
The Structures of Heterogeneous Membranes and Their Interactions with an Anticancer Peptide: A Molecular Dynamics Study

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
3. Results and Discussion
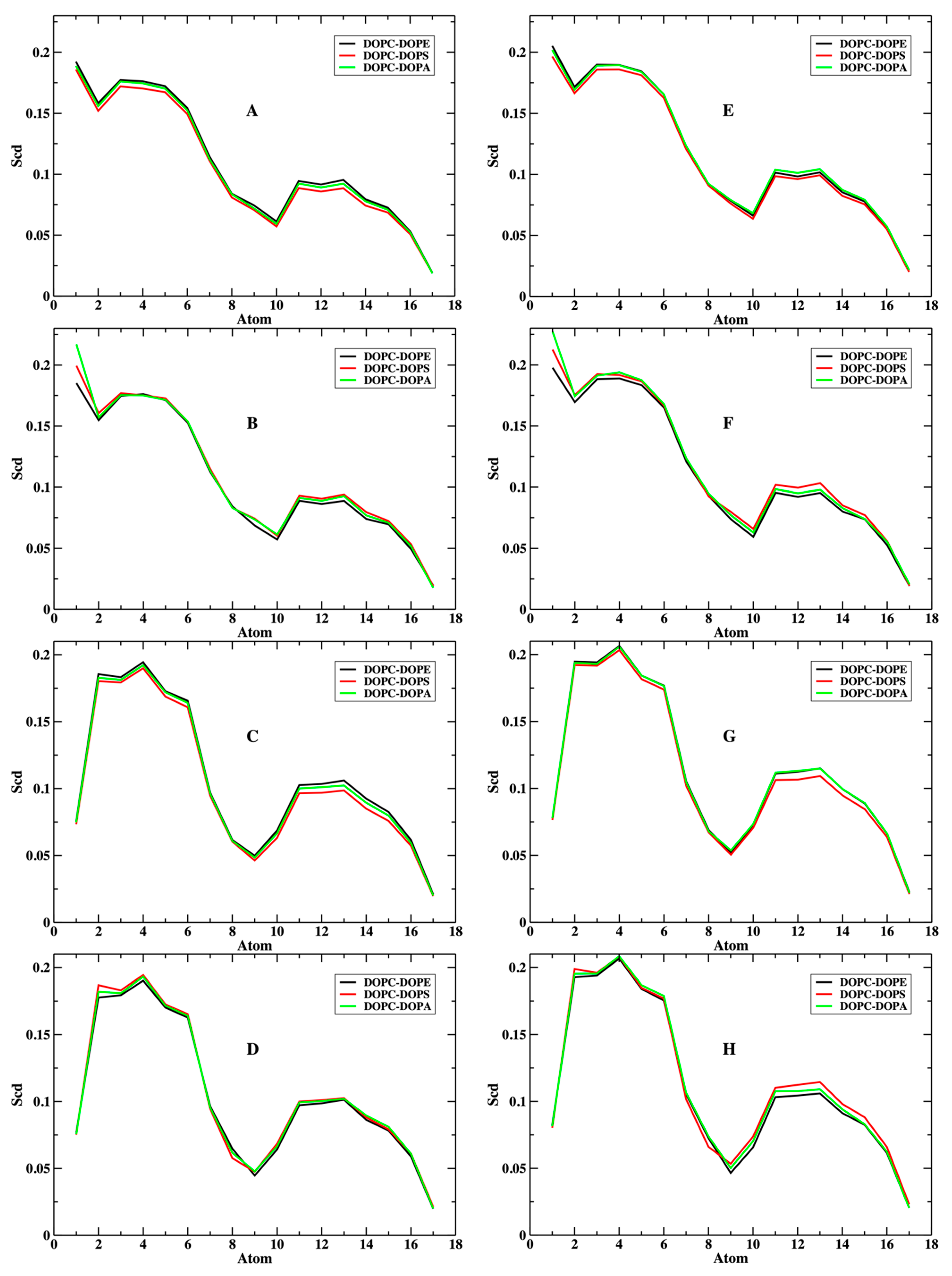
3.1. The Phospholipids
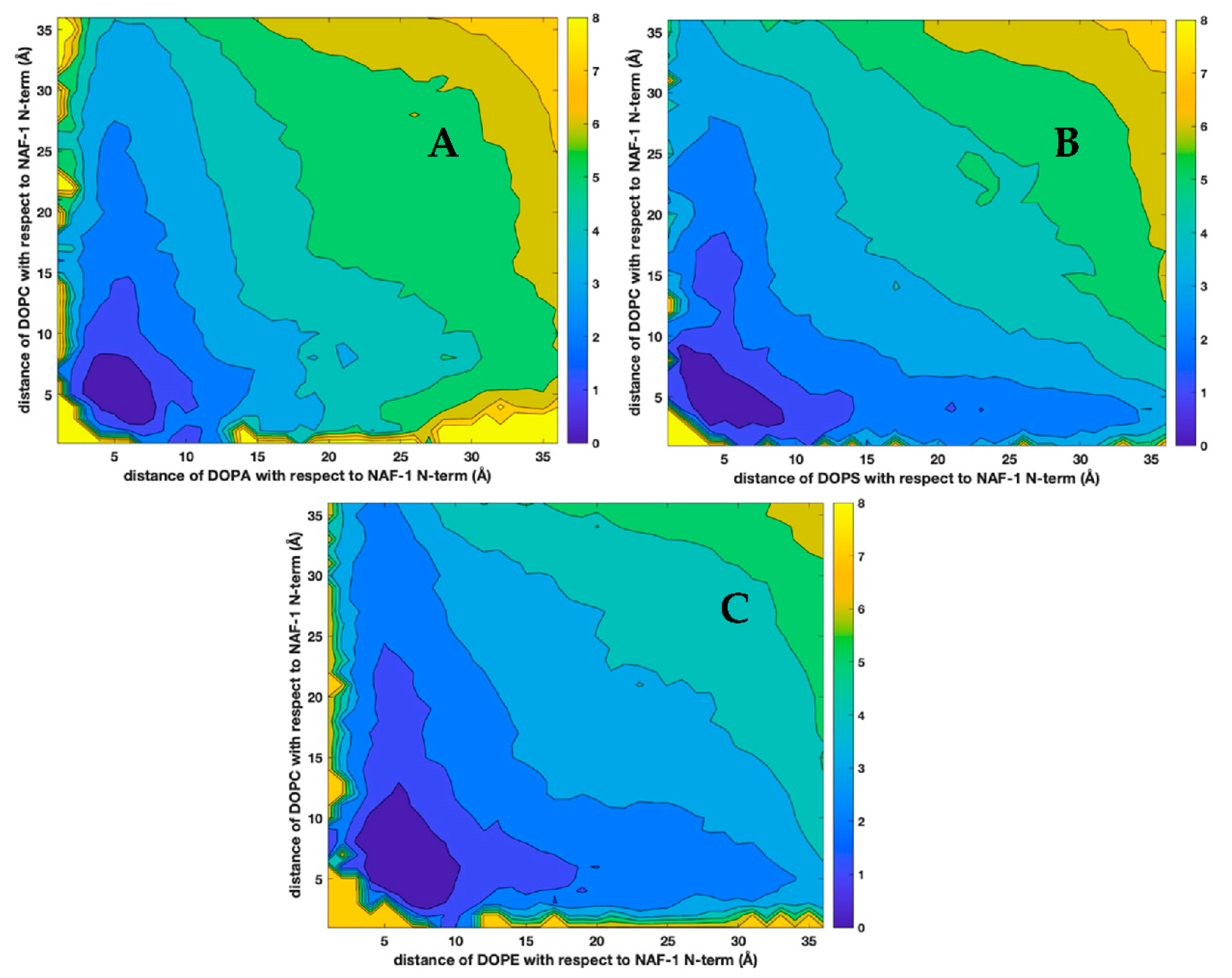
3.2. The Peptide and the Phospholipids
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hong, C.; Tieleman, D.P.; Wang, Y. Microsecond Molecular Dynamics Simulations of Lipid Mixing. Langmuir 2014, 30, 11993–12001. [Google Scholar] [CrossRef] [PubMed]
- Enkavi, G.; Javanainen, M.; Kulig, W.; Rog, T.; Vattulainen, I. Multiscale Simulations of Biological Membranes: The Challenge to Understand Biological Phenomena in a Living Substance. Chem. Rev. 2019, 119, 5607–5774. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; Monticelli, L.; Melo, M.N.; Alessandri, R.; Tieleman, D.P.; Souza, P.C.T. Two decades of Martini: Better beads, broader scope. Wiley Interdiscip. Rev.-Comput. Mol. Sci. 2022, e1620. [Google Scholar] [CrossRef]
- Marrink, S.J.; Corradi, V.; Souza, P.C.T.; Ingolfsson, H.I.; Tieleman, D.P.; Sansom, M.S.P. Computational Modeling of Realistic Cell Membranes. Chem. Rev. 2019, 119, 6184–6226. [Google Scholar] [CrossRef] [PubMed]
- Venable, R.M.; Kramer, A.; Pastor, R.W. Molecular Dynamics Simulations of Membrane Permeability. Chem. Rev. 2019, 119, 5954–5997. [Google Scholar] [CrossRef] [PubMed]
- Brown, F.L.H. Continuum simulations of biomembrane dynamics and the importance of hydrodynamic effects. Q. Rev. Biophys. 2011, 44, 391–432. [Google Scholar] [CrossRef]
- Ayton, G.; Voth, G.A. Bridging microscopic and mesoscopic simulations of lipid bilayers. Biophys. J. 2002, 83, 3357–3370. [Google Scholar] [CrossRef]
- Venturoli, M.; Sperotto, M.M.; Kranenburg, M.; Smit, B. Mesoscopic models of biological membranes. Phys. Rep.-Rev. Sect. Phys. Lett. 2006, 437, 1–54. [Google Scholar] [CrossRef]
- Ramakrishnan, N.; Kumar, P.B.S.; Radhakrishnan, R. Mesoscale computational studies of membrane bilayer remodeling by curvature-inducing proteins. Phys. Rep.-Rev. Sect. Phys. Lett. 2014, 543, 1–60. [Google Scholar] [CrossRef]
- Kusumi, A.; Fujiwara, T.K.; Tsunoyama, T.A.; Kasai, R.S.; Liu, A.A.; Hirosawa, K.M.; Kinoshita, M.; Matsumori, N.; Komura, N.; Ando, H.; et al. Defining raft domains in the plasma membrane. Traffic 2020, 21, 106–137. [Google Scholar] [CrossRef]
- Nicolson, G.L.; de Mattos, G.F. Fifty Years of the Fluid-Mosaic Model of Biomembrane Structure and Organization and Its Importance in Biomedicine with Particular Emphasis on Membrane Lipid Replacement. Biomedicines 2022, 10, 1711. [Google Scholar] [CrossRef] [PubMed]
- Nilmeier, J.P.; Crooks, G.E.; Minh, D.D.L.; Chodera, J.D. Nonequilibrium candidate Monte Carlo is an efficient tool for equilibrium simulation. Proc. Natl. Acad. Sci. USA 2011, 108, E1009–E1018. [Google Scholar] [CrossRef] [PubMed]
- Fathizadeh, A.; Elber, R. A mixed alchemical and equilibrium dynamics to simulate hetrogeneous dense fluids: Illustration for Lennard-Jones mixtures and phospholipid membranes. J. Chem. Phys. 2018, 149, 072325. [Google Scholar] [CrossRef] [PubMed]
- Cherniavskyi, Y.K.; Fathizadeh, A.; Elber, R.; Tieleman, D.P. Computer simulations of a heterogeneous membrane with enhanced sampling techniques. J. Chem. Phys. 2020, 153, 144110. [Google Scholar] [CrossRef]
- Kumar, N.; Sastry, G.N. Study of lipid heterogeneity on bilayer membranes using molecular dynamics simulations. J. Mol. Graph. Model. 2021, 108, 108000. [Google Scholar] [CrossRef]
- Piggot, T.J.; Pineiro, A.; Khalid, S. Molecular Dynamics Simulations of Phosphatidylcholine Membranes: A Comparative Force Field Study. J. Chem. Theory Comput. 2012, 8, 4593–4609. [Google Scholar] [CrossRef]
- Botan, A.; Favela-Rosales, F.; Fuchs, P.F.J.; Javanainen, M.; Kanduc, M.; Kulig, W.; Lamber, A.; Loison, C.; Lyubartsev, A.; Miettinen, M.S.; et al. Toward Atomistic Resolution Structure of Phosphatidylcholine Headgroup and Glycerol Backbone at Different Ambient Conditions. J. Phys. Chem. B 2015, 119, 15075–15088. [Google Scholar] [CrossRef]
- Venable, R.M.; Luo, Y.; Gawrisch, K.; Roux, B.; Pastor, R.W. Simulations of Anionic Lipid Membranes: Development of Interaction-Specific Ion Parameters and Validation Using NMR Data. J. Phys. Chem. B 2013, 117, 10183–10192. [Google Scholar] [CrossRef]
- Jurkiewicz, P.; Cwiklik, L.; Vojtiskova, A.; Jungwirth, P.; Hof, M. Structure, dynamics, and hydration of POPC/POPS bilayers suspended in NaCl, KCl, and CsCl solutions. BBA-Biomembr. 2012, 1818, 609–616. [Google Scholar] [CrossRef]
- Conlan, A.R.; Axelrod, H.L.; Cohen, A.E.; Abresch, E.C.; Zuris, J.; Yee, D.; Nechushtai, R.; Jennings, P.A.; Paddock, M.L. Crystal Structure of Miner1: The Redox-active 2Fe-2S Protein Causative in Wolfram Syndrome 2. J. Mol. Biol. 2009, 392, 143–153. [Google Scholar] [CrossRef] [Green Version]
- Tamir, S.; Paddock, M.L.; Darash-Yahana-Baram, M.; Holt, S.H.; Sohn, Y.S.; Agranat, L.; Michaeli, D.; Stofleth, J.T.; Lipper, C.H.; Morcos, F.; et al. Structure-function analysis of NEET proteins uncovers their role as key regulators of iron and ROS homeostasis in health and disease. BBA-Mol. Cell Res. 2015, 1853, 1294–1315. [Google Scholar] [CrossRef] [PubMed]
- Sohn, Y.S.; Losub-Amir, A.; Cardenas, A.E.; Karmi, O.; Yahana, M.D.; Gruman, T.; Rowland, L.; Marjault, H.B.; Webb, L.J.; Mittler, R.; et al. A peptide-derived strategy for specifically targeting the mitochondria and ER of cancer cells: A new approach in fighting cancer. Chem. Sci. 2022, 13, 6929–6941. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, A.E.; Drexler, C.I.; Nechushtai, R.; Mittler, R.; Friedler, A.; Webb, L.J.; Elber, R. Peptide Permeation across a Phosphocholine Membrane: An Atomically Detailed Mechanism Determined through Simulations and Supported by Experimentation. J. Phys. Chem. B 2022, 126, 2834–2849. [Google Scholar] [CrossRef] [PubMed]
- Desale, K.; Kuche, K.; Jain, S. Cell-penetrating peptides (CPPs): An overview of applications for improving the potential of nanotherapeutics. Biomater. Sci. 2021, 9, 1153–1188. [Google Scholar] [CrossRef]
- Green, M.; Loewenstein, P.M. Autonomous Functional Domains of Chemically Synthesized Human Immunodeficiency Virus Tat Trans-Activator Protein. Cell 1988, 55, 1179–1188. [Google Scholar] [CrossRef]
- Reddy, K.V.R.; Yedery, R.D.; Aranha, C. Antimicrobial peptides: Premises and promises. Int. J. Antimicrob. Agents 2004, 24, 536–547. [Google Scholar] [CrossRef]
- Milletti, F. Cell-penetrating peptides: Classes, origin, and current landscape. Drug Discov. Today 2012, 17, 850–860. [Google Scholar] [CrossRef]
- Montgomerie, S.; Cruz, J.A.; Shrivastava, S.; Arndt, D.; Berjanskii, M.; Wishart, D.S. PROTEUS2: A web server for comprehensive protein structure prediction and structure-based annotation. Nucleic Acids Res. 2008, 36, W202–W209. [Google Scholar] [CrossRef]
- Alves, A.C.; Ribeiro, D.; Nunes, C.; Reis, S. Biophysics in cancer: The relevance of drug-membrane interaction studies. BBA-Biomembr. 2016, 1858, 2231–2244. [Google Scholar] [CrossRef]
- Zwaal, R.F.A.; Comfurius, P.; Bevers, E.M. Surface exposure of phosphatidylserine in pathological cells. Cell. Mol. Life Sci. 2005, 62, 971–988. [Google Scholar] [CrossRef] [PubMed]
- Riedl, S.; Rinner, B.; Asslaber, M.; Schaider, H.; Walzer, S.; Novak, A.; Lohner, K.; Zweytick, D. In search of a novel target—Phosphatidylserine exposed by non-apoptotic tumor cells and metastases of malignancies with poor treatment efficacy. BBA-Biomembr. 2011, 1808, 2638–2645. [Google Scholar] [CrossRef]
- Jo, S.; Lim, J.B.; Klauda, J.B.; Im, W. CHARMM-GUI Membrane Builder for Mixed Bilayers and Its Application to Yeast Membranes. Biophys. J. 2009, 97, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.F.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmuller, H.; MacKerell, A.D. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical Dynamics—Equilibrium Phase-Space Distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Nose, S. A Molecular-Dynamics Method for Simulations in the Canonical Ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single-Crystals—A New Molecular-Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Miyamoto, S.; Kollman, P.A. Settle—An Analytical Version of the Shake and Rattle Algorithm for Rigid Water Models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef]
- Van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Rosetti, C.M.; Mangiarotti, A.; Wilke, N. Sizes of lipid domains: What do we know from artificial lipid membranes? What are the possible shared features with membrane rafts in cells? BBA-Biomembr. 2017, 1859, 789–802. [Google Scholar] [CrossRef]
- Cardenas, A.E.; Shrestha, R.; Webb, L.J.; Elber, R. Membrane Permeation of a Peptide: It Is Better to be Positive. J. Phys. Chem. B 2015, 119, 6412–6420. [Google Scholar] [CrossRef]
- Povilaitis, S.C.; Fathizadeh, A.; Kogan, M.; Elber, R.; Webb, L.J. Design of Peptides for Membrane Insertion: The Critical Role of Charge Separation. J. Phys. Chem. B 2022, 126, 6454–6463. [Google Scholar] [CrossRef]
- Fathizadeh, A.; Elber, R. Ion Permeation through a Phospholipid Membrane: Transition State, Path Splitting, and Calculation of Permeability. J. Chem. Theory Comput. 2019, 15, 720–730. [Google Scholar] [CrossRef]
- Hanafusa, K.; Hotta, T.; Iwabuchi, K. Glycolipids: Linchpins in the Organization and Function of Membrane Microdomains. Front. Cell Dev. Biol. 2020, 8, 589799. [Google Scholar] [CrossRef] [PubMed]
- Fathizadeh, A.; Kogan, M.; Anderson, C.M.; Webb, L.J.; Elber, R. Defect-Assisted Permeation Through a Phospholipid Membrane: Experimental and Computational Study of the Peptide WKW. J. Phys. Chem. B 2019, 123, 6792–6798. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abbas, G.; Cardenas, A.E.; Elber, R. The Structures of Heterogeneous Membranes and Their Interactions with an Anticancer Peptide: A Molecular Dynamics Study. Life 2022, 12, 1473. https://doi.org/10.3390/life12101473
Abbas G, Cardenas AE, Elber R. The Structures of Heterogeneous Membranes and Their Interactions with an Anticancer Peptide: A Molecular Dynamics Study. Life. 2022; 12(10):1473. https://doi.org/10.3390/life12101473
Chicago/Turabian StyleAbbas, Ghulam, Alfredo E. Cardenas, and Ron Elber. 2022. "The Structures of Heterogeneous Membranes and Their Interactions with an Anticancer Peptide: A Molecular Dynamics Study" Life 12, no. 10: 1473. https://doi.org/10.3390/life12101473