H.; Steegh, K.; et al. Anticancer Chemotherapy-Induced Intratumoral Recruitment and Differentiationof Antigen-Presenting Cells. Immunity 2013, 38, 729–741. [Google Scholar] [CrossRef] [Green Version]
- Quann, E.J.; Liu, X.; Altan-Bonnet, G.; Huse, M. A cascade of protein kinase C isozymes promotes cytoskeletal polarization in T cells. Nat. Publ. Group 2011, 12, 647–654. [Google Scholar] [CrossRef]
- Thomas, J.D.; Lee, T.; Suh, N.P. A function-based framework for understanding biological systems. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 75–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, S.D.M.; Verveer, P.J.; Bastiaens, P.I.H. Growth factor-induced MAPK network topology shapes Erk response determining PC-12 cell fate. Nat. Cell Biol. 2007, 9, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Orton, R.J.; Adriaens, M.E.; Gormand, A.; Sturm, O.E.; Kolch, W.; Gilbert, D.R. Computational modelling of cancerous mutations in the EGFR/ERK signalling pathway. BMC Syst. Biol. 2009, 3, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, J.; Floros, I. Adaptive information processing of network modules to dynamic and spatial stimuli. BMC Syst. Biol. 2019, 13, 32. [Google Scholar] [CrossRef]
- Zhang, R.; Lahens, N.F.; Ballance, H.I.; Hughes, M.E.; Hogenesch, J.B. A circadian gene expression atlas in mammals: Implications for biology and medicine. Proc. Natl. Acad. Sci. USA 2014, 111, 16219–16224. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.J.; Ewald, A.J. A collective route to metastasis: Seeding by tumor cell clusters. Science 2016, 352, 167–169. [Google Scholar] [CrossRef] [Green Version]
- Klein, E.A.; Cooperberg, M.R.; Magi-Galluzzi, C.; Simko, J.P.; Falzarano, S.M.; Maddala, T.; Chan, J.M.; Li, J.; Cowan, J.E.; Tsiatis, A.C.; et al. A 17-gene Assay to Predict Prostate Cancer Aggressiveness in the Context of Gleason Grade Heterogeneity, Tumor Multifocality, and Biopsy Undersampling. Eur. Urol. 2014, 66, 550–560. [Google Scholar] [CrossRef] [Green Version]
- Kitano, H. Computational systems biology. Nature 2002, 420, 206–210. [Google Scholar] [CrossRef]
- Wang, D.Y.; Cardelli, L.; Phillips, A.; Piterman, N.; Fisher, J. Computational modeling of the EGFR network elucidates control mechanisms regulating signal dynamics. BMC Syst. Biol. 2009, 3, 118. [Google Scholar] [CrossRef] [Green Version]
- Klingmüller, U. Heterogeneous kinetics of AKT signaling in individual cells are accounted for by variable protein concentration. Front. Physiol. 2012, 3, 451. [Google Scholar]
- Kozer, N.; Barua, D.; Orchard, S.; Nice, E.C.; Burgess, A.W.; Hlavacek, W.S.; Clayton, A.H.A. Exploring higher-order EGFR oligomerisation and phosphorylation--a combined experimental and theoretical approach. Mol. BioSyst. 2013, 9, 1849–1863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Camillo, B.; Toffolo, G.; Cobelli, C. A Gene Network Simulator to Assess Reverse Engineering Algorithms. Ann. N. Y. Acad. Sci. 2009, 1158, 125–142. [Google Scholar] [CrossRef] [PubMed]
- Stoevesandt, O.; Kohler, K.; Wolf, S.; Andre, T.; Hummel, W.; Brock, R. A Network Analysis of Changes in Molecular Interactions in Cellular Signaling. Mol. Cell. Proteom. 2006, 6, 503–513. [Google Scholar] [CrossRef] [Green Version]
- Murphy, L.O.; MacKeigan, J.P.; Blenis, J. A Network of Immediate Early Gene Products Propagates Subtle Differences in Mitogen-Activated Protein Kinase Signal Amplitude and Duration. Mol. Cell. Biol. 2003, 24, 144–153. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Elbaum, M. A Simple Kinetic Model with Explicit Predictions for Nuclear Transport. Biophys. J. 2013, 105, 565–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sontag, E.; Kiyatkin, A.; Kholodenko, B.N. Inferring dynamic architecture of cellular networks using time series of gene expression, protein and metabolite data. Bioinformatics 2004, 20, 1877–1886. [Google Scholar] [CrossRef] [PubMed]
- Vera, J.; Rath, O.; Balsa-Canto, E.; Banga, J.R.; Kolch, W.; Wolkenhauer, O. Investigating dynamics of inhibitory and feedback loops in ERK signalling using power-law models. Mol. BioSyst. 2010, 6, 2174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, J.M.; Portillo, M.C.; Piñeiro-Vidal, M. Latitude-dependent underestimation of microbial extracellular enzyme activity in soils. Int. J. Environ. Sci. Technol. 2015, 12, 2427–2434. [Google Scholar] [CrossRef] [Green Version]
- Varusai, T.M.; Kolch, W.; Kholodenko, B.N.; Nguyen, L.K. Molecular BioSystems. Mol. BioSyst. 2015, 11, 2750–2762. [Google Scholar] [CrossRef]
- Mobasheri, A. Biosensors for the Multiplex Detection of Inflammatory Disease Biomarkers. Biosensors 2020, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Anwer, S.T.; Mobashir, M.; Fantoukh, O.I.; Khan, B.; Imtiyaz, K.; Naqvi, I.H.; Rizvi, M.M.A. Synthesis of Silver Nano Particles Using Myricetin and the In-Vitro Assessment of Anti-Colorectal Cancer Activity: In-Silico Integration. Int. J. Mol. Sci. 2022, 23, 11024. [Google Scholar] [CrossRef]
- Baud, V.; Karin, M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Disc. 2009, 8, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Bai, L.; Chen, W.; Xu, S. The NF-kappaB activation pathways, emerging molecular targets for cancer prevention and therapy. Expert Opin. Ther. Targets 2010, 14, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Mobashir, M. The Understanding of the Potential Linkage between COVID-19, Type-2 Diabetes, and Cancer (s) Could Help in Better Drug Targets and Therapeutics. Comb. Chem. High Throughput Screen. 2022, 25, 2370–2371. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almowallad, S.; Alqahtani, L.S.; Mobashir, M. NF-kB in Signaling Patterns and Its Temporal Dynamics Encode/Decode Human Diseases. Life 2022, 12, 2012. https://doi.org/10.3390/life12122012
Almowallad S, Alqahtani LS, Mobashir M. NF-kB in Signaling Patterns and Its Temporal Dynamics Encode/Decode Human Diseases. Life. 2022; 12(12):2012. https://doi.org/10.3390/life12122012
Chicago/Turabian StyleAlmowallad, Sanaa, Leena S. Alqahtani, and Mohammad Mobashir. 2022. "NF-kB in Signaling Patterns and Its Temporal Dynamics Encode/Decode Human Diseases" Life 12, no. 12: 2012. https://doi.org/10.3390/life12122012