
Ritonavir and xk263 Binding-Unbinding with HIV-1 Protease: Pathways, Energy and Comparison


Abstract
:1. Introduction
2. Materials and Methods
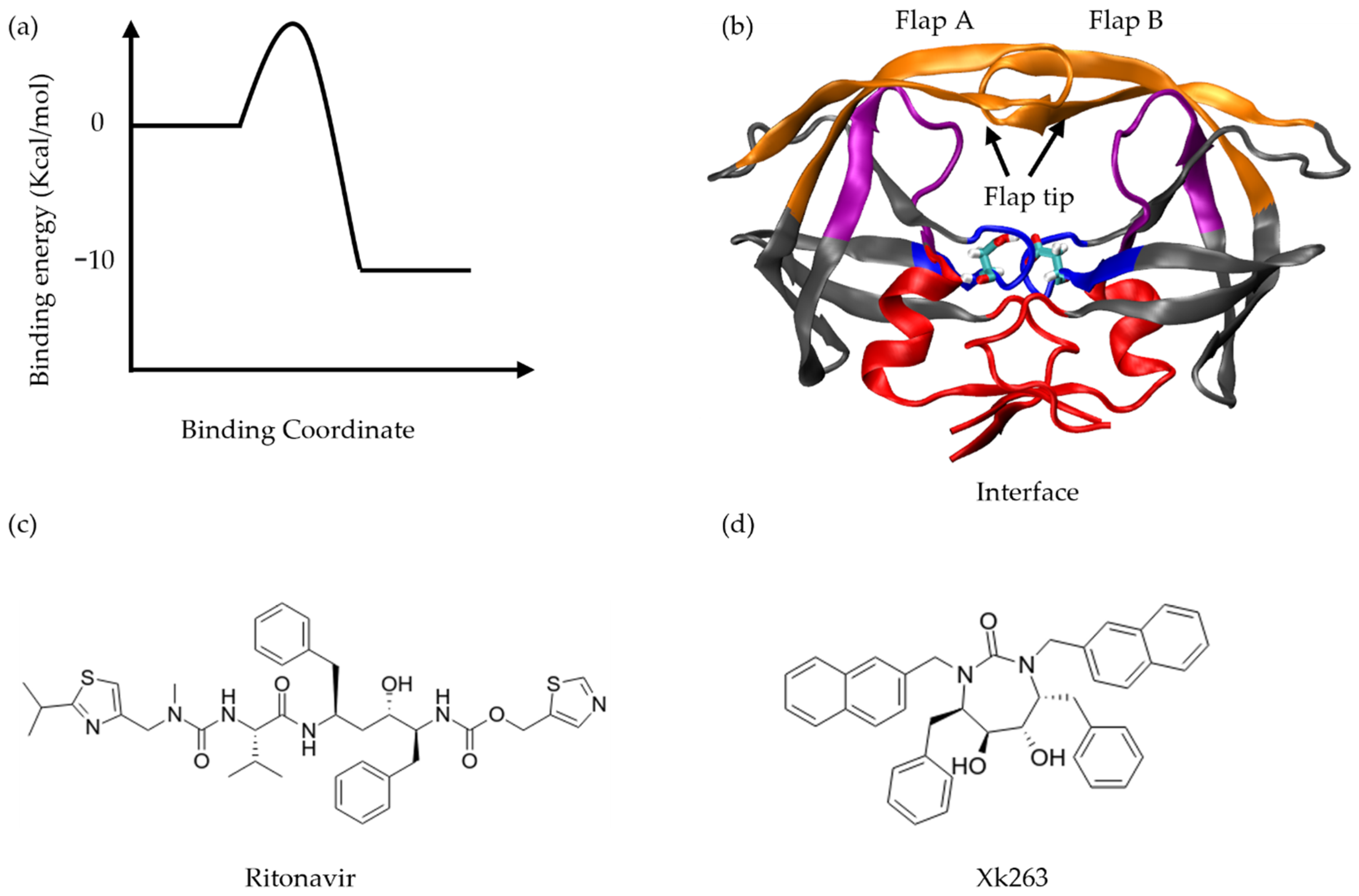
2.1. Target Molecular Systems
2.2. MD Simulations
2.3. Accelerated MD (aMD) Simulation
2.4. Re-Seeding Approach
2.5. Hydrogen Bond Analysis
2.6. MM/PBSA Interaction Energy
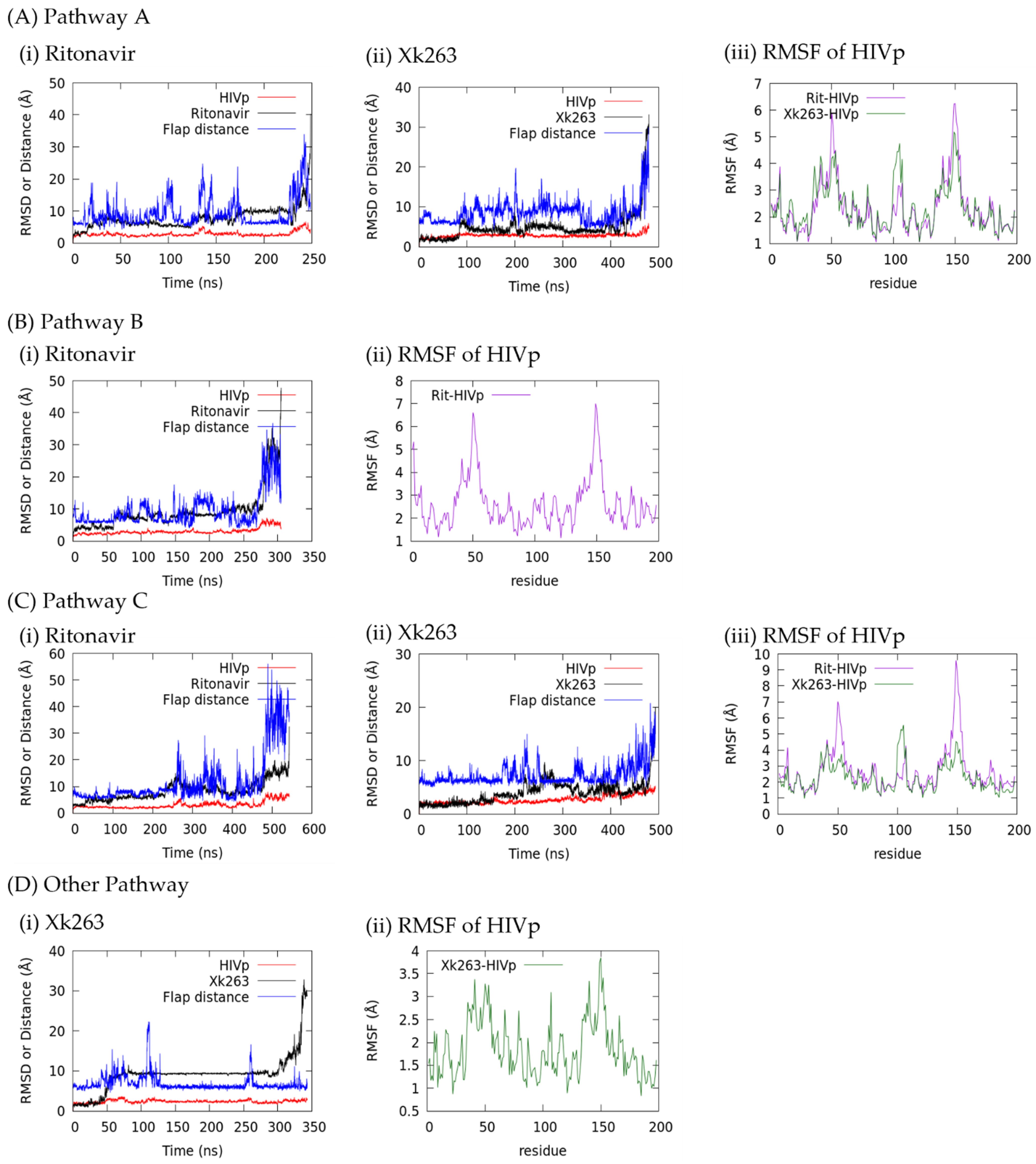
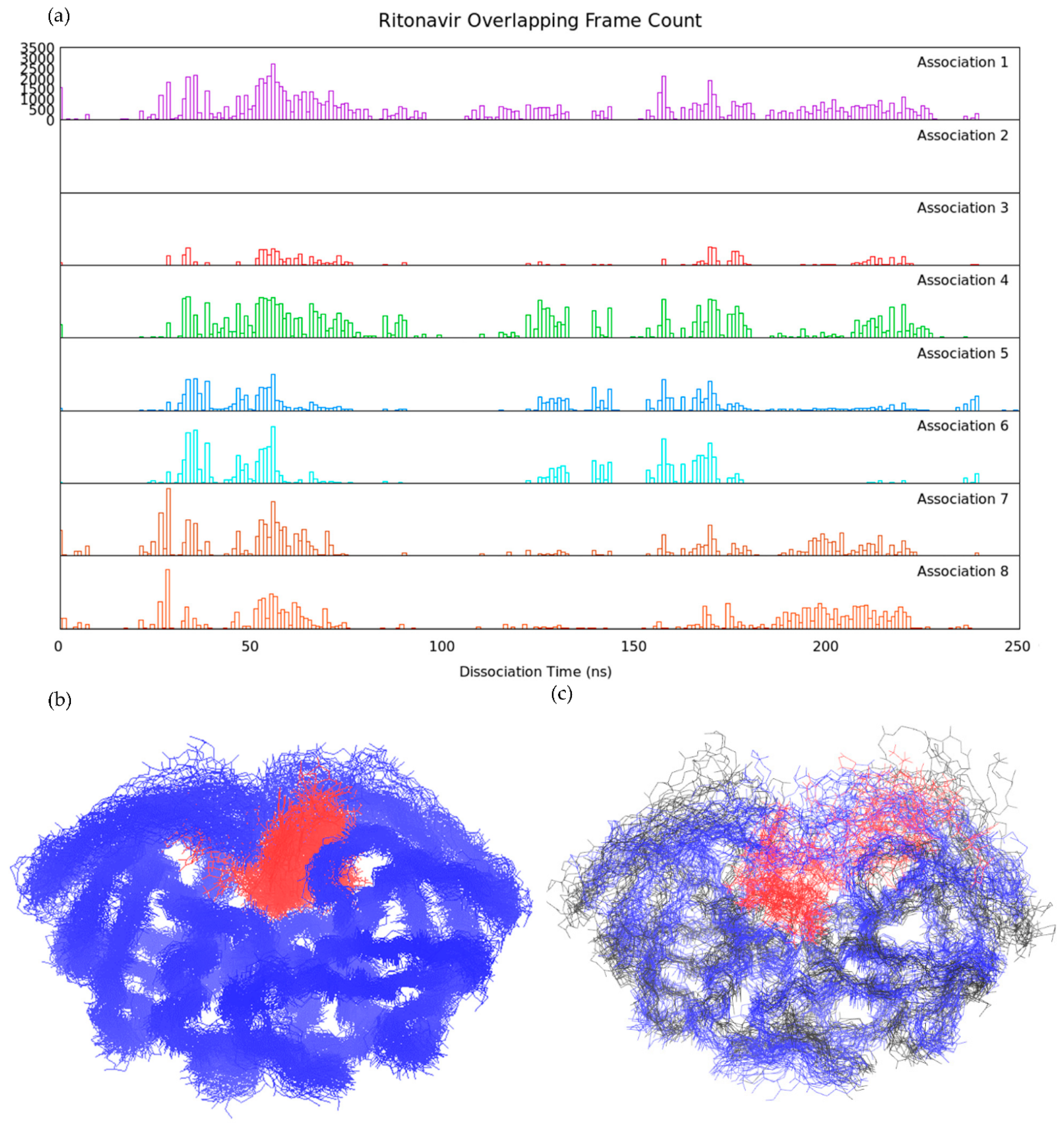
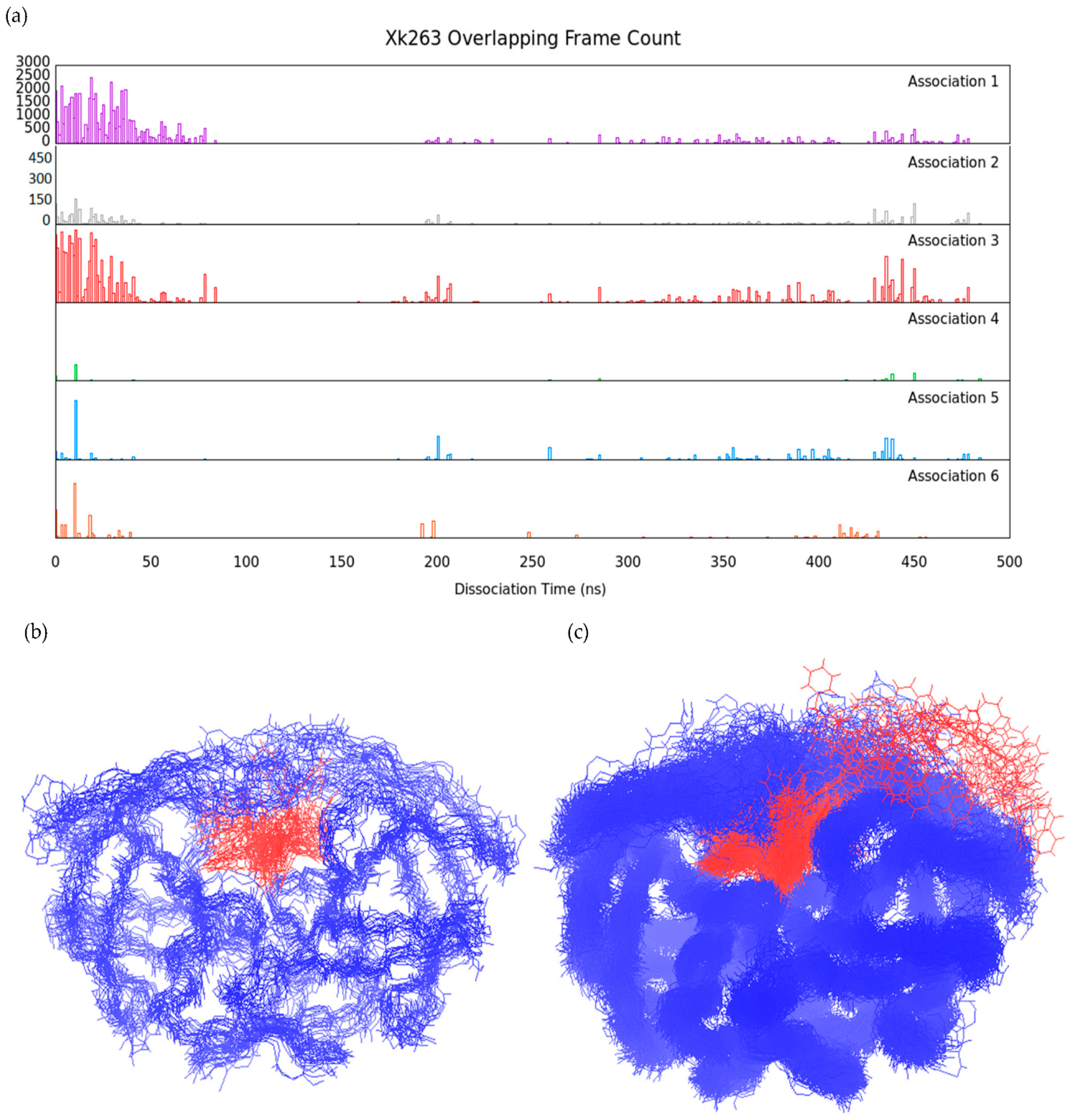
2.7. RMSD-Based Dissociation-Association Trajectory Comparison
3. Results
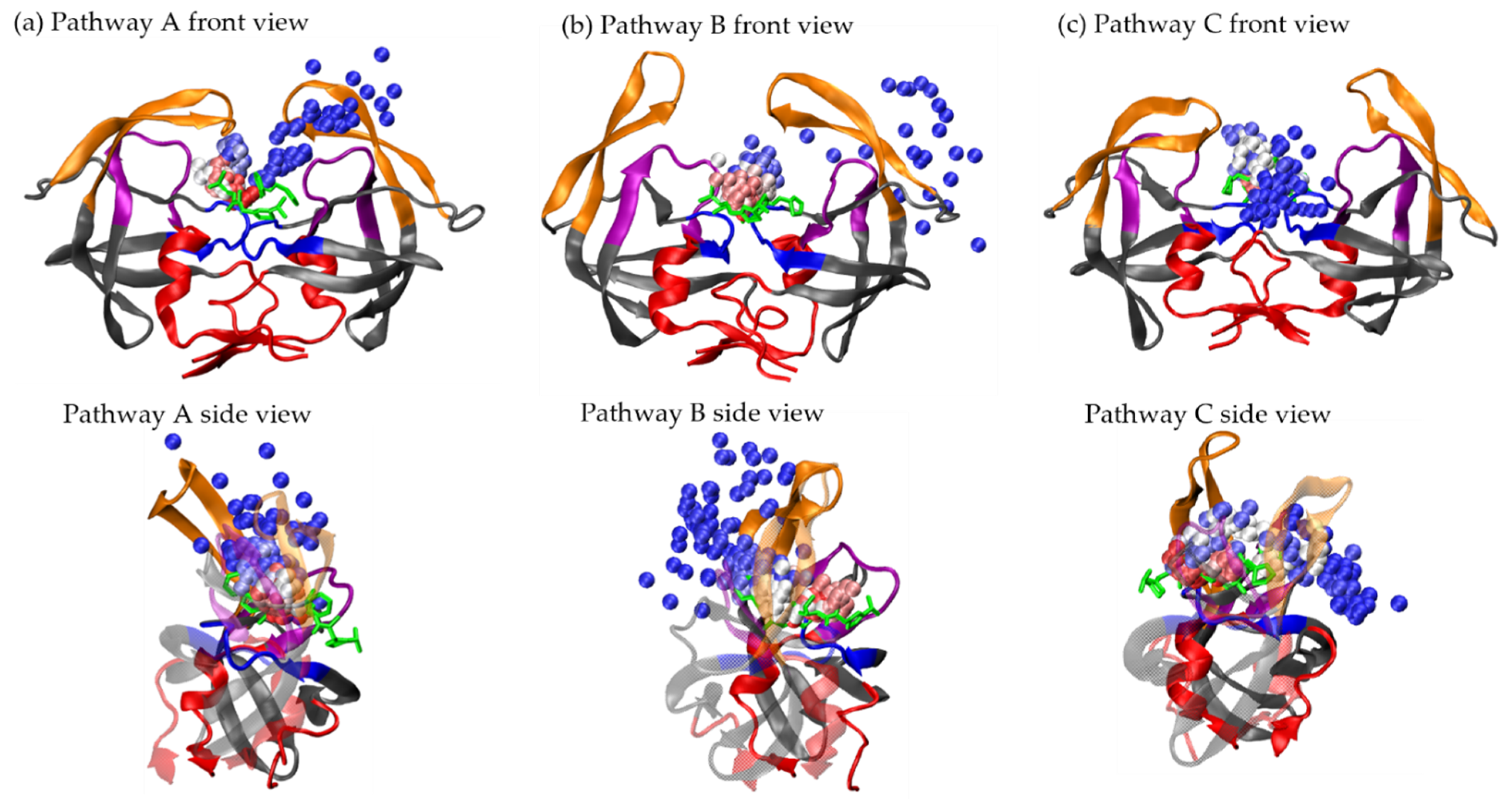
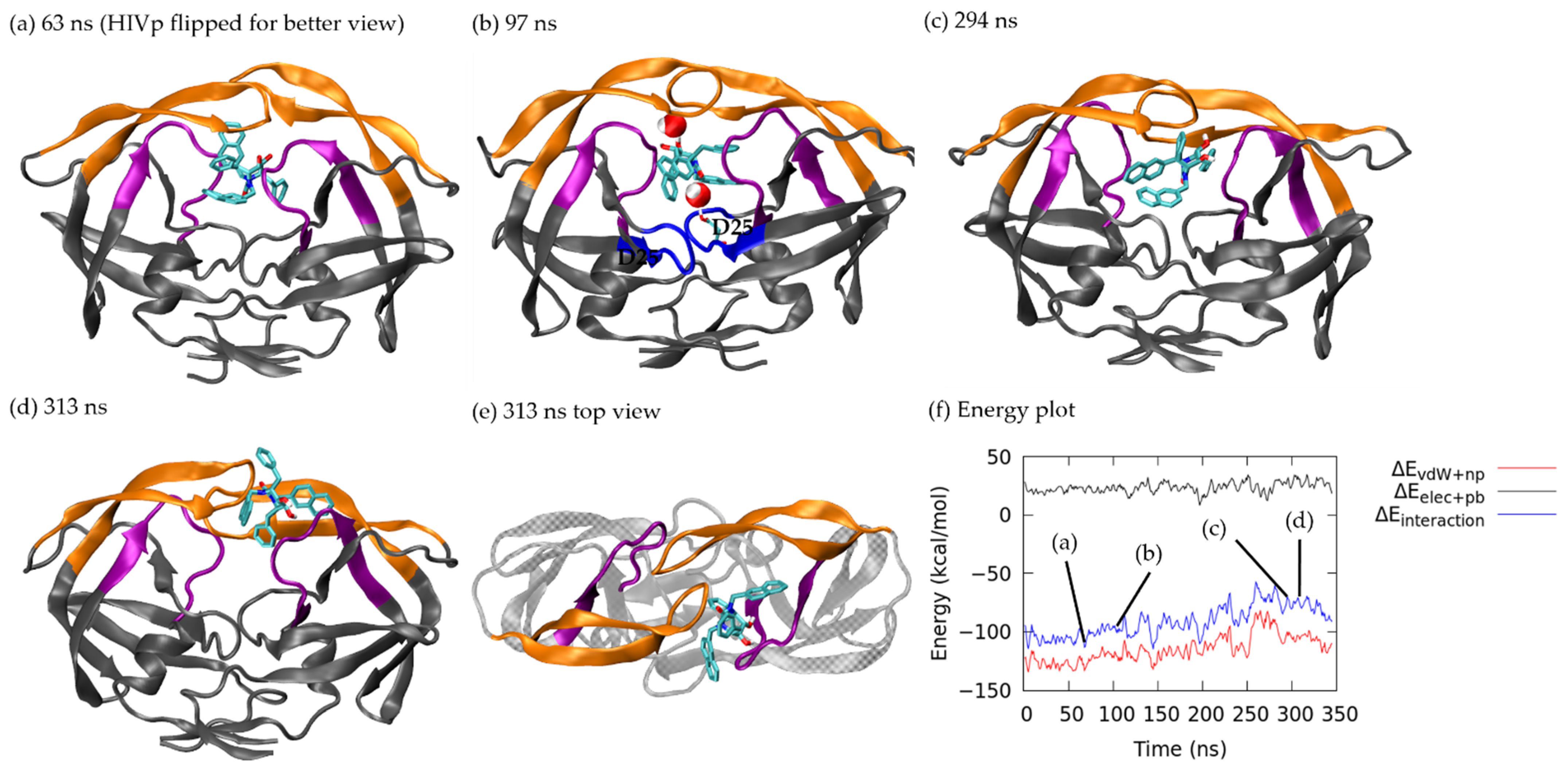
3.1. Ligand Unbinding Pathways
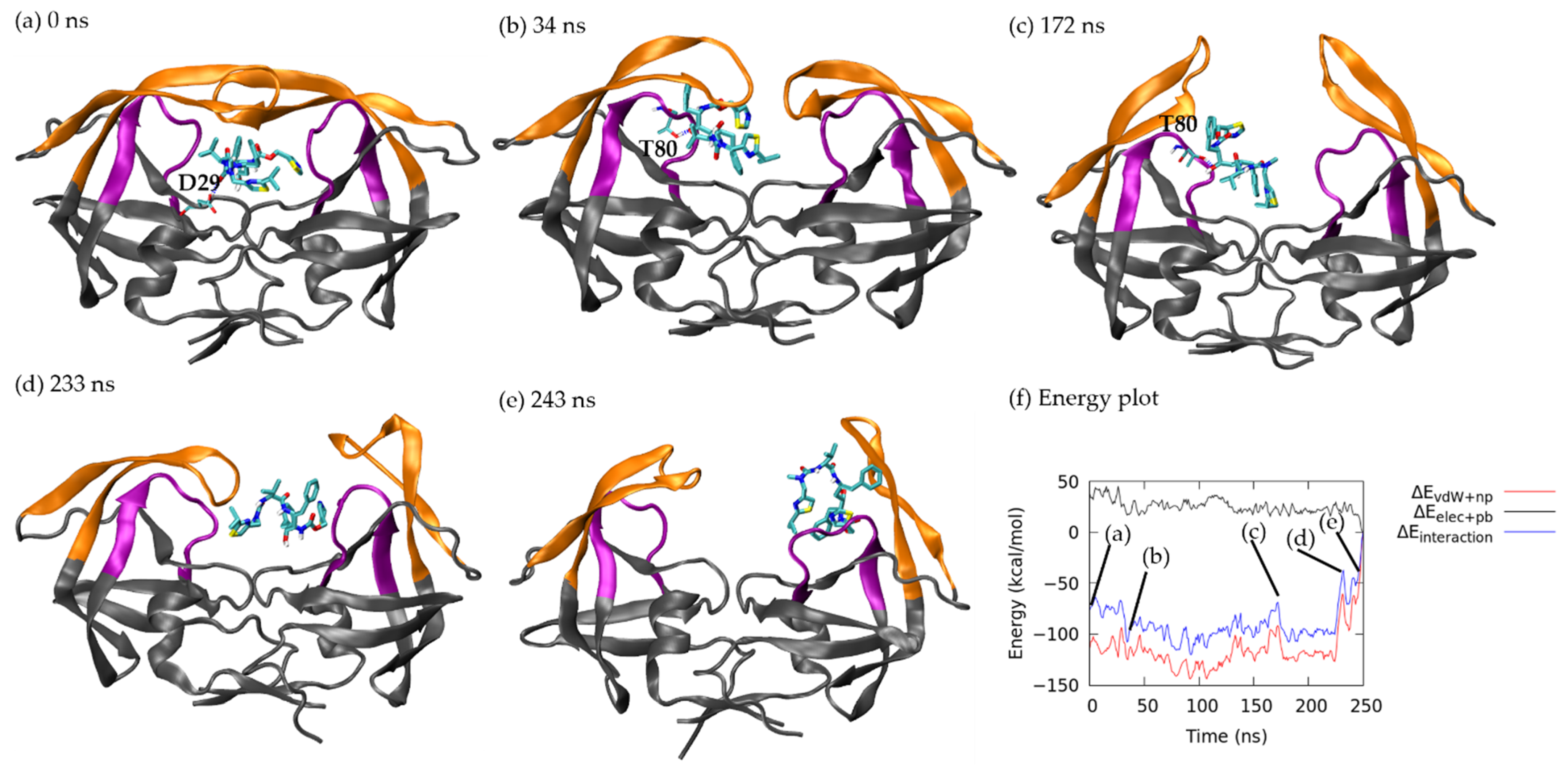
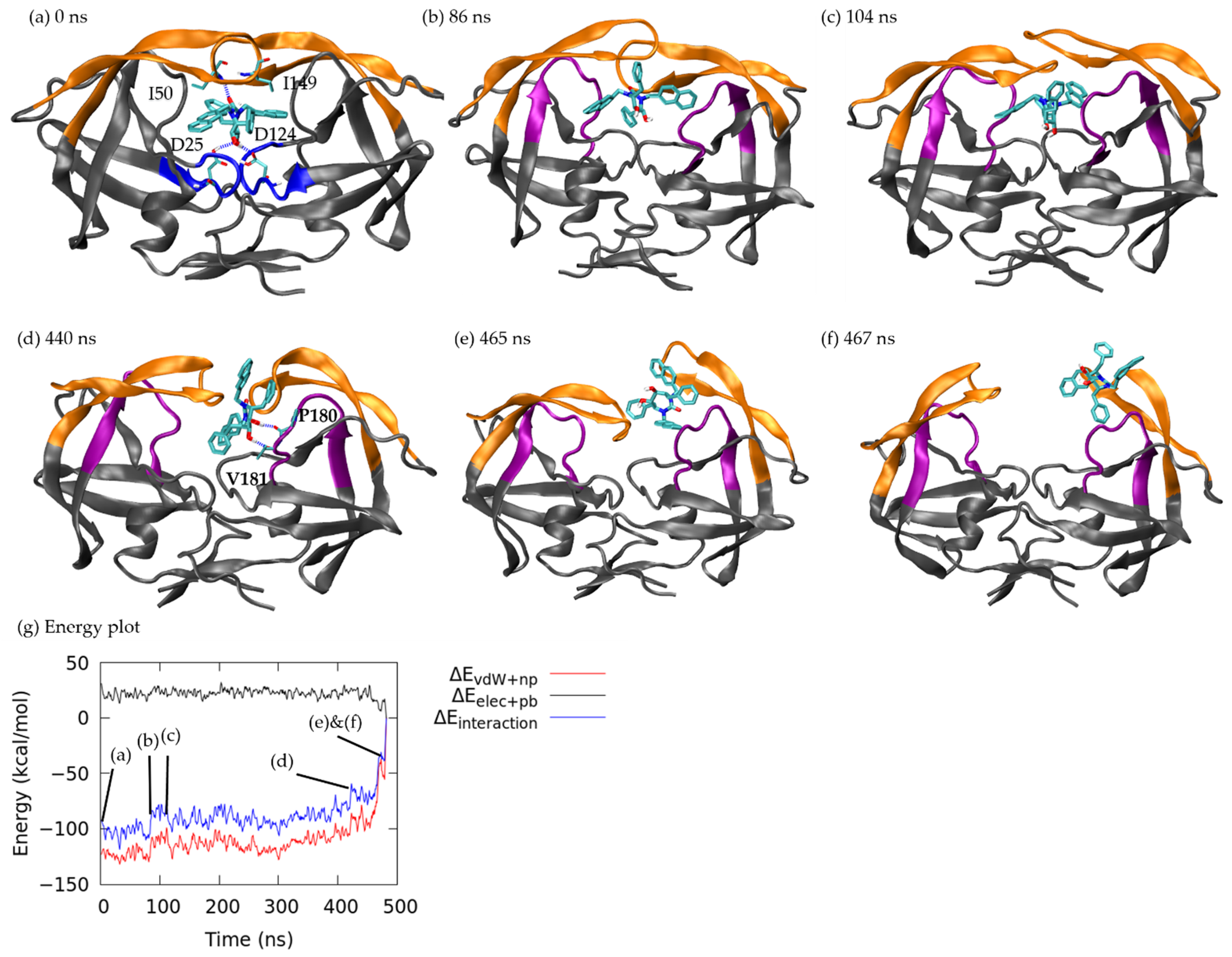
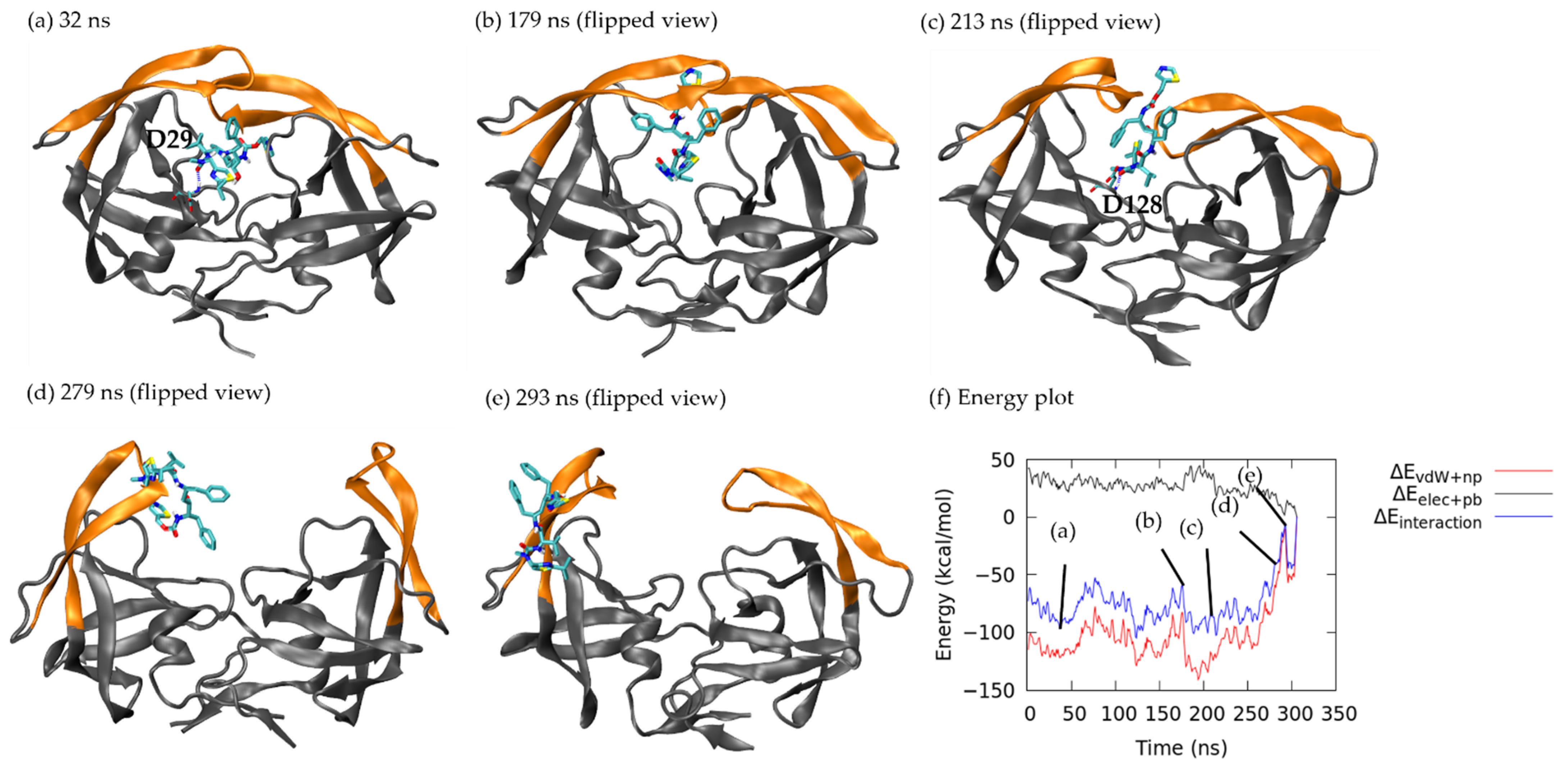
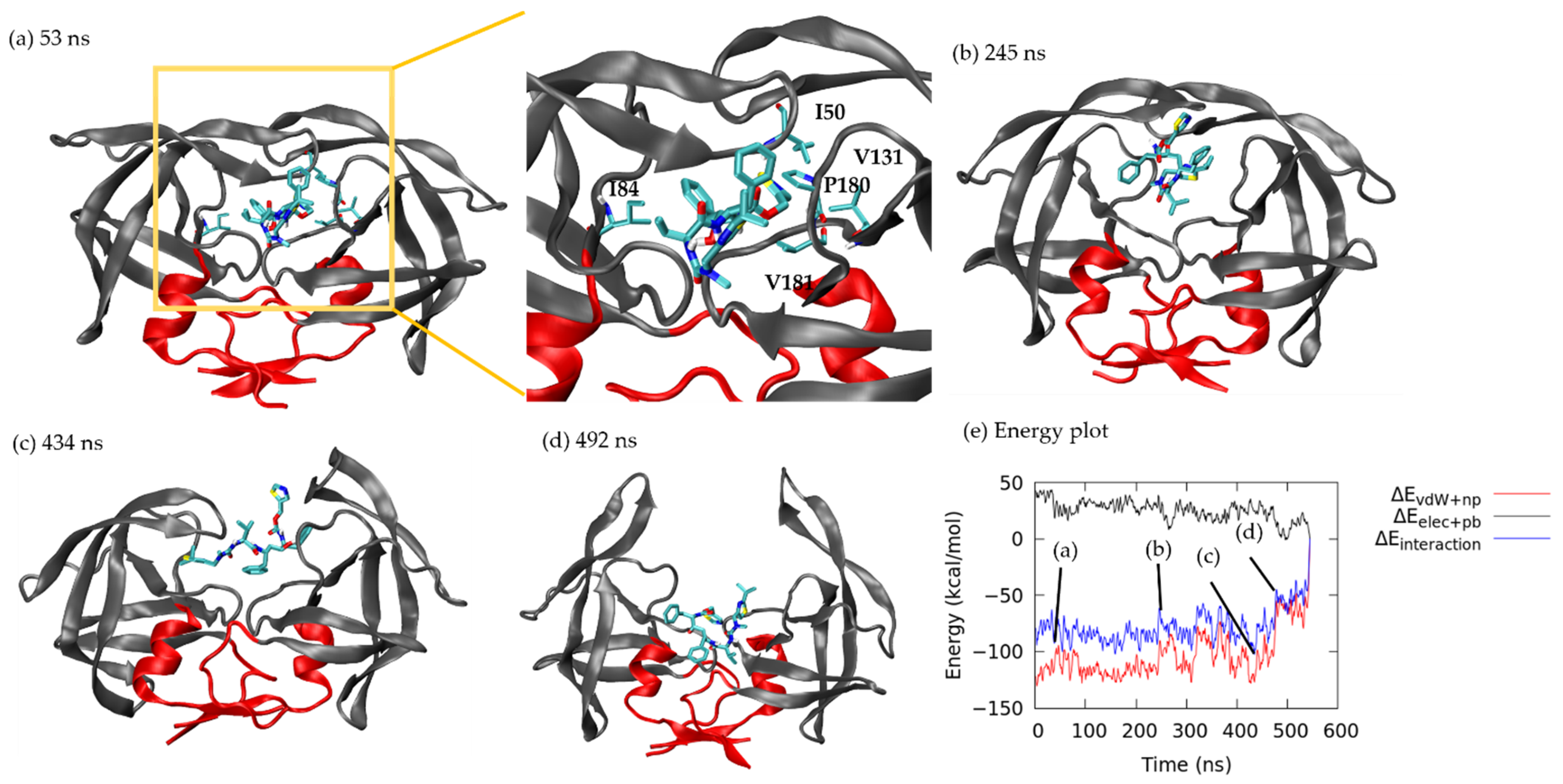
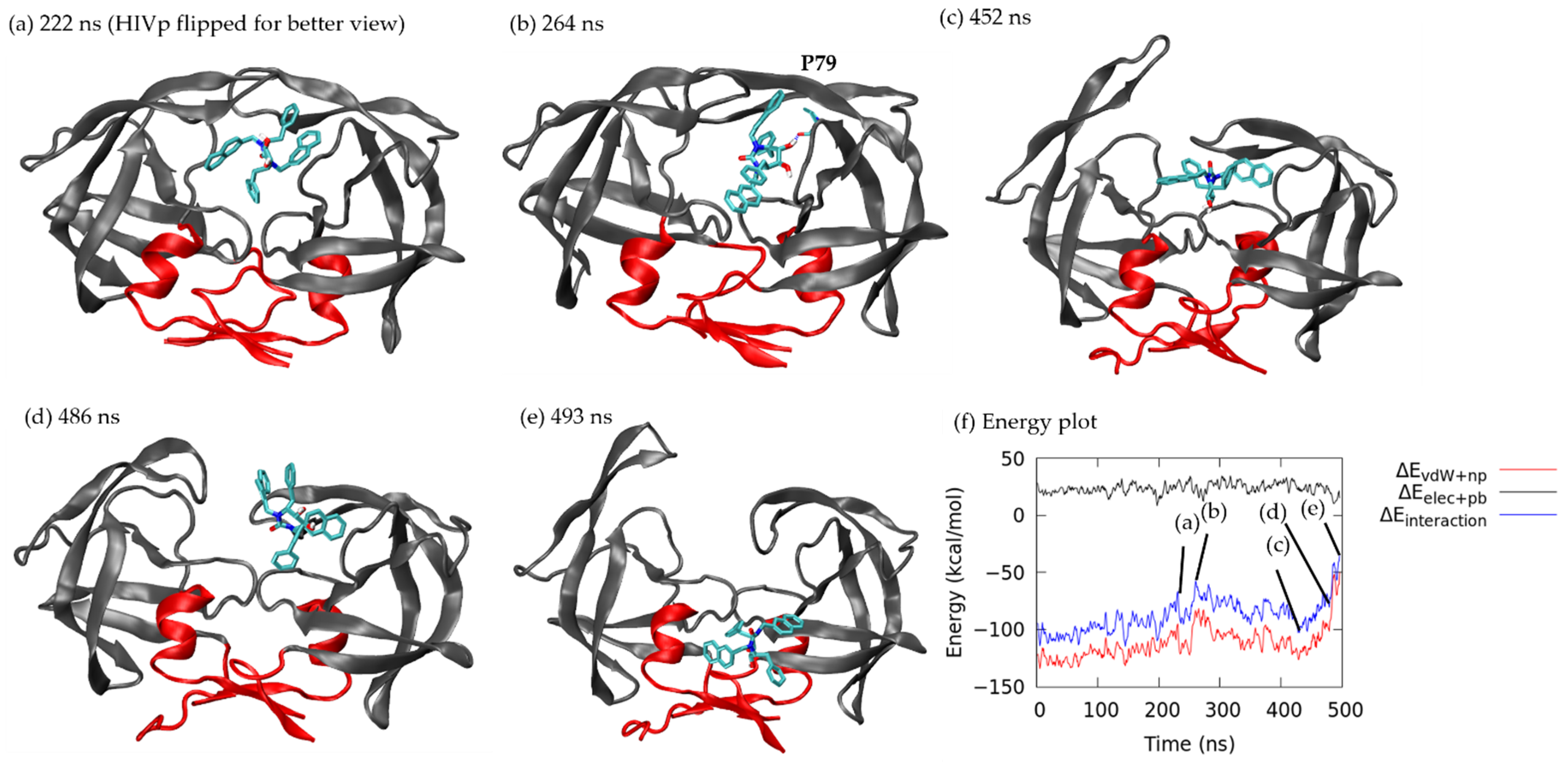
3.1.1. Pathway A: Dissociation between Flap/Loop Region
3.1.2. Pathway B: Dissociation with Surface Diffusion through the Flap Region
3.1.3. Pathway C: Dissociation with Surface Diffusion through Interface Region
3.1.4. Other Pathways
3.2. Association–Dissociation Trajectories Comparison
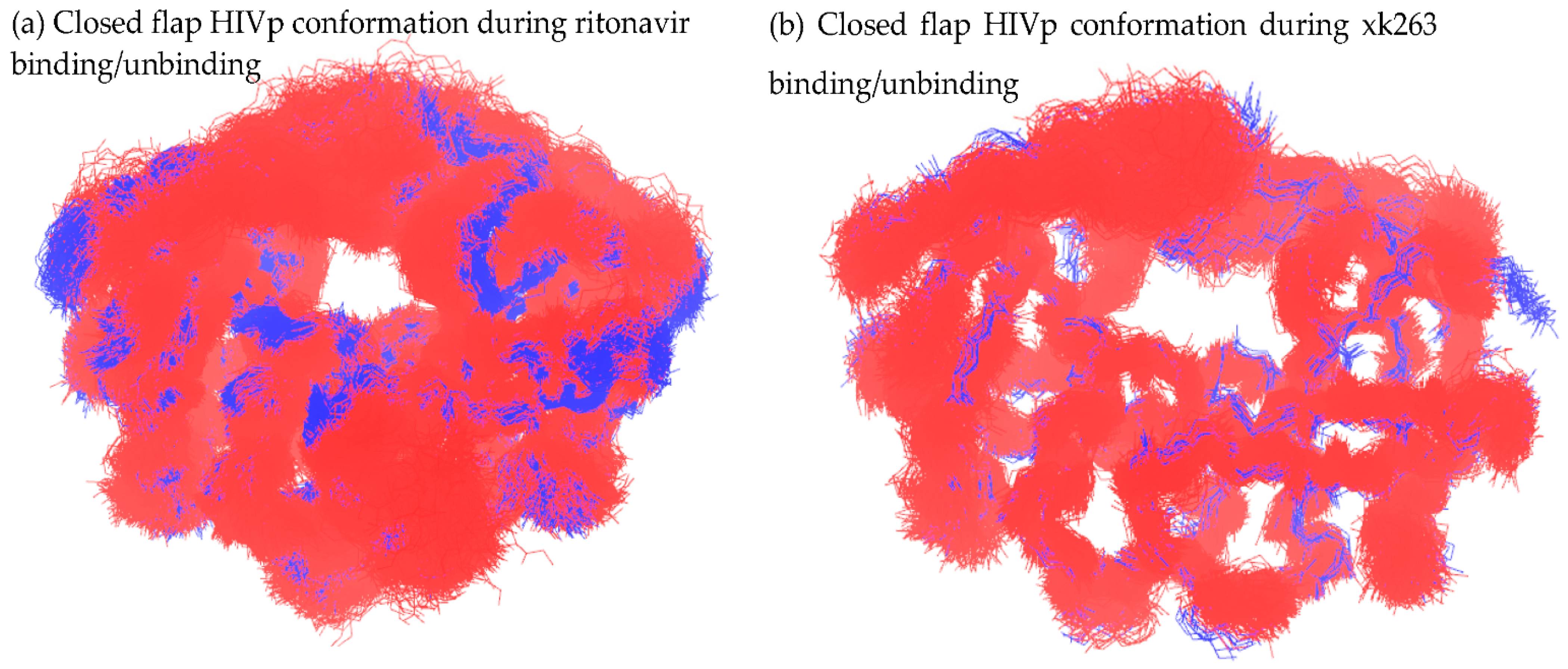
3.3. Mutual Conformations in Association/Dissociation
3.3.1. Closed Flap Conformation
3.3.2. Open Flap Configuration
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kohl, N.E.; Emini, E.A.; Schleif, W.A.; Davis, L.J.; Heimbach, J.C.; Dixon, R.A.F.; Scolnick, E.M.; Sigal, I.S. Active Human Immunodeficiency Virus Protease Is Required for Viral Infectivity. Proc. Nat. Acad. Sci. USA 1988, 85, 4686–4690. [Google Scholar] [CrossRef] [Green Version]
- Tomasselli, A.G.; Heinrikson, R.L. Targeting the HIV-Protease in AIDS Therapy: A Current Clinical Perspective. Biochim. Biophys. Acta-Protein Struct. Mol. Enzymol. 2000, 1477, 189–214. [Google Scholar] [CrossRef]
- Weber, I.T.; Wang, Y.F.; Harrison, R.W. HIV Protease: Historical Perspective and Current Research. Viruses 2021, 13, 839. [Google Scholar] [CrossRef] [PubMed]
- Zuo, X.; Huo, Z.; Kang, D.; Wu, G.; Zhou, Z.; Liu, X.; Zhan, P. Current Insights into Anti-HIV Drug Discovery and Development: A Review of Recent Patent Literature (2014–2017). Expert Opin. Ther. Pat. 2018, 28, 299–316. [Google Scholar] [CrossRef] [PubMed]
- Voshavar, C. Protease Inhibitors for the Treatment of HIV/AIDS: Recent Advances and Future Challenges. Curr. Top. Med. Chem. 2019, 19, 1571–1598. [Google Scholar] [CrossRef]
- Pawar, S.D.; Freas, C.; Weber, I.T.; Harrison, R.W. Analysis of Drug Resistance in HIV Protease. BMC Bioinform. 2018, 19, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, T.W.; Ragland, D.A.; Zeldovich, K.B.; Schiffer, C.A. Characterizing Protein-Ligand Binding Using Atomistic Simulation and Machine Learning: Application to Drug Resistance in HIV-1 Protease. J. Chem. Theory Comput. 2020, 16, 1284–1299. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, A.; Fernandes, R.P.; Quijia, C.; Araujo, V.H.S.; Pereira, J.; Garcia, J.S.; Trevisan, M.G.; Chorilli, M. Pharmacokinetic Parameters of HIV-1 Protease Inhibitors. Chemmedchem 2020, 15, 1018–1029. [Google Scholar] [CrossRef]
- Trylska, J.; Tozzini, V.; Chang, C.-E.A.; McCammon, J.A. HIV-1 Protease Substrate Binding and Product Release Pathways Explored with Coarse-Grained Molecular Dynamics. Biophys. J. 2007, 92, 4179–4187. [Google Scholar] [CrossRef] [Green Version]
- Lexa, K.W.; Carlson, H.A. Binding to the Open Conformation of HIV-1 Protease. Proteins-Struct. Funct. Bioinform. 2011, 79, 2282–2290. [Google Scholar] [CrossRef] [Green Version]
- Brik, A.; Wong, C.H. HIV-1 Protease: Mechanism and Drug Discovery. Org. Biomol. Chem. 2003, 1, 5–14. [Google Scholar] [CrossRef]
- Copeland, R.A. The Drug-Target Residence Time Model: A 10-Year Retrospective. Nat. Rev. Drug Discov. 2016, 15, 87–95. [Google Scholar] [CrossRef]
- Bernetti, M.; Masetti, M.; Rocchia, W.; Cavalli, A. Kinetics of Drug Binding and Residence Time. Annu. Rev. Phys. Chem. 2019, 70, 143–171. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Iuliano, J.N.; Tonge, P.J. Structure-Kinetic Relationships That Control the Residence Time of Drug-Target Complexes: Insights from Molecular Structure and Dynamics. Curr. Opin. Chem. Biol. 2018, 44, 101–109. [Google Scholar] [CrossRef]
- Ribeiro, J.M.L.; Tsai, S.T.; Pramanik, D.; Wang, Y.H.; Tiwary, P. Kinetics of Ligand-Protein Dissociation from All-Atom Simulations: Are We There Yet? Biochemistry 2019, 58, 156–165. [Google Scholar] [CrossRef]
- Lazim, R.; Suh, D.; Choi, S. Advances in Molecular Dynamics Simulations and Enhanced Sampling Methods for the Study of Protein Systems. Int. J. Mol. Sci. 2020, 21, 6339. [Google Scholar] [CrossRef]
- Huang, Y.M.M.; Kang, M.; Chang, C.E.A. Switches of Hydrogen Bonds during Ligand-Protein Association Processes Determine Binding Kinetics. J. Mol. Recognit. 2014, 27, 537–548. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.X.; Liu, W.T.; Li, H.Y.; Wang, W.; Sun, H.B.; Zhang, L.L.; Wu, S.L. Decoding Molecular Mechanism Underlying Binding of Drugs to HIV-1 Protease with Molecular Dynamics Simulations and MM-GBSA Calculations. Sar Qsar Environ. Res. 2021, 32, 889–915. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Wang, J.A.; Xu, Z.J.; Cai, T.T.; Zhu, W.L. Accurate Prediction of Relative Binding Affinities of a Series of HIV-1 Protease Inhibitors Using Semi-Empirical Quantum Mechanical Charge. J. Comput. Chem. 2020, 41, 1773–1780. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Senapati, S. Mechanism of Inhibition of Drug-Resistant HIV-1 Protease Clinical Isolates by TMC310911: A Molecular Dynamics Study. J. Mol. Struct. 2019, 1198, 126893. [Google Scholar] [CrossRef]
- Li, D.C.; Ji, B.H.; Hwang, K.C.; Huang, Y.G. Strength of Hydrogen Bond Network Takes Crucial Roles in the Dissociation Process of Inhibitors from the HIV-1 Protease Binding Pocket. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Huang, S.H.; Zhang, D.; Mei, H.; Kevin, M.; Qu, S.J.; Pan, X.C.; Lu, L.C. SMD-Based Interaction-Energy Fingerprints Can Predict Accurately the Dissociation Rate Constants of HIV-1 Protease Inhibitors. J. Chem. Inf. Model. 2019, 59, 159–169. [Google Scholar] [CrossRef]
- Ngo, S.T.; Nguyen, M.T. Determination of the Absolute Binding Free Energies of HIV-1 Protease Inhibitors Using Non-Equilibrium Molecular Dynamics Simulations. Chem. Phys. Lett. 2017, 676, 12–17. [Google Scholar] [CrossRef]
- Bhattarai, A.; Miao, Y.L. Gaussian Accelerated Molecular Dynamics for Elucidation of Drug Pathways. Expert Opin. Drug Discov. 2018, 13, 1055–1065. [Google Scholar] [CrossRef]
- Furfine, E.S.; D’Souza, E.; Ingold, K.J.; Leban, J.J.; Spector, T.; Porter, D.J. Two-Step Binding Mechanism for HIV Protease Inhibitors. 1992, 31, 7886–7891. Biochemistry 1992, 31, 7886–7891. [Google Scholar] [CrossRef]
- Katoh, E.; Louis, J.; Yamazaki, T.; Gronenborn, A.; Torchia, D.; Ishima, R. A Solution NMR Study of the Binding Kinetics and the Internal Dynamics of an HIV-1 Protease-Substrate Complex. Protein Sci. 2003, 12, 1376–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.M.M.; Raymundo, M.A.V.; Chen, W.; Chang, C.E.A. Mechanism of the Association Pathways for a Pair of Fast and Slow Binding Ligands of HIV-1 Protease. Biochemistry 2017, 56, 1311–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qu, S.J.; Huang, S.H.; Pang, X.C.; Yang, L.; Mei, H. Constructing Interconsistent, Reasonable, and Predictive Models for Both the Kinetic and Thermodynamic Properties of HIV-1 Protease Inhibitors. J. Chem. Inf. Model. 2016, 56, 2061–2068. [Google Scholar] [CrossRef] [PubMed]
- Shuman, C.F.; Hamalainen, M.D.; Danielson, U.H. Kinetic and Thermodynamic Characterization of HIV-1 Protease Inhibitors. J. Mol. Recognit. 2004, 17, 106–119. [Google Scholar] [CrossRef]
- Miao, Y.L.; Huang, Y.M.; Walker, R.; McCammon, J.A.; Chang, C.E. Ligand Binding Pathways and Conformational Transitions of the HIV Protease. Abstr. Pap. Am. Chem.Soc. 2018, 57, 1533–1541. [Google Scholar] [CrossRef] [Green Version]
- Acevedo, O.; Ambrose, Z.; Flaherty, P.T.; Aamer, H.; Jain, P.; Sambasivarao, S.V. Identification of HIV Inhibitors Guided by Free Energy Perturbation Calculations. Curr. Pharm. Des. 2012, 18, 1199–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karnati, K.R.; Wang, Y.X. Structural and Binding Insights into HIV-1 Protease and P2-Ligand Interactions through Molecular Dynamics Simulations, Binding Free Energy and Principal Component Analysis. J. Mol. Gr. Model. 2019, 92, 112–122. [Google Scholar] [CrossRef]
- Kempf, D.J.; Marsh, K.C.; Denissen, J.F.; McDonald, E.; Vasavanonda, S.; Flentge, C.A.; Green, B.E.; Fino, L.; Park, C.H.; Kong, X.P.; et al. ABT-538 Is a Potent Inhibitor of Human-Immunodeficiency-Virus Protease and Has High Oral Bioavailability in Humans. Proc. Nat. Acad. Sci. USA 1995, 92, 2484–2488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, P.Y.S.; Jadhav, P.K.; Eyermann, C.J.; Hodge, C.N.; Ru, Y.; Bacheler, L.T.; Meek, J.L.; Otto, M.J.; Rayner, M.M.; Wong, Y.N.; et al. Rational Design of Potent, Bioavailable, Nonpeptide Cyclic Ureas as Hiv Protease Inhibitors. Science 1994, 263, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Z.; Yang, M.Y.; Hu, G.D.; Shi, S.H.; Yi, C.H.; Zhang, Q.G. Insights into the Functional Role of Protonation States in the HIV-1 Protease-BEA369 Complex: Molecular Dynamics Simulations and Free Energy Calculations. J. Mol. Model. 2009, 15, 1245–1252. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. Amber 18; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Gilson, M.K.; Gilson, H.S.R.; Potter, M.J. Fast Assignment of Accurate Partial Atomic Charges: An Electronegativity Equalization Method That Accounts for Alternate Resonance Forms. J. Chem. Inf. Comput. Sci. 2003, 43, 1982–1997. [Google Scholar] [CrossRef]
- Kholmurodov, K.; Smith, W.; Yasuoka, K.; Darden, T.; Ebisuzaki, T. A Smooth-Particle Mesh Ewald Method for DL_POLY Molecular Dynamics Simulation Package on the Fujitsu VPP700. J. Comput. Chem. 2000, 21, 1187–1191. [Google Scholar] [CrossRef]
- Jean-Paul, R.; Giovanni, C.; Herman, J.C.B. Numerical integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar]
- Hamelberg, D.; Mongan, J.; McCammon, J.A. Accelerated Molecular Dynamics: A Promising and Efficient Simulation Method for Biomolecules. J. Chem. Phys. 2004, 120, 11919–11929. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.L.; Oezguen, N.; Urvil, P.; Ferguson, C.; Dann, S.M.; Savidge, T.C. Regulation of Protein-Ligand Binding Affinity by Hydrogen Bond Pairing. Sci. Adv. 2016, 2, e1501240. [Google Scholar] [CrossRef] [Green Version]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Huang, X.; Britto, M.D.; Kear-Scott, J.L.; Boone, C.D.; Rocca, J.R.; Simmerling, C.; McKenna, R.; Bieri, M.; Gooley, P.R.; Dunn, B.M.; et al. The Role of Select Subtype Polymorphisms on HIV-1 Protease Conformational Sampling and Dynamics. J. Biol. Chem. 2014, 289, 17203–17214. [Google Scholar] [CrossRef] [Green Version]
- Karthik, S.; Senapati, S. Dynamic Flaps in HIV-1 Protease Adopt Unique Ordering at Different Stages in the Catalytic Cycle. Proteins 2011, 79, 1830–1840. [Google Scholar] [CrossRef]
- Ali, A.; Bandaranayake, R.M.; Cai, Y.F.; King, N.M.; Kolli, M.; Mittal, S.; Murzycki, J.F.; Nalam, M.N.L.; Nalivaika, E.A.; Ozen, A.; et al. Molecular Basis for Drug Resistance in HIV-1 Protease. Viruses 2010, 2, 2509–2535. [Google Scholar] [CrossRef]
- Yilmaz, N.K.; Swanstrom, R.; Schiffer, C.A. Improving Viral Protease Inhibitors to Counter Drug Resistance. Trends Microbiol. 2016, 24, 547–557. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.Y.; Roberts, C.C.; Chang, C.E.A. Understanding Ligand-Receptor Non-Covalent Binding Kinetics Using Molecular Modeling. Front. Biosci.-Landmark 2017, 22, 960–981. [Google Scholar] [CrossRef] [Green Version]
- Li, D.C.; Ji, B.H.; Liu, M.S.; Hwang, K.C.; Huang, Y.G. Coarse-Grained Molecular Dynamics of Inhibitors Binding into HIV-1 Protease. In Proceedings of the 11th International Congress of the IUPESM/World Congress on Medical Physics and Biomedical Engineering, Munich, Germany, 7–12 September 2009; pp. 623–625. [Google Scholar]
- Toth, G.; Borics, A. Flap Opening Mechanism of HIV-1 Protease. J. Mol. Gr. Model. 2006, 24, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Sadiq, S.K.; De Fabritiis, G. Explicit Solvent Dynamics and Energetics of HIV-1 Protease Flap Opening and Closing. Proteins-Struct. Funct. Bioinform. 2010, 78, 2873–2885. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ritonavir | xk263 | |
|---|---|---|
| Total dissociations | 20 | 15 |
| Pathway A: Unbind between flap/loop | 11 | 8 |
| Pathway B: Diffusion on flap | 4 | Not observed |
| Pathway C: Diffusion on interface | 3 | 3 |
| Others | 2 | 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, J.; Raymundo, M.A.V.; Chang, C.-E.A. Ritonavir and xk263 Binding-Unbinding with HIV-1 Protease: Pathways, Energy and Comparison. Life 2022, 12, 116. https://doi.org/10.3390/life12010116
Sun J, Raymundo MAV, Chang C-EA. Ritonavir and xk263 Binding-Unbinding with HIV-1 Protease: Pathways, Energy and Comparison. Life. 2022; 12(1):116. https://doi.org/10.3390/life12010116
Chicago/Turabian StyleSun, Jianan, Mark Anthony V. Raymundo, and Chia-En A. Chang. 2022. "Ritonavir and xk263 Binding-Unbinding with HIV-1 Protease: Pathways, Energy and Comparison" Life 12, no. 1: 116. https://doi.org/10.3390/life12010116