
Secreted Effectors Modulating Immune Responses to Toxoplasma gondii

 ,
,
Abstract
:1. Introduction
2. Defending Home Base: Effectors on the Parasitophorous Vacuole Membrane
2.1. ROP18 Virulence Complex: An Inhibitor of IFNγ-Induced IRG Loading (ROP18/ROP17/ROP5/GRA7)
2.2. GRA60, GRA12, ROP54: IRG/GBP Modulators
2.3. GRA15: An Activator of NF-κB Signaling
2.4. GRA15: A Mediator of IFNγ-Induced Cell-Autonomous Immune Reaction
2.5. ROP38: An Inhibitor of NF-κB Signaling
2.6. GRA6: An Activator of NFAT4 Signaling and CCL2 Secretion
2.7. GRA25: An Additional CCL2 Inducer
3. Taking It on the Road: Effectors in the Endoplasmic Reticulum
ROP18 and the ER
4. Taking It on the Road: Effectors in the Cytoplasm
4.1. GRA18: ß-Catenin Induced M2 Cytokines Secretion
4.2. MAG1: An Effector, Matrix, and Cyst Wall Protein
5. Attacking Enemy Headquarters: Effectors Found in the Host Nucleus
5.1. ROP16: An Inducer of M2 Polarization
5.2. GRA28: An Inducer of CCL22 Chemokine Secretion
5.3. GRA24: A Modulator of p38 α Mitogen-Activated Protein
5.4. GRA16: A Modulator of HAUSP and PP2A
5.5. HCE1/TEEGR: A Modulation of E2F Activation
5.6. TgIST: A Suppressor of Interferon Signaling
5.7. TgNSM: A Suppressor of IFNγ-Induced Necroptosis
6. Translocation of GRA Effectors
7. Effector Translocation in T. gondii Tissue Cysts
8. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| EZH2 | enhancer of zeste homolog 2 |
| GBP | p65 guanylate binding protein |
| IRG | immunity-related GTPase |
| IVN | intravacuolar network |
| NF-κB | nuclear factor kappa B |
| NLS | nuclear localization signal |
| PVM | parasitophorous vacuolar membrane |
| QTL | quantitative trait locus |
| STAT | signal transducer and activator of transcription |
| TRAF | TNF receptor-associated factor |
References
- Derouin, F.; Garin, Y.J. Toxoplasma gondii: Blood and tissue kinetics during acute and chronic infections in mice. Exp. Parasitol. 1991, 73, 460–468. [Google Scholar] [CrossRef]
- Di Cristina, M.; Marocco, D.; Galizi, R.; Proietti, C.; Spaccapelo, R.; Crisanti, A. Temporal and spatial distribution of Toxoplasma gondii differentiation into Bradyzoites and tissue cyst formation in vivo. Infect. Immun. 2008, 76, 3491–3501. [Google Scholar] [CrossRef] [Green Version]
- Bohne, W.; Heesemann, J.; Gross, U. Induction of bradyzoite-specific Toxoplasma gondii antigens in gamma interferon-treated mouse macrophages. Infect. Immun. 1993, 61, 1141–1145. [Google Scholar] [CrossRef] [Green Version]
- Bohne, W.; Heesemann, J.; Gross, U. Reduced replication of Toxoplasma gondii is necessary for induction of bradyzoite-specific antigens: A possible role for nitric oxide in triggering stage conversion. Infect. Immun. 1994, 62, 1761–1767. [Google Scholar] [CrossRef] [Green Version]
- Weiss, L.M.; Laplace, D.; Takvorian, P.M.; Tanowitz, H.B.; Cali, A.; Wittner, M. A cell culture system for study of the development of Toxoplasma gondii bradyzoites. J. Eukaryot. Microbiol. 1995, 42, 150–157. [Google Scholar] [CrossRef]
- Elsheikha, H.M.; Marra, C.M.; Zhu, X.-Q. Epidemiology, Pathophysiology, Diagnosis, and Management of Cerebral Toxoplasmosis. Clin. Microbiol. Rev. 2021, 34. [Google Scholar] [CrossRef]
- Mordue, D.G.; Desai, N.; Dustin, M.; Sibley, L.D. Invasion by Toxoplasma gondii establishes a moving junction that selectively excludes host cell plasma membrane proteins on the basis of their membrane anchoring. J. Exp. Med. 1999, 190, 1783–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sibley, L.D. Intracellular parasite invasion strategies. Science 2004, 304, 248–253. [Google Scholar] [CrossRef]
- Carruthers, V.B.; Sibley, L.D. Sequential protein secretion from three distinct organelles of Toxoplasma gondii accompanies invasion of human fibroblasts. Eur. J. Cell Biol. 1997, 73, 114–123. [Google Scholar] [PubMed]
- Lebrun, M.; Michelin, A.; El Hajj, H.; Poncet, J.; Bradley, P.J.; Vial, H.; Dubremetz, J.F. The rhoptry neck protein RON4 re-localizes at the moving junction during Toxoplasma gondii invasion. Cell. Microbiol. 2005, 7, 1823–1833. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.L.; Mital, J.; Ward, G.E.; Bradley, P.; Boothroyd, J.C. Identification of the moving junction complex of Toxoplasma gondii: A collaboration between distinct secretory organelles. PLoS Pathog. 2005, 1, e17. [Google Scholar] [CrossRef] [PubMed]
- Morisaki, J.H.; Heuser, J.E.; Sibley, L.D. Invasion of Toxoplasma gondii occurs by active penetration of the host cell. J. Cell Sci. 1995, 108, 2457–2464. [Google Scholar] [CrossRef] [PubMed]
- Gazzinelli, R.T.; Mendonça-Neto, R.; Lilue, J.; Howard, J.; Sher, A. Innate Resistance against Toxoplasma gondii: An Evolutionary Tale of Mice, Cats, and Men. Cell Host Microbe 2014, 15, 132–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarovinsky, F.; Zhang, D.; Andersen, J.F.; Bannenberg, G.L.; Serhan, C.N.; Hayden, M.S.; Hieny, S.; Sutterwala, F.S.; Flavell, R.A.; Ghosh, S.; et al. TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science 2005, 308, 1626–1629. [Google Scholar] [CrossRef] [Green Version]
- Raetz, M.; Kibardin, A.; Sturge, C.R.; Pifer, R.; Li, H.; Burstein, E.; Ozato, K.; Larin, S.; Yarovinsky, F. Cooperation of TLR12 and TLR11 in the IRF8-dependent IL-12 response to Toxoplasma gondii profilin. J. Immunol. 2013, 191, 4818–4827. [Google Scholar] [CrossRef] [Green Version]
- Taylor, G.A.; Feng, C.G.; Sher, A. Control of IFN-gamma-mediated host resistance to intracellular pathogens by immunity-related GTPases (p47 GTPases). Microbes Infect. 2007, 9, 1644–1651. [Google Scholar] [CrossRef]
- Bekpen, C.; Hunn, J.P.; Rohde, C.; Parvanova, I.; Guethlein, L.; Dunn, D.M.; Glowalla, E.; Leptin, M.; Howard, J.C. The interferon-inducible p47 (IRG) GTPases in vertebrates: Loss of the cell autonomous resistance mechanism in the human lineage. Genome Biol. 2005, 6, R92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butcher, B.A.; Greene, R.I.; Henry, S.C.; Annecharico, K.L.; Weinberg, J.B.; Denkers, E.Y.; Sher, A.; Taylor, G.A. p47 GTPases regulate Toxoplasma gondii survival in activated macrophages. Infect. Immun. 2005, 73, 3278–3286. [Google Scholar] [CrossRef] [Green Version]
- Andrade, W.A.; Souza, M.D.C.; Ramos-Martinez, E.; Nagpal, K.; Dutra, M.S.; Melo, M.B.; Bartholomeu, D.C.; Ghosh, S.; Golenbock, D.T.; Gazzinelli, R.T. Combined Action of Nucleic Acid-Sensing Toll-like Receptors and TLR11/TLR12 Heterodimers Imparts Resistance to Toxoplasma gondii in Mice. Cell Host Microbe 2013, 13, 42–53. [Google Scholar] [CrossRef] [Green Version]
- Gov, L.; Schneider, C.A.; Lima, T.S.; Pandori, W.; Lodoen, M.B. NLRP3 and Potassium Efflux Drive Rapid IL-1β Release from Primary Human Monocytes during Toxoplasma gondii Infection. J. Immunol. 2017, 199, 2855–2864. [Google Scholar] [CrossRef] [Green Version]
- Gov, L.; Karimzadeh, A.; Ueno, N.; Lodoen, M.B. Human innate immunity to Toxoplasma gondii is mediated by host caspase-1 and ASC and parasite GRA15. mBio 2013, 4, e00255-13. [Google Scholar] [CrossRef] [Green Version]
- Nagineni, C.N.; Pardhasaradhi, K.; Martins, M.C.; Detrick, B.; Hooks, J.J. Mechanisms of interferon-induced inhibition of Toxoplasma gondii replication in human retinal pigment epithelial cells. Infect. Immun. 1996, 64, 4188–4196. [Google Scholar] [CrossRef] [Green Version]
- Pfefferkorn, E.R. Interferon gamma blocks the growth of Toxoplasma gondii in human fibroblasts by inducing the host cells to degrade tryptophan. Proc. Natl. Acad. Sci. USA 1984, 81, 908–912. [Google Scholar] [CrossRef] [Green Version]
- Andrade, R.M.; Wessendarp, M.; Gubbels, M.-J.; Striepen, B.; Subauste, C.S. CD40 induces macrophage anti–Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. J. Clin. Investig. 2006, 116, 2366–2377. [Google Scholar] [CrossRef] [Green Version]
- Howe, D.K.; Sibley, L.D. Toxoplasma gondii comprises three clonal lineages: Correlation of parasite genotype with human disease. J. Infect. Dis. 1995, 172, 1561–1566. [Google Scholar] [CrossRef] [PubMed]
- Sidik, S.M.; Huet, D.; Ganesan, S.M.; Huynh, M.-H.; Wang, T.; Nasamu, A.S.; Thiru, P.; Saeij, J.P.J.; Carruthers, V.B.; Niles, J.C.; et al. A Genome-wide CRISPR Screen in Toxoplasma Identifies Essential Apicomplexan Genes. Cell 2016, 166, 1423–1435.e12. [Google Scholar] [CrossRef] [Green Version]
- Behnke, M.S.; Khan, A.; Wootton, J.C.; Dubey, J.P.; Tang, K.; Sibley, L.D. Virulence differences in Toxoplasma mediated by amplification of a family of polymorphic pseudokinases. Proc. Natl. Acad. Sci. USA 2011, 108, 9631–9636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reese, M.L.; Zeiner, G.M.; Saeij, J.P.J.; Boothroyd, J.C.; Boyle, J.P. Polymorphic family of injected pseudokinases is paramount in Toxoplasma virulence. Proc. Natl. Acad. Sci. USA 2011, 108, 9625–9630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etheridge, R.D.; Alaganan, A.; Tang, K.; Lou, H.J.; Turk, B.E.; Sibley, L.D. The Toxoplasma pseudokinase ROP5 forms complexes with ROP18 and ROP17 kinases that synergize to control acute virulence in mice. Cell Host Microbe 2014, 15, 537–550. [Google Scholar] [CrossRef] [Green Version]
- Fentress, S.J.; Behnke, M.S.; Dunay, I.R.; Mashayekhi, M.; Rommereim, L.M.; Fox, B.A.; Bzik, D.J.; Taylor, G.A.; Turk, B.E.; Lichti, C.F.; et al. Phosphorylation of immunity-related GTPases by a Toxoplasma gondii-secreted kinase promotes macrophage survival and virulence. Cell Host Microbe 2010, 8, 484–495. [Google Scholar] [CrossRef] [Green Version]
- Alaganan, A.; Fentress, S.J.; Tang, K.; Wang, Q.; Sibley, L.D. Toxoplasma GRA7 effector increases turnover of immunity-related GTPases and contributes to acute virulence in the mouse. Proc. Natl. Acad. Sci. USA 2014, 111, 1126–1131. [Google Scholar] [CrossRef] [Green Version]
- Nyonda, M.A.; Hammoudi, P.-M.; Ye, S.; Maire, J.; Marq, J.-B.; Yamamoto, M.; Soldati-Favre, D. Toxoplasma gondii GRA60 is an effector protein that modulates host cell autonomous immunity and contributes to virulence. Cell. Microbiol. 2021, 23, e13278. [Google Scholar] [CrossRef]
- Kim, E.W.; Nadipuram, S.M.; Tetlow, A.L.; Barshop, W.D.; Liu, P.T.; Wohlschlegel, J.A.; Bradley, P.J. The Rhoptry Pseudokinase ROP54 Modulates Toxoplasma gondii Virulence and Host GBP2 Loading. mSphere 2016, 1, e00045-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, B.A.; Guevara, R.B.; Rommereim, L.M.; Falla, A.; Bellini, V.; Pètre, G.; Rak, C.; Cantillana, V.; Dubremetz, J.-F.; Cesbron-Delauw, M.-F.; et al. Toxoplasma gondii Parasitophorous Vacuole Membrane-Associated Dense Granule Proteins Orchestrate Chronic Infection and GRA12 Underpins Resistance to Host Gamma Interferon. mBio 2019, 10, e00589-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosowski, E.E.; Lu, D.; Julien, L.; Rodda, L.; Gaiser, R.A.; Jensen, K.D.C.; Saeij, J.P.J. Strain-specific activation of the NF-κB pathway by GRA15, a novel Toxoplasma gondii dense granule protein. J. Exp. Med. 2011, 208, 195–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Li, S.; Zhao, Y.; Zhang, B.; Li, Y.; Liu, S.; Du, H.; Cao, L.; Ou, M.; Ye, X.; et al. The GRA15 protein from Toxoplasma gondii enhances host defense responses by activating the interferon stimulator STING. J. Biol. Chem. 2019, 294, 16494–16508. [Google Scholar] [CrossRef] [Green Version]
- Sangaré, L.O.; Yang, N.; Konstantinou, E.K.; Lu, D.; Mukhopadhyay, D.; Young, L.H.; Saeij, J.P.J. Toxoplasma GRA15 Activates the NF-κB Pathway through Interactions with TNF Receptor-Associated Factors. mBio 2019, 10, e00808-19. [Google Scholar] [CrossRef] [Green Version]
- Mukhopadhyay, D.; Sangaré, L.O.; Braun, L.; Hakimi, M.-A.; Saeij, J.P. Toxoplasma GRA15 limits parasite growth in IFNγ-activated fibroblasts through TRAF ubiquitin ligases. EMBO J. 2020, 39, e103758. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, X.; Liu, J.; Fu, Y.; Xu, J.; Liu, Q. Toxoplasma gondii rhoptry protein38 (TgROP38) affects parasite invasion, egress, and induces IL-18 secretion during early infection. Acta Biochim. Biophys. Sin. 2018, 50, 766–775. [Google Scholar] [CrossRef] [Green Version]
- Fox, B.A.; Rommereim, L.M.; Guevara, R.B.; Falla, A.; Hortua Triana, M.A.; Sun, Y.; Bzik, D.J. The Toxoplasma gondii Rhoptry Kinome Is Essential for Chronic Infection. mBio 2016, 7, e00193-16. [Google Scholar] [CrossRef] [Green Version]
- Melo, M.B.; Nguyen, Q.P.; Cordeiro, C.; Hassan, M.A.; Yang, N.; McKell, R.; Rosowski, E.E.; Julien, L.; Butty, V.; Dardé, M.-L.; et al. Transcriptional analysis of murine macrophages infected with different Toxoplasma strains identifies novel regulation of host signaling pathways. PLoS Pathog. 2013, 9, e1003779. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, L.; Chen, F.; Harb, O.S.; Davis, P.H.; Beiting, D.P.; Brownback, C.S.; Ouloguem, D.; Roos, D.S. Integrative genomic approaches highlight a family of parasite-specific kinases that regulate host responses. Cell Host Microbe 2010, 8, 208–218. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.S.; Sasai, M.; Ohshima, J.; Lee, Y.; Bando, H.; Takeda, K.; Yamamoto, M. Selective and strain-specific NFAT4 activation by the Toxoplasma gondii polymorphic dense granule protein GRA6. J. Exp. Med. 2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shastri, A.J.; Marino, N.D.; Franco, M.; Lodoen, M.B.; Boothroyd, J.C. GRA25 is a novel virulence factor of Toxoplasma gondii and influences the host immune response. Infect. Immun. 2014, 82, 2595–2605. [Google Scholar] [CrossRef] [Green Version]
- He, H.; Brenier-Pinchart, M.-P.; Braun, L.; Kraut, A.; Touquet, B.; Couté, Y.; Tardieux, I.; Hakimi, M.-A.; Bougdour, A. Characterization of a Toxoplasma effector uncovers an alternative GSK3/β-catenin-regulatory pathway of inflammation. eLife 2018, 7, e39887. [Google Scholar] [CrossRef]
- Tomita, T.; Mukhopadhyay, D.; Han, B.; Yakubu, R.; Tu, V.; Mayoral, J.; Sugi, T.; Ma, Y.; Saeij, J.P.J.; Weiss, L.M. Toxoplasma gondii Matrix Antigen 1 Is a Secreted Immunomodulatory Effector. mBio 2021, 12, e00603-21. [Google Scholar] [CrossRef] [PubMed]
- Saeij, J.P.J.; Boyle, J.P.; Coller, S.; Taylor, S.; Sibley, L.D.; Brooke-Powell, E.T.; Ajioka, J.W.; Boothroyd, J.C. Polymorphic secreted kinases are key virulence factors in toxoplasmosis. Science 2006, 314, 1780–1783. [Google Scholar] [CrossRef] [Green Version]
- Jensen, K.D.C.; Hu, K.; Whitmarsh, R.J.; Hassan, M.A.; Julien, L.; Lu, D.; Chen, L.; Hunter, C.A.; Saeij, J.P.J. Toxoplasma gondii rhoptry 16 kinase promotes host resistance to oral infection and intestinal inflammation only in the context of the dense granule protein GRA15. Infect. Immun. 2013, 81, 2156–2167. [Google Scholar] [CrossRef] [Green Version]
- Rudzki, E.N.; Ander, S.E.; Coombs, R.S.; Alrubaye, H.I.; Cabo, L.F.; Blank, M.L.; Gutierrez-Melo, N.; Dubey, J.P.; Coyne, C.B.; Boyle, J.P. Toxoplasma gondii GRA28 is required for specific induction of the regulatory chemokine CCL22 in human and mouse cells. bioRxiv 2020. [Google Scholar] [CrossRef]
- Braun, L.; Brenier-Pinchart, M.-P.; Yogavel, M.; Curt-Varesano, A.; Curt-Bertini, R.-L.; Hussain, T.; Kieffer-Jaquinod, S.; Coute, Y.; Pelloux, H.; Tardieux, I.; et al. A Toxoplasma dense granule protein, GRA24, modulates the early immune response to infection by promoting a direct and sustained host p38 MAPK activation. J. Exp. Med. 2013, 210, 2071–2086. [Google Scholar] [CrossRef] [PubMed]
- Bougdour, A.; Durandau, E.; Brenier-Pinchart, M.-P.; Ortet, P.; Barakat, M.; Kieffer, S.; Curt-Varesano, A.; Curt-Bertini, R.-L.; Bastien, O.; Coute, Y.; et al. Host cell subversion by Toxoplasma GRA16, an exported dense granule protein that targets the host cell nucleus and alters gene expression. Cell Host Microbe 2013, 13, 489–500. [Google Scholar] [CrossRef]
- Braun, L.; Brenier-Pinchart, M.-P.; Hammoudi, P.-M.; Cannella, D.; Kieffer-Jaquinod, S.; Vollaire, J.; Josserand, V.; Touquet, B.; Couté, Y.; Tardieux, I.; et al. The Toxoplasma effector TEEGR promotes parasite persistence by modulating NF-κB signalling via EZH2. Nat. Microbiol. 2019, 4, 1208–1220. [Google Scholar] [CrossRef] [PubMed]
- Panas, M.W.; Naor, A.; Cygan, A.M.; Boothroyd, J.C. Toxoplasma Controls Host Cyclin E Expression through the Use of a Novel MYR1-Dependent Effector Protein, HCE1. mBio 2019, 10, e00674-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gay, G.; Braun, L.; Brenier-Pinchart, M.-P.; Vollaire, J.; Josserand, V.; Bertini, R.-L.; Varesano, A.; Touquet, B.; De Bock, P.-J.; Coute, Y.; et al. Toxoplasma gondii TgIST co-opts host chromatin repressors dampening STAT1-dependent gene regulation and IFN-γ–mediated host defenses. J. Exp. Med. 2016. [Google Scholar] [CrossRef]
- Olias, P.; Etheridge, R.D.; Zhang, Y.; Holtzman, M.J.; Sibley, L.D. Toxoplasma Effector Recruits the Mi-2/NuRD Complex to Repress STAT1 Transcription and Block IFN-γ-Dependent Gene Expression. Cell Host Microbe 2016, 20, 72–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, A.; David Sibley, L. Toxoplasma gondii secreted effectors co-opt host repressor complexes to inhibit necroptosis. Cell Host Microbe 2021. [Google Scholar] [CrossRef]
- Martens, S.; Howard, J. The interferon-inducible GTPases. Annu. Rev. Cell Dev. Biol. 2006, 22, 559–589. [Google Scholar] [CrossRef]
- Khaminets, A.; Hunn, J.P.; Könen-Waisman, S.; Zhao, Y.O.; Preukschat, D.; Coers, J.; Boyle, J.P.; Ong, Y.-C.; Boothroyd, J.C.; Reichmann, G.; et al. Coordinated loading of IRG resistance GTPases on to the Toxoplasma gondii parasitophorous vacuole. Cell. Microbiol. 2010, 12, 939–961. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.; Barragan, A.; Su, C.; Fux, B.; Fentress, S.J.; Tang, K.; Beatty, W.L.; Hajj, H.E.; Jerome, M.; Behnke, M.S.; et al. A secreted serine-threonine kinase determines virulence in the eukaryotic pathogen Toxoplasma gondii. Science 2006, 314, 1776–1780. [Google Scholar] [CrossRef] [Green Version]
- Fleckenstein, M.C.; Reese, M.L.; Könen-Waisman, S.; Boothroyd, J.C.; Howard, J.C.; Steinfeldt, T. A Toxoplasma gondii Pseudokinase Inhibits Host IRG Resistance Proteins. PLoS Biol. 2012, 10, e1001358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murillo-León, M.; Müller, U.B.; Zimmermann, I.; Singh, S.; Widdershooven, P.; Campos, C.; Alvarez, C.; Könen-Waisman, S.; Lukes, N.; Ruzsics, Z.; et al. Molecular mechanism for the control of virulent Toxoplasma gondii infections in wild-derived mice. Nat. Commun. 2019, 10, 1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermanns, T.; Müller, U.B.; Könen-Waisman, S.; Howard, J.C.; Steinfeldt, T. The Toxoplasma gondii rhoptry protein ROP18 is an Irga6-specific kinase and regulated by the dense granule protein GRA7. Cell. Microbiol. 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelin, A.; Bittame, A.; Bordat, Y.; Travier, L.; Mercier, C.; Dubremetz, J.-F.; Lebrun, M. GRA12, a Toxoplasma dense granule protein associated with the intravacuolar membranous nanotubular network. Int. J. Parasitol. 2009, 39, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-L.; Bai, M.-J.; Elsheikha, H.M.; Liang, Q.-L.; Li, T.-T.; Cao, X.-Z.; Zhu, X.-Q. Novel roles of dense granule protein 12 (GRA12) in Toxoplasma gondii infection. FASEB J. 2020, 34, 3165–3178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.-H.; Sun, S.-C. Tumor Necrosis Factor Receptor-Associated Factor Regulation of Nuclear Factor κB and Mitogen-Activated Protein Kinase Pathways. Front. Immunol. 2018, 9, 1849. [Google Scholar] [CrossRef]
- Pandori, W.J.; Lima, T.S.; Mallya, S.; Kao, T.H.; Gov, L.; Lodoen, M.B. Toxoplasma gondii activates a Syk-CARD9-NF-κB signaling axis and gasdermin D-independent release of IL-1β during infection of primary human monocytes. PLoS Pathog. 2019, 15, e1007923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgado, P.; Sudarshana, D.M.; Gov, L.; Harker, K.S.; Lam, T.; Casali, P.; Boyle, J.P.; Lodoen, M.B. Type II Toxoplasma gondii induction of CD40 on infected macrophages enhances interleukin-12 responses. Infect. Immun. 2014, 82, 4047–4055. [Google Scholar] [CrossRef] [Green Version]
- Bando, H.; Lee, Y.; Sakaguchi, N.; Pradipta, A.; Ma, J.S.; Tanaka, S.; Cai, Y.; Liu, J.; Shen, J.; Nishikawa, Y.; et al. Inducible Nitric Oxide Synthase Is a Key Host Factor for Toxoplasma GRA15-Dependent Disruption of the Gamma Interferon-Induced Antiparasitic Human Response. mBio 2018, 9, e01738-18. [Google Scholar] [CrossRef] [Green Version]
- Clough, B.; Wright, J.D.; Pereira, P.M.; Hirst, E.M.; Johnston, A.C.; Henriques, R.; Frickel, E.-M. K63-Linked Ubiquitination Targets Toxoplasma gondii for Endo-lysosomal Destruction in IFNγ-Stimulated Human Cells. PLoS Pathog. 2016, 12, e1006027. [Google Scholar] [CrossRef]
- Mercier, C.; Dubremetz, J.-F.; Rauscher, B.; Lecordier, L.; Sibley, L.D.; Cesbron-Delauw, M.-F. Biogenesis of nanotubular network in Toxoplasma parasitophorous vacuole induced by parasite proteins. Mol. Biol. Cell 2002, 13, 2397–2409. [Google Scholar] [CrossRef] [Green Version]
- Tu, V.; Tomita, T.; Sugi, T.; Mayoral, J.; Han, B.; Yakubu, R.R.; Williams, T.; Horta, A.; Ma, Y.; Weiss, L.M. The Toxoplasma gondii Cyst Wall Interactome. mBio 2020, 11, e02699-19. [Google Scholar] [CrossRef] [Green Version]
- Dunay, I.R.; Damatta, R.A.; Fux, B.; Presti, R.; Greco, S.; Colonna, M.; Sibley, L.D. Gr1(+) inflammatory monocytes are required for mucosal resistance to the pathogen Toxoplasma gondii. Immunity 2008, 29, 306–317. [Google Scholar] [CrossRef] [Green Version]
- Robben, P.M.; LaRegina, M.; Kuziel, W.A.; Sibley, L.D. Recruitment of Gr-1+ monocytes is essential for control of acute toxoplasmosis. J. Exp. Med. 2005, 201, 1761–1769. [Google Scholar] [CrossRef]
- Goldszmid, R.S.; Coppens, I.; Lev, A.; Caspar, P.; Mellman, I.; Sher, A. Host ER-parasitophorous vacuole interaction provides a route of entry for antigen cross-presentation in Toxoplasma gondii-infected dendritic cells. J. Exp. Med. 2009, 206, 399–410. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Ma, J.S.; Mueller, C.; Kamiyama, N.; Saiga, H.; Kubo, E.; Kimura, T.; Okamoto, T.; Okuyama, M.; Kayama, H.; et al. ATF6{beta} is a host cellular target of the Toxoplasma gondii virulence factor ROP18. J. Exp. Med. 2011, 208, 1533–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, R.; Tang, Y.; Chen, L.; Cai, H.; Lai, D.-H.; Liu, K.; Wan, L.; Gong, L.; Yu, L.; Luo, Q.; et al. Encephalitis is mediated by ROP18 of Toxoplasma gondii, a severe pathogen in AIDS patients. Proc. Natl. Acad. Sci. USA 2018, 115, E5344–E5352. [Google Scholar] [CrossRef] [Green Version]
- Kahali, S.; Sarcar, B.; Prabhu, A.; Seto, E.; Chinnaiyan, P. Class I histone deacetylases localize to the endoplasmic reticulum and modulate the unfolded protein response. FASEB J. 2012, 26, 2437–2445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolotti, A.; Zhang, Y.; Hendershot, L.M.; Harding, H.P.; Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell Biol. 2000, 2, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Steinhart, Z.; Angers, S. Wnt signaling in development and tissue homeostasis. Development 2018, 145, dev146589. [Google Scholar] [CrossRef] [Green Version]
- Romagnani, S. Cytokines and chemoattractants in allergic inflammation. Mol. Immunol. 2002, 38, 881–885. [Google Scholar] [CrossRef]
- Rapp, M.; Wintergerst, M.W.M.; Kunz, W.G.; Vetter, V.K.; Knott, M.M.L.; Lisowski, D.; Haubner, S.; Moder, S.; Thaler, R.; Eiber, S.; et al. CCL22 controls immunity by promoting regulatory T cell communication with dendritic cells in lymph nodes. J. Exp. Med. 2019, 216, 1170–1181. [Google Scholar] [CrossRef] [Green Version]
- Parmley, S.F.; Yang, S.; Harth, G.; Sibley, L.D.; Sucharczuk, A.; Remington, J.S. Molecular characterization of a 65-kilodalton Toxoplasma gondii antigen expressed abundantly in the matrix of tissue cysts. Mol. Biochem. Parasitol. 1994, 66, 283–296. [Google Scholar] [CrossRef]
- Ewald, S.E.; Chavarria-Smith, J.; Boothroyd, J.C. NLRP1 is an inflammasome sensor for Toxoplasma gondii. Infect. Immun. 2014, 82, 460–468. [Google Scholar] [CrossRef] [Green Version]
- Gorfu, G.; Cirelli, K.M.; Melo, M.B.; Mayer-Barber, K.; Crown, D.; Koller, B.H.; Masters, S.; Sher, A.; Leppla, S.H.; Moayeri, M.; et al. Dual role for inflammasome sensors NLRP1 and NLRP3 in murine resistance to Toxoplasma gondii. mBio 2014, 5, e01117-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeij, J.P.J.; Coller, S.; Boyle, J.P.; Jerome, M.E.; White, M.W.; Boothroyd, J.C. Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature 2007, 445, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Ong, Y.-C.; Reese, M.L.; Boothroyd, J.C. Toxoplasma rhoptry protein 16 (ROP16) subverts host function by direct tyrosine phosphorylation of STAT6. J. Biol. Chem. 2010, 285, 28731–28740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, M.; Standley, D.M.; Takashima, S.; Saiga, H.; Okuyama, M.; Kayama, H.; Kubo, E.; Ito, H.; Takaura, M.; Matsuda, T.; et al. A single polymorphic amino acid on Toxoplasma gondii kinase ROP16 determines the direct and strain-specific activation of Stat3. J. Exp. Med. 2009, 206, 2747–2760. [Google Scholar] [CrossRef]
- Kaplan, M.H.; Schindler, U.; Smiley, S.T.; Grusby, M.J. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity 1996, 4, 313–319. [Google Scholar] [CrossRef] [Green Version]
- Jensen, K.D.C.; Wang, Y.; Wojno, E.D.T.; Shastri, A.J.; Hu, K.; Cornel, L.; Boedec, E.; Ong, Y.-C.; Chien, Y.-H.; Hunter, C.A.; et al. Toxoplasma polymorphic effectors determine macrophage polarization and intestinal inflammation. Cell Host Microbe 2011, 9, 472–483. [Google Scholar] [CrossRef] [Green Version]
- Nadipuram, S.M.; Kim, E.W.; Vashisht, A.A.; Lin, A.H.; Bell, H.N.; Coppens, I.; Wohlschlegel, J.A.; Bradley, P.J. In Vivo Biotinylation of the Toxoplasma Parasitophorous Vacuole Reveals Novel Dense Granule Proteins Important for Parasite Growth and Pathogenesis. mBio 2016, 7, e00808-16. [Google Scholar] [CrossRef] [Green Version]
- Franco, M.; Panas, M.W.; Marino, N.D.; Lee, M.C.W.; Buchholz, K.R.; Kelly, F.D.; Bednarski, J.J.; Sleckman, B.P.; Pourmand, N.; Boothroyd, J.C. A novel secreted protein, MYR1, is central to Toxoplasma’s manipulation of host cells. mBio 2016, 7, e02231-15. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, E.; Palencia, A.; Braun, L.; Kapp, U.; Bougdour, A.; Belrhali, H.; Bowler, M.W.; Hakimi, M.-A. Structural Basis for the Subversion of MAP Kinase Signaling by an Intrinsically Disordered Parasite Secreted Agonist. Structure 2017, 25, 16–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, H.L.; Snyder, L.M.; Doherty, C.M.; Fox, B.A.; Bzik, D.J.; Denkers, E.Y. Toxoplasma gondii dense granule protein GRA24 drives MyD88-independent p38 MAPK activation, IL-12 production and induction of protective immunity. PLoS Pathog. 2020, 16, e1008572. [Google Scholar] [CrossRef]
- Mukhopadhyay, D.; Arranz-Solís, D.; Saeij, J.P.J. Toxoplasma GRA15 and GRA24 are important activators of the host innate immune response in the absence of TLR11. PLoS Pathog. 2020, 16, e1008586. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, D.; Shiloh, A.; Luo, J.; Nikolaev, A.Y.; Qin, J.; Gu, W. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 2002, 416, 648–653. [Google Scholar] [CrossRef]
- Kim, S.-G.; Seo, S.-H.; Shin, J.-H.; Yang, J.-P.; Lee, S.H.; Shin, E.-H. Increase in the nuclear localization of PTEN by the Toxoplasma GRA16 protein and subsequent induction of p53-dependent apoptosis and anticancer effect. J. Cell. Mol. Med. 2019, 23, 3234–3245. [Google Scholar] [CrossRef] [Green Version]
- Seo, S.-H.; Kim, S.-G.; Shin, J.-H.; Ham, D.-W.; Shin, E.-H. Toxoplasma GRA16 Inhibits NF-κB Activation through PP2A-B55 Upregulation in Non-Small-Cell Lung Carcinoma Cells. Int. J. Mol. Sci. 2020, 21, 6642. [Google Scholar] [CrossRef] [PubMed]
- Panas, M.W.; Boothroyd, J.C. Toxoplasma Uses GRA16 To Upregulate Host c-Myc. mSphere 2020, 5, e00402-20. [Google Scholar] [CrossRef]
- Kent, L.N.; Leone, G. The broken cycle: E2F dysfunction in cancer. Nat. Rev. Cancer 2019, 19, 326–338. [Google Scholar] [CrossRef]
- Kouznetsova, V.L.; Tchekanov, A.; Li, X.; Yan, X.; Tsigelny, I.F. Polycomb repressive 2 complex-Molecular mechanisms of function. Protein Sci. 2019, 28, 1387–1399. [Google Scholar] [CrossRef] [PubMed]
- Matta, S.K.; Olias, P.; Huang, Z.; Wang, Q.; Park, E.; Yokoyama, W.M.; Sibley, L.D. Toxoplasma gondii effector TgIST blocks type I interferon signaling to promote infection. Proc. Natl. Acad. Sci. USA 2019, 116, 17480–17491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yap, G.S.; Sher, A. Effector cells of both nonhemopoietic and hemopoietic origin are required for interferon (IFN)-gamma- and tumor necrosis factor (TNF)-alpha-dependent host resistance to the intracellular pathogen, Toxoplasma gondii. J. Exp. Med. 1999, 189, 1083–1092. [Google Scholar] [CrossRef] [Green Version]
- Han, S.-J.; Melichar, H.J.; Coombes, J.L.; Chan, S.W.; Koshy, A.A.; Boothroyd, J.C.; Barton, G.M.; Robey, E.A. Internalization and TLR-dependent type I interferon production by monocytes in response to Toxoplasma gondii. Immunol. Cell Biol. 2014, 92, 872–881. [Google Scholar] [CrossRef] [Green Version]
- Torchy, M.P.; Hamiche, A.; Klaholz, B.P. Structure and function insights into the NuRD chromatin remodeling complex. Cell. Mol. Life Sci. 2015, 72, 2491–2507. [Google Scholar] [CrossRef] [PubMed]
- Mayoral, J.; Shamamian, P., Jr.; Weiss, L.M. In Vitro Characterization of Protein Effector Export in the Bradyzoite Stage of Toxoplasma gondii. mBio 2020, 11, e00046-20. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, S.; Saeij, J.P.J. Toxoplasma Does Not Secrete the GRA16 and GRA24 Effectors Beyond the Parasitophorous Vacuole Membrane of Tissue Cysts. Front. Cell. Infect. Microbiol. 2018, 8, 366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, P.J.; Fairall, L.; Schwabe, J.W.R. Nuclear hormone receptor co-repressors: Structure and function. Mol. Cell. Endocrinol. 2012, 348, 440–449. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Chan, F.K.-M.; Kroemer, G. Necroptosis: Mechanisms and Relevance to Disease. Annu. Rev. Pathol. 2017, 12, 103–130. [Google Scholar] [CrossRef]
- Rastogi, S.; Cygan, A.M.; Boothroyd, J.C. Translocation of effector proteins into host cells by Toxoplasma gondii. Curr. Opin. Microbiol. 2019, 52, 130–138. [Google Scholar] [CrossRef]
- Wang, Y.; Sangaré, L.O.; Paredes-Santos, T.C.; Hassan, M.A.; Krishnamurthy, S.; Furuta, A.M.; Markus, B.M.; Lourido, S.; Saeij, J.P.J. Genome-wide screens identify Toxoplasma gondii determinants of parasite fitness in IFNγ-activated murine macrophages. Nat. Commun. 2020, 11, 5258. [Google Scholar] [CrossRef]
- Gold, D.A.; Kaplan, A.D.; Lis, A.; Bett, G.C.L.; Rosowski, E.E.; Cirelli, K.M.; Bougdour, A.; Sidik, S.M.; Beck, J.R.; Lourido, S.; et al. The Toxoplasma Dense Granule Proteins GRA17 and GRA23 Mediate the Movement of Small Molecules between the Host and the Parasitophorous Vacuole. Cell Host Microbe 2015, 17, 642–652. [Google Scholar] [CrossRef] [Green Version]
- Marino, N.D.; Panas, M.W.; Franco, M.; Theisen, T.C.; Naor, A.; Rastogi, S.; Buchholz, K.R.; Lorenzi, H.A.; Boothroyd, J.C. Identification of a novel protein complex essential for effector translocation across the parasitophorous vacuole membrane of Toxoplasma gondii. PLoS Pathog. 2018, 14, e1006828. [Google Scholar] [CrossRef]
- Panas, M.W.; Ferrel, A.; Naor, A.; Tenborg, E.; Lorenzi, H.A.; Boothroyd, J.C. Translocation of Dense Granule Effectors across the Parasitophorous Vacuole Membrane in Toxoplasma-Infected Cells Requires the Activity of ROP17, a Rhoptry Protein Kinase. mSphere 2019, 4, e00276-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, V.; Mayoral, J.; Sugi, T.; Tomita, T.; Han, B.; Ma, Y.F.; Weissa, L.M. Enrichment and proteomic characterization of the cyst wall from in vitro Toxoplasma gondii cysts. MBio 2019, 10, e00469-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayoral, J.; Tomita, T.; Tu, V.; Aguilan, J.T.; Sidoli, S.; Weiss, L.M. Toxoplasma gondii PPM3C, a secreted protein phosphatase, affects parasitophorous vacuole effector export. PLoS Pathog. 2020, 16, e1008771. [Google Scholar] [CrossRef] [PubMed]
- Tomita, T.; Bzik, D.J.; Ma, Y.F.; Fox, B.A.; Markillie, L.M.; Taylor, R.C.; Kim, K.; Weiss, L.M. The Toxoplasma gondii Cyst Wall Protein CST1 Is Critical for Cyst Wall Integrity and Promotes Bradyzoite Persistence. PLoS Pathog. 2013, 9, e1003823. [Google Scholar] [CrossRef] [Green Version]
- Rastogi, S.; Xue, Y.; Quake, S.R.; Boothroyd, J.C. Differential Impacts on Host Transcription by ROP and GRA Effectors from the Intracellular Parasite Toxoplasma gondii. mBio 2020, 11, e00182-20. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Theisen, T.C.; Rastogi, S.; Ferrel, A.; Quake, S.R.; Boothroyd, J.C. A single-parasite transcriptional atlas of Toxoplasma gondii reveals novel control of antigen expression. eLife 2020, 9, e54129. [Google Scholar] [CrossRef] [Green Version]
- Patir, A.; Gossner, A.; Ramachandran, P.; Alves, J.; Freeman, T.C.; Henderson, N.C.; Watson, M.; Hassan, M.A. Single-cell RNA-seq reveals CD16- monocytes as key regulators of human monocyte transcriptional response to Toxoplasma. Sci. Rep. 2020, 10, 21047. [Google Scholar] [CrossRef]
- Coffey, M.J.; Sleebs, B.E.; Uboldi, A.D.; Garnham, A.L.; Franco, M.; Marino, N.D.; Panas, M.W.; Ferguson, D.J.; Enciso, M.; O’Neill, M.T.; et al. An aspartyl protease defines a novel pathway for export of Toxoplasma proteins into the host cell. eLife 2015, 4, e10809. [Google Scholar] [CrossRef] [PubMed]
- Hammoudi, P.-M.; Jacot, D.; Mueller, C.; Di Cristina, M.; Dogga, S.K.; Marq, J.-B.; Romano, J.; Tosetti, N.; Dubrot, J.; Emre, Y.; et al. Fundamental Roles of the Golgi-Associated Toxoplasma Aspartyl Protease, ASP5, at the Host-Parasite Interface. PLoS Pathog. 2015, 11, e1005211. [Google Scholar] [CrossRef] [PubMed] [Green Version]


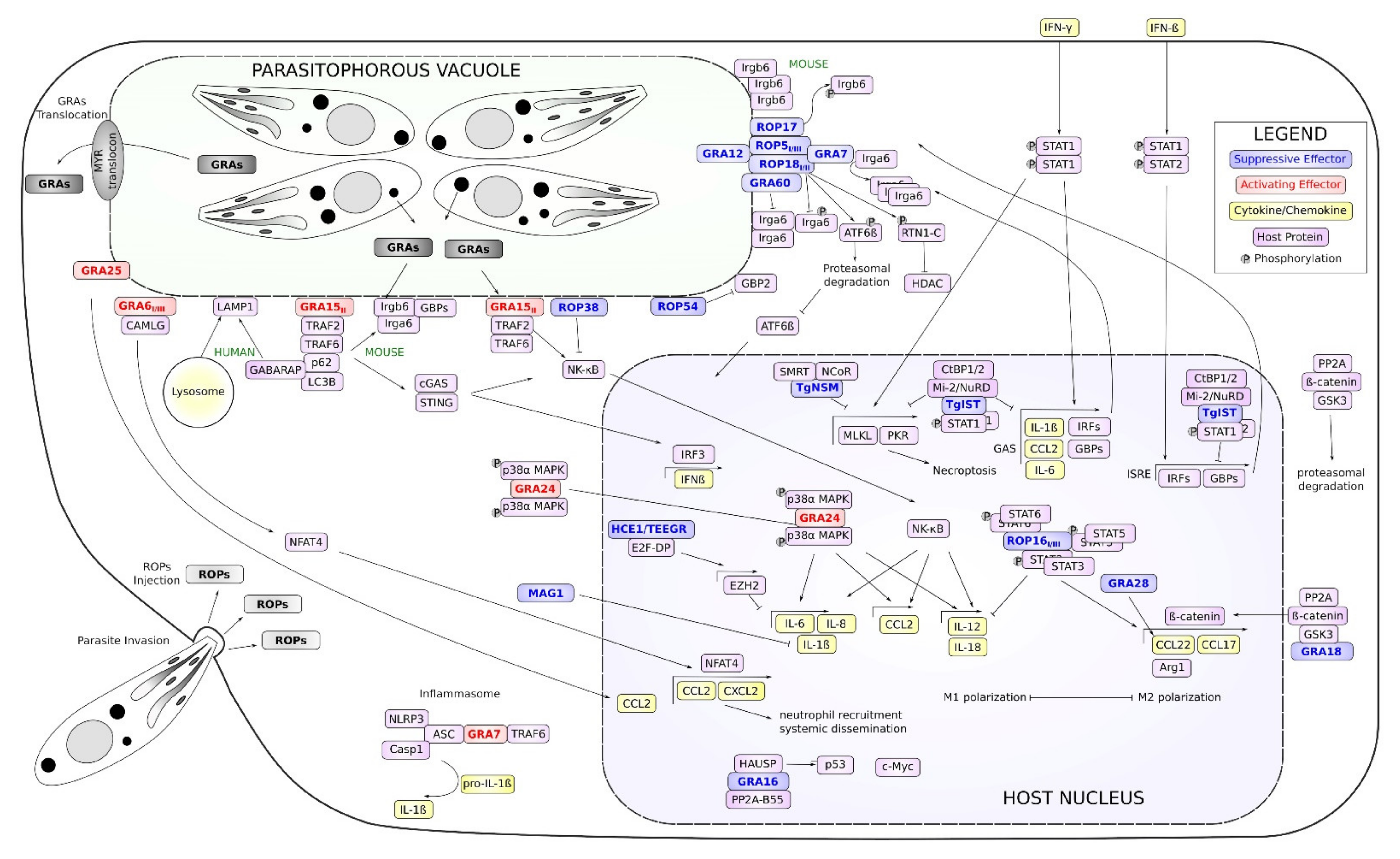{kind=link}
| Name | Location | Functions | Interacting Partner | Mouse Virulence of KO Parasites | Gene Name | Ortholog in Cystoisospora | Ortholog in Sarcocystis | Ortholog in Eimeria | Essentiality Score [26] | NonSyn/Snyn SNP | References |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ROP5I/III | PVM | Pseudokinase inhibits IRG coating by preparing Irga6 for phosphorylation by ROP18. | Irga6 | Highly avirulent [27,28] | TGME49_308090 | Y | Y | Y | n/a | 3 | [27,28] |
| ROP18I/II | PVM/ER | Serine-threonine kinase inhibits IRG coating by phosphorylating monomeric Irga6. Suppresses unfolded protein response and antigen presentation. Induces apoptosis in neurons by RTN1-C. | IRGs (preferentially Irga6), ATF6ß, RTN1-C | Slightly reduced, ∆rop18/17 double mutant is highly avirulent [29] | TGME49_205250 | Y | Y | Y | 0.89 | 4.5 | [30] |
| GRA7 | PVM | Alter the turnover kinetics of Irga6. Activate inflammasome. | Irga6, TRAF6, ASC | Slightly reduced, ∆rop18/gra7 double mutant is highly avirulent [31] | TGME49_203310 | N | N | N | 1.9 | 4 | [31] |
| ROP17 | PVM | Serine-threonine kinase disassembles polymerized Irgb6. | IRGs (preferentially Irgb6) | Slightly reduced, ∆rop18/17 double mutant is highly avirulent [29] | TGME49_258580 | Y | Y | Y | 1.29 | 3.8 | [29] |
| GRA60 | PVM | Prevents Irga6 and Irgb10 coating on PVM. | n/a | Slightly reduced [32] | TGME49_204270 | N | N | N | -0.59 | 2.41 | [32] |
| ROP54 | PVM | Reduces GBP2 loading on PVM. | n/a | Reduced [33] | TGME49_210370 | N | N | N | 1.07 | 1.58 | [33] |
| GRA12 | IVN/PVM | Prevents IFNγ-induced parasite destruction. | n/a | Highly avirulent [34] | TGME49_288650 | Y | Y | Y | 1.75 | 0.64 | [34] |
| GRA15II | PVM | Activates NF-κB, induce secretion of IL-12 and IL-1ß. Mediates IFNγ-induced PV-lysosome fusion and IRG loading. | TRAF2/TRAF6 | No difference [35], more virulent [36] | TGME49_275470 | N | N | N | 2.33 | 3.75 | [35,37,38] |
| ROP38 | IVN/PVM | Inhibits NF-κB pathway and IL-12 production. | n/a | Reduced [39], no difference [40] | TGME49_242110 | Y | Y | Y | n/a | 4.5 | [41,42] |
| GRA6 | IVN/PVM | Induces CXCL2/CCL2 by activation NFAT4, recruits monocytes/neutrophils, and disseminates parasites. | CAMLG | Reduced [43] | TGME49_275440 | N | N | N | 1.84 | 8.25 | [43] |
| GRA25 | PV | Induces CCL2 secretion. | n/a | Reduced [44] | TGME49_290700 | N | N | N | 1.31 | 3.08 | [44] |
| GRA18 | Cytoplasm | Activates ß-catenin and release anti-inflammatory chemokines CCL22 and CCL17. | GSK3/PP2A-B56 | Reduced [45] | TGME49_288840 | N | N | N | 1.66 | 1.93 | [45] |
| MAG1 | PVM/Cytoplasm | Suppresses IL-1ß secretion in macrophages. | n/a | Reduced [46] | TGME49_270240 | Y | N | N | 1.25 | 1 | [46] |
| ROP16I/III | Nucleus | Suppresses Th1/M1 response, induces Th2/M2 response. Protects mice from lethal ileitis. | STAT3/5/6 | More virulent [47,48] | TGME49_262730 | N | N | N | 1.11 | 2.37 | [47,48] |
| GRA28 | Nucleus | Induces CCL22 secretion. | n/a | No difference [49] | TGME49_231960 | N | N | N | 1.48 | 5.25 | [49] |
| GRA24 | Nucleus | Activates p38α MAPK and induces Th1/M1 cytokines and chemokines secretion (e.g., IL-12, IL-6, CCL2, CCL5). | p38α MAPK | No difference [50] | TGME49_230180 | N | N | N | 2.86 | 2.69 | [50] |
| GRA16 | Nucleus | Interferes p53-dependent apoptosis and inhibits NF-κB pathway (in parasite-free tumor model). | HAUSP, PP2A-B55 | Reduced in type II strain but not in type I [51] | TGME49_208830 | N | N | N | 2.28 | 3.13 | [51] |
| HCE1/TEEGR | Nucleus | Represses NF-κB-induced gene expression (e.g., IL-6, IL-8, IL-1ß). | E2F | Reduced [52] | TGME49_239010 | N | N | N | 1.46 | 5.46 | [52,53] |
| TgIST | Nucleus | Represses IFN-induced gene expression (e.g., IRGs loading). | STAT1/2, NuRD | Highly avirulent [54,55] | TGME49_240060 | N | N | N | 1.44 | 5.67 | [54,55] |
| TgNSM | Nucleus | Represses IFN-induced gene expression and necroptosis. | NCoR/SMRT | n/a | TGME49_235140 | N | N | N | 1.41 | 6.73 | [56] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomita, T.; Guevara, R.B.; Shah, L.M.; Afrifa, A.Y.; Weiss, L.M. Secreted Effectors Modulating Immune Responses to Toxoplasma gondii. Life 2021, 11, 988. https://doi.org/10.3390/life11090988
Tomita T, Guevara RB, Shah LM, Afrifa AY, Weiss LM. Secreted Effectors Modulating Immune Responses to Toxoplasma gondii. Life. 2021; 11(9):988. https://doi.org/10.3390/life11090988
Chicago/Turabian StyleTomita, Tadakimi, Rebekah B. Guevara, Lamisha M. Shah, Andrews Y. Afrifa, and Louis M. Weiss. 2021. "Secreted Effectors Modulating Immune Responses to Toxoplasma gondii" Life 11, no. 9: 988. https://doi.org/10.3390/life11090988