
The Role of Metabolism in Migraine Pathophysiology and Susceptibility

 , , ,
, , , 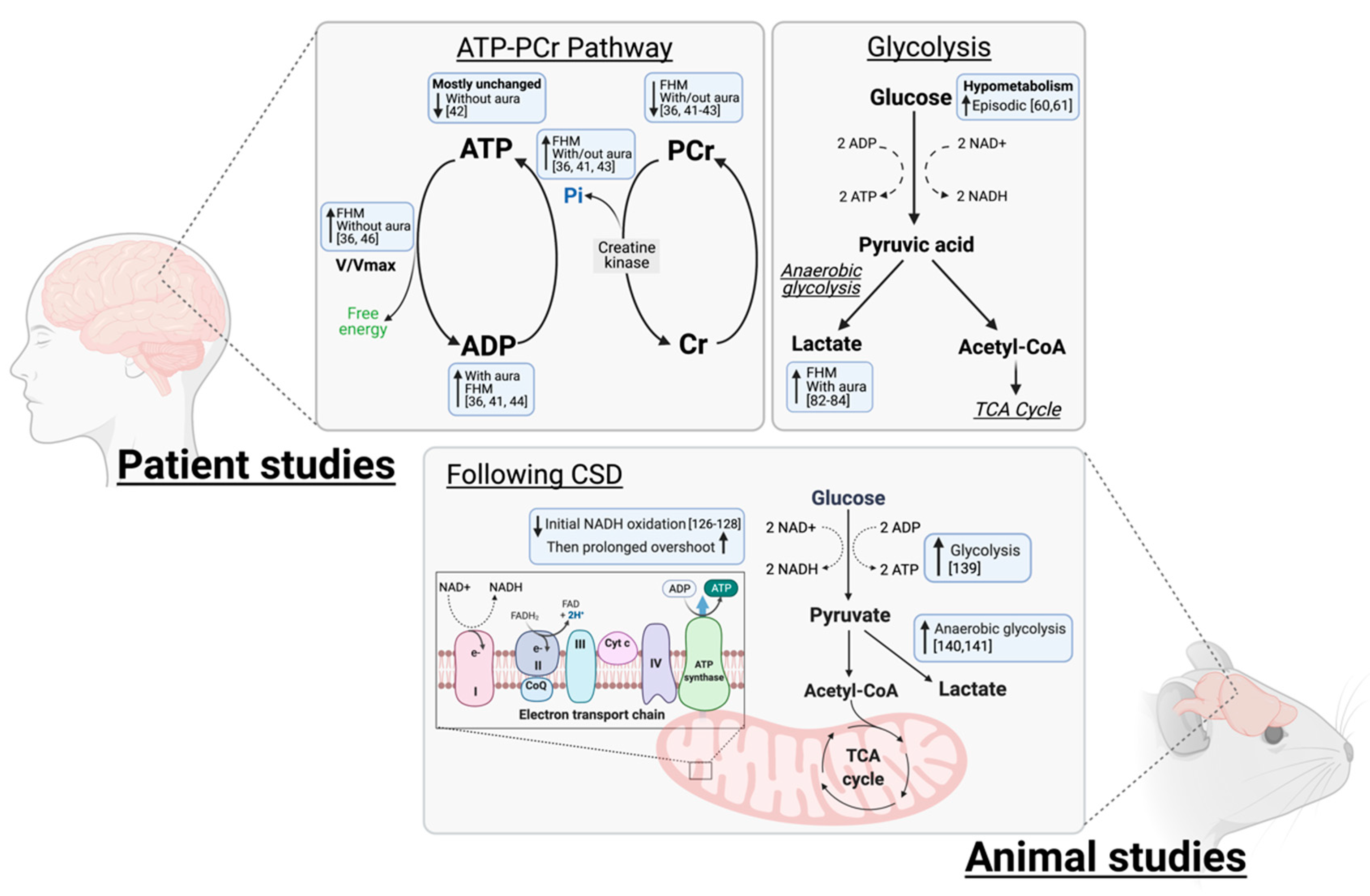{kind=link}
Abstract
:1. Introduction
1.1. Background
1.2. Headache Mechanisms
1.3. Energy Metabolism in Headache
2. Patient Studies
2.1. ATP-PCR System
2.2. Magnesium Availability
2.3. Glycolysis and Glucose Metabolism
2.4. Ketogenesis
2.5. Anaerobic Metabolism
2.6. Prophylactic Supplementation of Patients
3. Animal Data
3.1. Imaging and Labelling Studies
3.1.1. NADH and Oxidative Metabolism
3.1.2. Glycolysis and Glucose Metabolism
3.2. Mitochondrial Studies in Animals
3.3. Supplementation of Animals
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- James, S.L.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1789–1858. [Google Scholar] [CrossRef] [Green Version]
- Stovner, L.J.; Nichols, E.; Steiner, T.J.; Abd-Allah, F.; Abdelalim, A.; Al-Raddadi, R.M.; Ansha, M.G.; Barac, A.; Bensenor, I.M.; Doan, L.P.; et al. Global, regional, and national burden of migraine and tension-type headache, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 954–976. [Google Scholar] [CrossRef] [Green Version]
- Steiner, T.J.; Stovner, L.J.; Birbeck, G.L. Migraine: The seventh disabler. J. Headache Pain 2013, 14, 1. [Google Scholar] [CrossRef] [Green Version]
- Matharu, N.A.B.; Surat, T.; Giorgio, L.; Mariam, T.; Marjan, J.; Manjit, M. Quality of life in primary headache disorders: A review. Cephalalgia 2015, 36, 67–91. [Google Scholar] [CrossRef]
- Lipton, R.B.; Bigal, M.E.; Kolodner, K.; Stewart, W.F.; Liberman, J.N.; Steiner, T.J. The family impact of migraine: Population-based studies in the USA and UK. Cephalalgia 2003, 23, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Stewart, W.F.; Lipton, R.B.; Simon, D. Work-related disability: Results from the American migraine study. Cephalalgia 1996, 16, 231–238. [Google Scholar] [CrossRef]
- Foundation, W. Migraine’s Impact on Employment in Europe. What Can be Done to Improve Work Outcomes for People with Migraine? Lancaster University: Lancaster, UK, 2019; p. 54. Available online: https://www.lancaster.ac.uk/media/lancaster-university/content-assets/documents/lums/work-foundation/Migraines-impact-on-employment-in-Europe-FINAL-pub-vA-accessible.pdf (accessed on 15 April 2021).
- Kernick, D.; Stapley, S.; Hamilton, W. GPs’ classification of headache: Is primary headache underdiagnosed? Br. J. Gen. Pract. J. R. Coll. Gen. Pract. 2008, 58, 102–104. [Google Scholar] [CrossRef] [Green Version]
- Loder, E. Triptan therapy in migraine. N. Engl. J. Med. 2010, 363, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, 3rd edition. Cephalalgia 2018, 38, 1–211. [Google Scholar] [CrossRef]
- Wolff, H.G. Headache and other head pain. In Headache and Other Head Pain; Oxford University Press: Oxford, UK, 2007; p. 648. [Google Scholar]
- Goadsby, P.J.; Lipton, R.B.; Ferrari, M.D. Migraine—Current understanding and treatment. N. Engl. J. Med. 2002, 346, 257–270. [Google Scholar] [CrossRef] [Green Version]
- Moulton, E.A.; Burstein, R.; Tully, S.; Hargreaves, R.; Becerra, L.; Borsook, D. Interictal dysfunction of a brainstem descending modulatory center in migraine patients. PLoS ONE 2008, 3, e3799. [Google Scholar] [CrossRef] [PubMed]
- Eftekhari, S.; Warfvinge, K.; Blixt, F.W.; Edvinsson, L. Differentiation of nerve fibers storing CGRP and CGRP receptors in the peripheral trigeminovascular system. J. Pain 2013, 14, 1289–1303. [Google Scholar] [CrossRef] [Green Version]
- Goadsby, P.J.; Edvinsson, L.; Ekman, R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann. Neurol 1990, 28, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Lassen, L.H.; Haderslev, P.A.; Jacobsen, V.B.; Iversen, H.K.; Sperling, B.; Olesen, J. CGRP may play a causative role in migraine. Cephalalgia 2002, 22, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Hadjikhani, N.; Sanchez Del Rio, M.; Wu, O.; Schwartz, D.; Bakker, D.; Fischl, B.; Kwong, K.K.; Cutrer, F.M.; Rosen, B.R.; Tootell, R.B.; et al. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc. Natl. Acad. Sci. USA 2001, 98, 4687–4692. [Google Scholar] [CrossRef] [Green Version]
- Leao, A.A.P. Spreading depression of activity in the cerebral cortex. J. Neurophysiol. 1944, 7, 359–390. [Google Scholar] [CrossRef]
- Sugaya, E.; Takato, M.; Noda, Y. Neuronal and glial activity during spreading depression in cerebral cortex of cat. J. Neurophysiol. 1975, 38, 822–841. [Google Scholar] [CrossRef]
- Kraig, R.P.; Nicholson, C. Extracellular ionic variations during spreading depression. Neuroscience 1978, 3, 1045–1059. [Google Scholar] [CrossRef]
- Strong, A.J.; Fabricius, M.; Boutelle, M.G.; Hibbins, S.J.; Hopwood, S.E.; Jones, R.; Parkin, M.C.; Lauritzen, M. Spreading and synchronous depressions of cortical activity in acutely injured human brain. Stroke 2002, 33, 2738–2743. [Google Scholar] [CrossRef] [Green Version]
- Dohmen, C.; Sakowitz, O.W.; Fabricius, M.; Bosche, B.; Reithmeier, T.; Ernestus, R.I.; Brinker, G.; Dreier, J.P.; Woitzik, J.; Strong, A.J.; et al. Spreading depolarizations occur in human ischemic stroke with high incidence. Ann. Neurol. 2008, 63, 720–728. [Google Scholar] [CrossRef]
- Tozzi, A.; de Iure, A.; Di Filippo, M.; Costa, C.; Caproni, S.; Pisani, A.; Bonsi, P.; Picconi, B.; Cupini, L.M.; Materazzi, S.; et al. Critical role of calcitonin gene-related peptide receptors in cortical spreading depression. Proc. Natl. Acad. Sci. USA 2012, 109, 18985–18990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moskowitz, M.A.; Nozaki, K.; Kraig, R.P. Neocortical spreading depression provokes the expression of c-fos protein-like immunoreactivity within trigeminal nucleus caudalis via trigeminovascular mechanisms. J. Neurosci. 1993, 13, 1167–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, T.W.; Mannix, L.K.; Fan, X.; Assaid, C.; Furtek, C.; Jones, C.J.; Lines, C.R.; Rapoport, A.M. Randomized controlled trial of an oral CGRP receptor antagonist, MK-0974, in acute treatment of migraine. Neurology 2008, 70, 1304. [Google Scholar] [CrossRef] [PubMed]
- Hewitt, D.J.; Aurora, S.K.; Dodick, D.W.; Goadsby, P.J.; Ge, Y.; Bachman, R.; Taraborelli, D.; Fan, X.; Assaid, C.; Lines, C.; et al. Randomized controlled trial of the CGRP receptor antagonist MK-3207 in the acute treatment of migraine. Cephalalgia 2011, 31, 712–722. [Google Scholar] [CrossRef]
- Olesen, J.; Diener, H.C.; Husstedt, I.W.; Goadsby, P.J.; Hall, D.; Meier, U.; Pollentier, S.; Lesko, L.M. Calcitonin gene-related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N. Engl. J. Med. 2004, 350, 1104–1110. [Google Scholar] [CrossRef] [Green Version]
- Dodick, D.W.; Goadsby, P.J.; Silberstein, S.D.; Lipton, R.B.; Olesen, J.; Ashina, M.; Wilks, K.; Kudrow, D.; Kroll, R.; Kohrman, B.; et al. Safety and efficacy of ALD403, an antibody to calcitonin gene-related peptide, for the prevention of frequent episodic migraine: A randomised, double-blind, placebo-controlled, exploratory phase 2 trial. Lancet Neurol. 2014, 13, 1100–1107. [Google Scholar] [CrossRef]
- Dodick, D.W.; Goadsby, P.J.; Spierings, E.L.H.; Scherer, J.C.; Sweeney, S.P.; Grayzel, D.S. Safety and efficacy of LY2951742, a monoclonal antibody to calcitonin gene-related peptide, for the prevention of migraine: A phase 2, randomised, double-blind, placebo-controlled study. Lancet Neurol. 2014, 13, 885–892. [Google Scholar] [CrossRef]
- Ashina, H.; Iljazi, A.; Amin, F.M.; Ashina, M.; Lipton, R.B.; Schytz, H.W. Interrelations between migraine-like headache and persistent post-traumatic headache attributed to mild traumatic brain injury: A prospective diary study. J. Headache Pain 2020, 21, 134. [Google Scholar] [CrossRef]
- Mollan, S.P.; Hoffmann, J.; Sinclair, A.J. Advances in the understanding of headache in idiopathic intracranial hypertension. Curr. Opin. Neurol. 2019, 32, 92–98. [Google Scholar] [CrossRef] [Green Version]
- Yiangou, A.; Mitchell, J.L.; Fisher, C.; Edwards, J.; Vijay, V.; Alimajstorovic, Z.; Grech, O.; Lavery, G.G.; Mollan, S.P.; Sinclair, A.J. Erenumab for headaches in idiopathic intracranial hypertension: A prospective open-label evaluation. Headache J. Head Face Pain 2021, 61, 157–169. [Google Scholar] [CrossRef]
- Yiangou, A.; Mitchell, J.L.; Vijay, V.; Grech, O.; Bilton, E.; Lavery, G.G.; Fisher, C.; Edwards, J.; Mollan, S.P.; Sinclair, A.J. Calcitonin gene related peptide monoclonal antibody treats headache in patients with active idiopathic intracranial hypertension. J. Headache Pain 2020, 21, 116. [Google Scholar] [CrossRef] [PubMed]
- Ashina, H.; Iljazi, A.; Al-Khazali, H.M.; Christensen, C.E.; Amin, F.M.; Ashina, M.; Schytz, H.W. Hypersensitivity to Calcitonin Gene-Related Peptide in Post-Traumatic Headache. Ann. Neurol. 2020, 88, 1220–1228. [Google Scholar] [CrossRef]
- Ashina, H.; Moskowitz, M.A. Shared biological foundations of post-traumatic headache and migraine. Headache J. Head Face Pain 2021. [Google Scholar] [CrossRef] [PubMed]
- Uncini, A.; Lodi, R.; Di Muzio, A.; Silvestri, G.; Servidei, S.; Lugaresi, A.; Iotti, S.; Zaniol, P.; Barbiroli, B. Abnormal brain and muscle energy metabolism shown by 31P-MRS in familial hemiplegic migraine. J. Neurol. Sci. 1995, 129, 214–222. [Google Scholar] [CrossRef]
- Bigal, M.E.; Bordini, C.A.; Tepper, S.J.; Speciali, J.G. Intravenous magnesium sulphate in the acute treatment of migraine without aura and migraine with aura. A randomized, double-blind, placebo-controlled study. Cephalalgia Int. J. Headache 2002, 22, 345–353. [Google Scholar] [CrossRef]
- Corbo, J.; Esses, D.; Bijur, P.E.; Iannaccone, R.; Gallagher, E.J. Randomized clinical trial of intravenous magnesium sulfate as an adjunctive medication for emergency department treatment of migraine headache. Ann. Emerg. Med. 2001, 38, 621–627. [Google Scholar] [CrossRef]
- Kemp, G.J. Non-invasive methods for studying brain energy metabolism: What they show and what it means. Dev. Neurosci. 2000, 22, 418–428. [Google Scholar] [CrossRef]
- Reyngoudt, H.; Achten, E.; Paemeleire, K. Magnetic resonance spectroscopy in migraine: What have we learned so far? Cephalalgia 2012, 32, 845–859. [Google Scholar] [CrossRef] [Green Version]
- Barbiroli, B.; Montagna, P.; Cortelli, P.; Funicello, R.; Iotti, S.; Monari, L.; Pierangeli, G.; Zaniol, P.; Lugaresi, E. Abnormal brain and muscle energy metabolism shown by 31P-magnetic resonance spectroscopy in patients affected by migraine with aura. Neurology 1992, 42, 1209. [Google Scholar] [CrossRef] [PubMed]
- Reyngoudt, H.; Paemeleire, K.; Descamps, B.; De Deene, Y.; Achten, E. 31P-MRS demonstrates a reduction in high-energy phosphates in the occipital lobe of migraine without aura patients. Cephalalgia Int. J. Headache 2011, 31, 1243–1253. [Google Scholar] [CrossRef] [Green Version]
- Welch, K.M.A.; Levine, S.R.; Andrea, G.; Schultz, L.R.; Helpern, J.A. Preliminary observations on brain energy metabolism in migraine studied by in vivo phosphorus 31 NMR spectroscopy. Neurology 1989, 39, 538. [Google Scholar] [CrossRef]
- Sacquegna, T.; Lodi, R.; De Carolis, P.; Tinuper, P.; Cortelli, P.; Zaniol, P.; Funicello, R.; Montagna, P.; Barbiroli, B. Brain energy metabolism studied by 31P-MR spectroscopy in a case of migraine with prolonged aura. Acta Neurol. Scand. 1992, 86, 376–380. [Google Scholar] [CrossRef]
- Schulz, U.G.; Blamire, A.M.; Corkill, R.G.; Davies, P.; Styles, P.; Rothwell, P.M. Association between cortical metabolite levels and clinical manifestations of migrainous aura: An MR-spectroscopy study. Brain 2007, 130, 3102–3110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montagna, P.; Cortelli, P.; Monari, L.; Pierangeli, G.; Parchi, P.; Lodi, R.; Iotti, S.; Frassineti, C.; Zaniol, P.; Lugaresi, E.; et al. 31P-magnetic resonance spectroscopy in migraine without aura. Neurology 1994, 44, 666–669. [Google Scholar] [CrossRef]
- Boska, M.D.; Welch, K.M.A.; Barker, P.B.; Nelson, J.A.; Schultz, L. Contrasts in cortical magnesium, phospholipid and energy metabolism between migraine syndromes. Neurology 2002, 58, 1227. [Google Scholar] [CrossRef] [PubMed]
- Barbiroli, B.; Montagna, P.; Martinelli, P.; Lodi, R.; Iotti, S.; Cortelli, P.; Funicello, R.; Zaniol, P. Defective brain energy metabolism shown by in vivo 31P MR spectroscopy in 28 patients with mitochondrial cytopathies. J. Cereb. Blood Flow Metab. 1993, 13, 469–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mody, I.; Lambert, J.D.; Heinemann, U. Low extracellular magnesium induces epileptiform activity and spreading depression in rat hippocampal slices. J. Neurophysiol. 1987, 57, 869–888. [Google Scholar] [CrossRef]
- Laurant, P.; Touyz, R.M.; Schiffrin, E.L. Effect of magnesium on vascular tone and reactivity in pressurized mesenteric resistance arteries from spontaneously hypertensive rats. Can. J. Physiol. Pharm. 1997, 75, 293–300. [Google Scholar] [CrossRef]
- Kuno, M.; Takahashi, T. Effects of calcium and magnesium on transmitter release at Ia synapses of rat spinal motoneurones in vitro. J. Physiol. 1986, 376, 543–553. [Google Scholar] [CrossRef] [Green Version]
- Lodi, R.; Lotti, S.; Cortelli, P.; Pierangeli, G.; Cevoli, S.; Clementi, V.; Soriani, S.; Montagna, P.; Barbiroli, B. Deficient energy metabolism is associated with low free magnesium in the brains of patients with migraine and cluster headache. Brain Res. Bull. 2001, 54, 437–441. [Google Scholar] [CrossRef]
- Ramadan, N.M.; Halvorson, H.; Vande-Linde, A.; Levine, S.R.; Helpern, J.A.; Welch, K.M. Low brain magnesium in migraine. Headache 1989, 29, 590–593. [Google Scholar] [CrossRef] [PubMed]
- Talebi, M.; Savadi-Oskouei, D.; Farhoudi, M.; Mohammadzade, S.; Ghaemmaghamihezaveh, S.; Hasani, A.; Hamdi, A. Relation between serum magnesium level and migraine attacks. Neurosciences 2011, 16, 320–323. [Google Scholar] [PubMed]
- Sarchielli, P.; Coata, G.; Firenze, C.; Morucci, P.; Abbritti, G.; Gallai, V. Serum and Salivary Magnesium Levels in Migraine and Tension-Type Headache. Results in a Group of Adult Patients. Cephalalgia 1992, 12, 21–27. [Google Scholar] [CrossRef]
- Mauskop, A.; Altura, B.T.; Cracco, R.Q.; Altura, B.M. Chronic daily headache—One disease or two? Diagnostic role of serum ionized magnesium. Cephalalgia 1994, 14, 24–28. [Google Scholar] [CrossRef]
- Peikert, A.; Wilimzig, C.; Köhne-Volland, R. Prophylaxis of Migraine with Oral Magnesium: Results From A Prospective, Multi-Center, Placebo-Controlled and Double-Blind Randomized Study. Cephalalgia 1996, 16, 257–263. [Google Scholar] [CrossRef]
- Köseoglu, E.; Talaslioglu, A.; Gönül, A.S.; Kula, M. The effects of magnesium prophylaxis in migraine without aura. Magnes Res. 2008, 21, 101–108. [Google Scholar] [PubMed]
- Pfaffenrath, V.; Wessely, P.; Meyer, C.; Isler, H.R.; Evers, S.; Grotemeyer, K.H.; Taneri, Z.; Soyka, D.; Göbel, H.; Fischer, M. Magnesium in the prophylaxis of migraine—A double-blind placebo-controlled study. Cephalalgia 1996, 16, 436–440. [Google Scholar] [CrossRef]
- Demirkaya, S.; Vural, O.; Dora, B.; Topçuoğlu, M.A. Efficacy of intravenous magnesium sulfate in the treatment of acute migraine attacks. Headache 2001, 41, 171–177. [Google Scholar] [CrossRef]
- Mauskop, A.; Altura, B.T.; Cracco, R.Q.; Altura, B.M. Intravenous magnesium sulphate relieves migraine attacks in patients with low serum ionized magnesium levels: A pilot study. Clin. Sci. 1995, 89, 633–636. [Google Scholar] [CrossRef] [Green Version]
- Frank, L.R.; Olson, C.M.; Shuler, K.B.; Gharib, S.F. Intravenous magnesium for acute benign headache in the emergency department: A randomized double-blind placebo-controlled trial. Can. J. Emerg. Med. 2004, 6, 327–332. [Google Scholar] [CrossRef] [Green Version]
- Kelman, L. The Triggers or Precipitants of the Acute Migraine Attack. Cephalalgia 2007, 27, 394–402. [Google Scholar] [CrossRef]
- Nadelson, C. Sport and exercise-induced migraines. Curr. Sports Med. Rep. 2006, 5, 29–33. [Google Scholar] [CrossRef]
- Rainero, I.; Limone, P.; Ferrero, M.; Valfrè, W.; Pelissetto, C.; Rubino, E.; Gentile, S.; Lo Giudice, R.; Pinessi, L. Insulin Sensitivity is Impaired in Patients with Migraine. Cephalalgia 2005, 25, 593–597. [Google Scholar] [CrossRef]
- Cavestro, C.; Rosatello, A.; Micca, G.; Ravotto, M.; Pia Marino, M.; Asteggiano, G.; Beghi, E. Insulin Metabolism is Altered in Migraineurs: A New Pathogenic Mechanism for Migraine? Headache J. Head Face Pain 2007, 47, 1436–1442. [Google Scholar] [CrossRef]
- Shaw, S.W.J.; Johnson, R.H.; Keogh, H.J. Metabolic changes during glucose tolerance tests in migraine attacks. J. Neurol. Sci. 1977, 33, 51–59. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, S.; Suh, S.I.; Koh, S.B.; Park, K.W.; Oh, K. Interictal metabolic changes in episodic migraine: A voxel-based FDG-PET study. Cephalalgia 2010, 30, 53–61. [Google Scholar] [CrossRef]
- Fumal, A.; Laureys, S.; Di Clemente, L.; Boly, M.; Bohotin, V.; Vandenheede, M.; Coppola, G.; Salmon, E.; Kupers, R.; Schoenen, J. Orbitofrontal cortex involvement in chronic analgesic-overuse headache evolving from episodic migraine. Brain 2006, 129, 543–550. [Google Scholar] [CrossRef]
- Magis, D.; D’Ostilio, K.; Thibaut, A.; De Pasqua, V.; Gerard, P.; Hustinx, R.; Laureys, S.; Schoenen, J. Cerebral metabolism before and after external trigeminal nerve stimulation in episodic migraine. Cephalalgia 2017, 37, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Edmond, J.; Robbins, R.A.; Bergstrom, J.D.; Cole, R.A.; de Vellis, J. Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. J. Neurosci. Res. 1987, 18, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Barbanti, P.; Fofi, L.; Aurilia, C.; Egeo, G.; Caprio, M. Ketogenic diet in migraine: Rationale, findings and perspectives. Neurol. Sci. 2017, 38, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Sirven, J.; Whedon, B.; Caplan, D.; Liporace, J.; Glosser, D.; O’Dwyer, J.; Sperling, M.R. The Ketogenic Diet for Intractable Epilepsy in Adults: Preliminary Results. Epilepsia 1999, 40, 1721–1726. [Google Scholar] [CrossRef] [PubMed]
- Strahlman, R.S. Can Ketosis Help Migraine Sufferers? A Case Report. Headache J. Head Face Pain 2006, 46, 182. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, C.; Currà, A.; Sirianni, G.; Coppola, G.; Bracaglia, M.; Cardillo, A.; De Nardis, L.; Pierelli, F. Diet transiently improves migraine in two twin sisters: Possible role of ketogenesis? Funct. Neurol. 2013, 28, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, C.; Coppola, G.; Sirianni, G.; Di Lorenzo, G.; Bracaglia, M.; Di Lenola, D.; Siracusano, A.; Rossi, P.; Pierelli, F. Migraine improvement during short lasting ketogenesis: A proof-of-concept study. Eur. J. Neurol. 2015, 22, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Di Lorenzo, C.; Coppola, G.; Bracaglia, M.; Di Lenola, D.; Evangelista, M.; Sirianni, G.; Rossi, P.; Di Lorenzo, G.; Serrao, M.; Parisi, V.; et al. Cortical functional correlates of responsiveness to short-lasting preventive intervention with ketogenic diet in migraine: A multimodal evoked potentials study. J. Headache Pain 2016, 17, 58. [Google Scholar] [CrossRef] [Green Version]
- Maggioni, F.; Margoni, M.; Zanchin, G. Ketogenic diet in migraine treatment: A brief but ancient history. Cephalalgia 2011, 31, 1150–1151. [Google Scholar] [CrossRef]
- Kossoff, E.H.; Huffman, J.; Turner, Z.; Gladstein, J. Use of the modified Atkins diet for adolescents with chronic daily headache. Cephalalgia 2010, 30, 1014–1016. [Google Scholar] [CrossRef]
- Arngrim, N.; Schytz, H.W.; Britze, J.; Amin, F.M.; Vestergaard, M.B.; Hougaard, A.; Wolfram, F.; de Koning, P.J.H.; Olsen, K.S.; Secher, N.H.; et al. Migraine induced by hypoxia: An MRI spectroscopy and angiography study. Brain 2016, 139, 723–737. [Google Scholar] [CrossRef] [Green Version]
- Amery, W.K. Brain hypoxia: The turning-point in the genesis of the migraine attack? Cephalalgia 1982, 2, 83–109. [Google Scholar] [CrossRef]
- Grimaldi, D.; Tonon, C.; Cevoli, S.; Pierangeli, G.; Malucelli, E.; Rizzo, G.; Soriani, S.; Montagna, P.; Barbiroli, B.; Lodi, R.; et al. Clinical and neuroimaging evidence of interictal cerebellar dysfunction in FHM2. Cephalalgia 2010, 30, 552–559. [Google Scholar] [CrossRef]
- Sándor, P.S.; Dydak, U.; Schoenen, J.; Kollias, S.S.; Hess, K.; Boesiger, P.; Agosti, R.M. MR-spectroscopic imaging during visual stimulation in subgroups of migraine with aura. Cephalalgia 2005, 25, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Kuwabara, T.; Ohkubo, M.; Tsuji, S.; Yuasa, T. Elevation of cerebral lactate detected by localized 1H-magnetic resonance spectroscopy in migraine during the interictal period. Neurology 1996, 47, 1093–1095. [Google Scholar] [CrossRef] [PubMed]
- Reyngoudt, H.; Paemeleire, K.; Dierickx, A.; Descamps, B.; Vandemaele, P.; De Deene, Y.; Achten, E. Does visual cortex lactate increase following photic stimulation in migraine without aura patients? A functional 1H-MRS study. J. Headache Pain 2011, 12, 295–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, E.; Sanchez-Porras, R.; Dohmen, C.; Hertle, D.; Unterberg, A.W.; Sakowitz, O.W. Spreading depolarizations in a case of migraine-related stroke. Cephalalgia 2012, 32, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Proia, P.; Amato, A.; Contrò, V.; Monaco, A.L.; Brusa, J.; Brighina, F.; Messina, G. Relevance of lactate level detection in migrane and fibromyalgia. Eur. J. Transl. Myol. 2019, 29, 8202. [Google Scholar] [CrossRef] [Green Version]
- Yavuz Altunkaynak, M.Ö. Devrimsel Harika Ertem, Betül Güveli, Filiz Uzun Okay, Zerrin Yıldırım, Belgin Mutluay, Ayten Ceyhan Dirican, Emine Altunkaynak, Ayhan Köksal, Sevim Baybaş. Serum lactic acid and pyruvic acid levels in patients with migraine and tension type headache. Dusunen Adam J. Psychiatry Neurol. Sci. 2013, 26, 276–280. [Google Scholar]
- Okada, H.; Araga, S.; Takeshima, T.; Nakashima, K. Plasma lactic acid and pyruvic acid levels in migraine and tension-type headache. Headache 1998, 38, 39–42. [Google Scholar] [CrossRef]
- Welch, K.M.; Levine, S.R.; D’Andrea, G.; Helpern, J.A. Brain pH in migraine: An in vivo phosphorus-31 magnetic resonance spectroscopy study. Cephalalgia 1988, 8, 273–277. [Google Scholar] [CrossRef]
- Abe, K.; Fujimura, H.; Nishikawa, Y.; Yorifuji, S.; Mezaki, T.; Hirono, N.; Nishitani, N.; Kameyama, M. Marked reduction in CSF lactate and pyruvate levels after CoQ therapy in a patient with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS). Acta Neurol. Scand. 1991, 83, 356–359. [Google Scholar] [CrossRef]
- Barbiroli, B.; Iotti, S.; Lodi, R. Improved brain and muscle mitochondrial respiration with CoQ. An in vivo study by ^{31}P-MR spectroscopy in patients with mitochondrial cytopathies. Biofactors 1999, 9, 253–260. [Google Scholar] [CrossRef]
- Sándor, P.S.; Di Clemente, L.; Coppola, G.; Saenger, U.; Fumal, A.; Magis, D.; Seidel, L.; Agosti, R.M.; Schoenen, J. Efficacy of coenzyme Q10 in migraine prophylaxis: A randomized controlled trial. Neurology 2005, 64, 713. [Google Scholar] [CrossRef] [PubMed]
- Shoeibi, A.; Olfati, N.; Soltani Sabi, M.; Salehi, M.; Mali, S.; Akbari Oryani, M. Effectiveness of coenzyme Q10 in prophylactic treatment of migraine headache: An open-label, add-on, controlled trial. Acta Neurol. Belg. 2017, 117, 103–109. [Google Scholar] [CrossRef]
- Rozen, T.D.; Oshinsky, M.L.; Gebeline, C.A.; Bradley, K.C.; Young, W.B.; Shechter, A.L.; Silberstein, S.D. Open Label Trial of Coenzyme Q10 as A Migraine Preventive. Cephalalgia 2002, 22, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Dahri, M.; Tarighat-Esfanjani, A.; Asghari-Jafarabadi, M.; Hashemilar, M. Oral coenzyme Q10 supplementation in patients with migraine: Effects on clinical features and inflammatory markers. Nutr. Neurosci. 2019, 22, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Hershey, A.D.; Powers, S.W.; Vockell, A.L.; Lecates, S.L.; Ellinor, P.L.; Segers, A.; Burdine, D.; Manning, P.; Kabbouche, M.A. Coenzyme Q10 deficiency and response to supplementation in pediatric and adolescent migraine. Headache 2007, 47, 73–80. [Google Scholar] [CrossRef]
- Arts, W.F.; Scholte, H.R.; Bogaard, J.M.; Kerrebijn, K.F.; Luyt-Houwen, I.E. NADH-CoQ reductase deficient myopathy: Successful treatment with riboflavin. Lancet 1983, 2, 581–582. [Google Scholar] [CrossRef]
- Boehnke, C.; Reuter, U.; Flach, U.; Schuh-Hofer, S.; Einhaupl, K.M.; Arnold, G. High-dose riboflavin treatment is efficacious in migraine prophylaxis: An open study in a tertiary care centre. Eur. J. Neurol. 2004, 11, 475–477. [Google Scholar] [CrossRef]
- Schoenen, J.; Jacquy, J.; Lenaerts, M. Effectiveness of high-dose riboflavin in migraine prophylaxis. A randomized controlled trial. Neurology 1998, 50, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Thys-Jacobs, S. Alleviation of migraines with therapeutic vitamin D and calcium. Headache 1994, 34, 590–592. [Google Scholar] [CrossRef]
- Togha, M.; Razeghi Jahromi, S.; Ghorbani, Z.; Martami, F.; Seifishahpar, M. Serum Vitamin D Status in a Group of Migraine Patients Compared With Healthy Controls: A Case–Control Study. Headache J. Head Face Pain 2018, 58, 1530–1540. [Google Scholar] [CrossRef]
- Mottaghi, T.; Khorvash, F.; Askari, G.; Maracy, M.R.; Ghiasvand, R.; Maghsoudi, Z.; Iraj, B. The relationship between serum levels of vitamin D and migraine. J. Res. Med. Sci. Off. J. Isfahan Univ. Med. Sci. 2013, 18, S66–S70. [Google Scholar]
- Song, T.J.; Chu, M.K.; Sohn, J.H.; Ahn, H.Y.; Lee, S.H.; Cho, S.J. Effect of Vitamin D Deficiency on the Frequency of Headaches in Migraine. J. Clin. Neurol. 2018, 14, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Hussein, M.; Fathy, W.; Abd Elkareem, R.M. The potential role of serum vitamin D level in migraine headache: A case-control study. J. Pain Res. 2019, 12, 2529–2536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mottaghi, T.; Askari, G.; Khorvash, F.; Maracy, M.R. Effect of Vitamin D supplementation on symptoms and C-reactive protein in migraine patients. J. Res. Med. Sci. Off. J. Isfahan Univ. Med. Sci. 2015, 20, 477–482. [Google Scholar] [CrossRef]
- Ghorbani, Z.; Rafiee, P.; Fotouhi, A.; Haghighi, S.; Rasekh Magham, R.; Ahmadi, Z.S.; Djalali, M.; Zareei, M.; Razeghi Jahromi, S.; Shahemi, S.; et al. The effects of vitamin D supplementation on interictal serum levels of calcitonin gene-related peptide (CGRP) in episodic migraine patients: Post hoc analysis of a randomized double-blind placebo-controlled trial. J. Headache Pain 2020, 21, 22. [Google Scholar] [CrossRef] [Green Version]
- Zandifar, A.; Masjedi, S.s.; Banihashemi, M.; Asgari, F.; Manouchehri, N.; Ebrahimi, H.; Haghdoost, F.; Saadatnia, M. Vitamin D Status in Migraine Patients: A Case-Control Study. Biomed. Res. Int. 2014, 2014, 514782. [Google Scholar] [CrossRef]
- Costa, B.; Comelli, F.; Bettoni, I.; Colleoni, M.; Giagnoni, G. The endogenous fatty acid amide, palmitoylethanolamide, has anti-allodynic and anti-hyperalgesic effects in a murine model of neuropathic pain: Involvement of CB1, TRPV1 and PPARγ receptors and neurotrophic factors. Pain 2008, 139, 541–550. [Google Scholar] [CrossRef]
- Mazzari, S.; Canella, R.; Petrelli, L.; Marcolongo, G.; Leon, A. N-(2-Hydroxyethyl)hexadecanamide is orally active in reducing edema formation and inflammatory hyperalgesia by down-modulating mast cell activation. Eur. J. Pharmacol. 1996, 300, 227–236. [Google Scholar] [CrossRef]
- Lo Verme, J.; Fu, J.; Astarita, G.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The Nuclear Receptor Peroxisome Proliferator-Activated Receptor-α Mediates the Anti-Inflammatory Actions of Palmitoylethanolamide. Mol. Pharmacol. 2005, 67, 15. [Google Scholar] [CrossRef]
- Minnich, A.; Tian, N.; Byan, L.; Bilder, G. A potent PPARα agonist stimulates mitochondrial fatty acid β-oxidation in liver and skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E270–E279. [Google Scholar] [CrossRef] [Green Version]
- Annunziata, C.; Lama, A.; Pirozzi, C.; Cavaliere, G.; Trinchese, G.; Di Guida, F.; Nitrato Izzo, A.; Cimmino, F.; Paciello, O.; De Biase, D.; et al. Palmitoylethanolamide counteracts hepatic metabolic inflexibility modulating mitochondrial function and efficiency in diet-induced obese mice. FASEB J. 2020, 34, 350–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Boronat, M.; Isorna, E.; Conde-Sieira, M.; Delgado, M.J.; Soengas, J.L.; de Pedro, N. First evidence on the role of palmitoylethanolamide in energy homeostasis in fish. Horm. Behav. 2020, 117, 104609. [Google Scholar] [CrossRef]
- Hesselink, J.M.K. Chronic idiopathic axonal neuropathy and pain, treated with the endogenous lipid mediator palmitoylethanolamide: A case collection. Int. Med. Case Rep. J. 2013, 6, 49–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chirchiglia, D.; Cione, E.; Caroleo, M.C.; Wang, M.; Di Mizio, G.; Faedda, N.; Giacolini, T.; Siviglia, S.; Guidetti, V.; Gallelli, L. Effects of Add-On Ultramicronized N-Palmitol Ethanol Amide in Patients Suffering of Migraine With Aura: A Pilot Study. Front. Neurol. 2018, 9, 674. [Google Scholar] [CrossRef] [PubMed]
- Papetti, L.; Sforza, G.; Tullo, G.; Alaimo di Loro, P.; Moavero, R.; Ursitti, F.; Ferilli, M.A.N.; Tarantino, S.; Vigevano, F.; Valeriani, M. Tolerability of Palmitoylethanolamide in a Pediatric Population Suffering from Migraine: A Pilot Study. Pain Res. Manag. 2020, 2020, 3938640. [Google Scholar] [CrossRef] [Green Version]
- Andreou, A.P.; Summ, O.; Charbit, A.R.; Romero-Reyes, M.; Goadsby, P.J. Animal models of headache: From bedside to bench and back to bedside. Expert Rev. Neurother. 2010, 10, 389–411. [Google Scholar] [CrossRef] [PubMed]
- Bergerot, A.; Holland, P.R.; Akerman, S.; Bartsch, T.; Ahn, A.H.; MaassenVanDenBrink, A.; Reuter, U.; Tassorelli, C.; Schoenen, J.; Mitsikostas, D.D.; et al. Animal models of migraine: Looking at the component parts of a complex disorder. Eur. J. Neurosci. 2006, 24, 1517–1534. [Google Scholar] [CrossRef]
- van den Maagdenberg, A.M.; Pietrobon, D.; Pizzorusso, T.; Kaja, S.; Broos, L.A.; Cesetti, T.; van de Ven, R.C.; Tottene, A.; van der Kaa, J.; Plomp, J.J.; et al. A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron 2004, 41, 701–710. [Google Scholar] [CrossRef]
- Leo, L.; Gherardini, L.; Barone, V.; De Fusco, M.; Pietrobon, D.; Pizzorusso, T.; Casari, G. Increased Susceptibility to Cortical Spreading Depression in the Mouse Model of Familial Hemiplegic Migraine Type 2. PLoS Genet. 2011, 7, e1002129. [Google Scholar] [CrossRef] [Green Version]
- Noseda, R.; Burstein, R. Migraine pathophysiology: Anatomy of the trigeminovascular pathway and associated neurological symptoms, CSD, sensitization and modulation of pain. Pain 2013, 13, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Yao, G.; Huang, Q.; Wang, M.; Yang, C.L.; Liu, C.F.; Yu, T.M. Behavioral study of a rat model of migraine induced by CGRP. Neurosci. Lett. 2017, 651, 134–139. [Google Scholar] [CrossRef]
- Vergen, J.; Hecht, C.; Zholudeva, L.V.; Marquardt, M.M.; Hallworth, R.; Nichols, M.G. Metabolic imaging using two-photon excited NADH intensity and fluorescence lifetime imaging. Microsc. Microanal. 2012, 18, 761–770. [Google Scholar] [CrossRef] [Green Version]
- Kasischke, K.A.; Lambert, E.M.; Panepento, B.; Sun, A.; Gelbard, H.A.; Burgess, R.W.; Foster, T.H.; Nedergaard, M. Two-photon NADH imaging exposes boundaries of oxygen diffusion in cortical vascular supply regions. J. Cereb. Blood Flow Metab. 2011, 31, 68–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galeffi, F.; Somjen, G.G.; Foster, K.A.; Turner, D.A. Simultaneous monitoring of tissue PO2 and NADH fluorescence during synaptic stimulation and spreading depression reveals a transient dissociation between oxygen utilization and mitochondrial redox state in rat hippocampal slices. J. Cereb. Blood Flow Metab. 2011, 31, 626–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takano, T.; Tian, G.F.; Peng, W.; Lou, N.; Lovatt, D.; Hansen, A.J.; Kasischke, K.A.; Nedergaard, M. Cortical spreading depression causes and coincides with tissue hypoxia. Nat. Neurosci. 2007, 10, 754–762. [Google Scholar] [CrossRef]
- Carlson, A.P.; Carter, R.E.; Shuttleworth, C.W. Vascular, electrophysiological, and metabolic consequences of cortical spreading depression in a mouse model of simulated neurosurgical conditions. Neurol. Res. 2012, 34, 223–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauritzen, M.; Jøsrgensen, M.B.; Diemer, N.H.; Gjedde, A.; Hansen, A.J. Persistent oligemia of rat cerebral cortex in the wake of spreading depression. Ann. Neurol. 1982, 12, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Fabricius, M.; Lauritzen, M. Transient hyperemia succeeds oligemia in the wake of cortical spreading depression. Brain Res. 1993, 602, 350–353. [Google Scholar] [CrossRef]
- Chang, J.C.; Shook, L.L.; Biag, J.; Nguyen, E.N.; Toga, A.W.; Charles, A.C.; Brennan, K.C. Biphasic direct current shift, haemoglobin desaturation and neurovascular uncoupling in cortical spreading depression. Brain 2010, 133, 996–1012. [Google Scholar] [CrossRef]
- Sonn, J.; Mayevsky, A. Responses to Cortical Spreading Depression under Oxygen Deficiency. Open Neurol. J. 2012, 6, 6–17. [Google Scholar] [CrossRef] [Green Version]
- Khennouf, L.; Gesslein, B.; Lind, B.L.; van den Maagdenberg, A.M.J.M.; Lauritzen, M. Activity-dependent calcium, oxygen, and vascular responses in a mouse model of familial hemiplegic migraine type 1. Ann. Neurol. 2016, 80, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, U.; Sukhotinsky, I.; Eikermann-Haerter, K.; Ayata, C. Glucose modulation of spreading depression susceptibility. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2013, 33, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Bures, J.; Buresova, O. Activation of latent foci of spreading cortical depression in rats. J. Neurophysiol. 1960, 23, 225–236. [Google Scholar] [CrossRef]
- Costa-Cruz, R.R.G.; Guedes, R.C.A. Cortical spreading depression during streptozotocin-induced hyperglycaemia in nutritionally normal and early-malnourished rats. Neurosci. Lett. 2001, 303, 177–180. [Google Scholar] [CrossRef]
- Astrup, J.; Norberg, K. Potassium activity in cerebral cortex in rats during progressive severe hypoglycemia. Brain Res. 1976, 103, 418–423. [Google Scholar] [CrossRef]
- Sprenger, T.; Ruether, K.V.; Boecker, H.; Valet, M.; Berthele, A.; Pfaffenrath, V.; Wöller, A.; Tölle, T.R. Altered Metabolism in Frontal Brain Circuits in Cluster Headache. Cephalalgia 2007, 27, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Feuerstein, D.; Backes, H.; Gramer, M.; Takagaki, M.; Gabel, P.; Kumagai, T.; Graf, R. Regulation of cerebral metabolism during cortical spreading depression. J. Cereb. Blood Flow Metab. 2016, 36, 1965–1977. [Google Scholar] [CrossRef]
- Csiba, L.; Paschen, W.; Mies, G. Regional changes in tissue pH and glucose content during cortical spreading depression in rat brain. Brain Res. 1985, 336, 167–170. [Google Scholar] [CrossRef]
- Scheller, D.; Kolb, J.; Tegtmeier, F. Lactate and pH change in close correlation in the extracellular space of the rat brain during cortical spreading depression. Neurosci. Lett. 1992, 135, 83–86. [Google Scholar] [CrossRef]
- Berthet, C.; Lei, H.; Thevenet, J.; Gruetter, R.; Magistretti, P.J.; Hirt, L. Neuroprotective role of lactate after cerebral ischemia. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2009, 29, 1780–1789. [Google Scholar] [CrossRef]
- Fried, N.T.; Moffat, C.; Seifert, E.L.; Oshinsky, M.L. Functional mitochondrial analysis in acute brain sections from adult rats reveals mitochondrial dysfunction in a rat model of migraine. Am. J. Physiol. Cell Physiol. 2014, 307, C1017–C1030. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Qiu, E.; Dong, Z.; Liu, R.; Wu, S.; Yu, S. Protection of flunarizine on cerebral mitochondria injury induced by cortical spreading depression under hypoxic conditions. J. Headache Pain 2011, 12, 47–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, X.; Guan, X.; Chen, K.; Jin, S.; Wang, C.; Yan, L.; Shi, Z.; Zhang, X.; Chen, L.; Wan, Q. Abnormal mitochondrial dynamics and impaired mitochondrial biogenesis in trigeminal ganglion neurons in a rat model of migraine. Neurosci. Lett. 2017, 636, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Bereiter, D.A.; Bereiter, D.F.; Hathaway, C.B. The NMDA receptor antagonist MK-801 reduces Fos-like immunoreactivity in central trigeminal neurons and blocks select endocrine and autonomic responses to corneal stimulation in the rat. Pain 1996, 64, 179–189. [Google Scholar] [CrossRef]
- Nowak, L.; Bregestovski, P.; Ascher, P.; Herbet, A.; Prochiantz, A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature 1984, 307, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Classey, J.D.; Knight, Y.E.; Goadsby, P.J. The NMDA receptor antagonist MK-801 reduces Fos-like immunoreactivity within the trigeminocervical complex following superior sagittal sinus stimulation in the cat. Brain Res. 2001, 907, 117–124. [Google Scholar] [CrossRef]
- van der Hel, W.S.; van den Bergh, W.M.; Nicolay, K.; Tulleken, K.A.; Dijkhuizen, R.M. Suppression of cortical spreading depressions after magnesium treatment in the rat. Neuroreport 1998, 9, 2179–2182. [Google Scholar] [CrossRef]
- Kass, I.S.; Cottrell, J.E.; Chambers, G. Magnesium and cobalt, not nimodipine, protect neurons against anoxic damage in the rat hippocampal slice. Anesthesiology 1988, 69, 710–715. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grech, O.; Mollan, S.P.; Wakerley, B.R.; Fulton, D.; Lavery, G.G.; Sinclair, A.J. The Role of Metabolism in Migraine Pathophysiology and Susceptibility. Life 2021, 11, 415. https://doi.org/10.3390/life11050415
Grech O, Mollan SP, Wakerley BR, Fulton D, Lavery GG, Sinclair AJ. The Role of Metabolism in Migraine Pathophysiology and Susceptibility. Life. 2021; 11(5):415. https://doi.org/10.3390/life11050415
Chicago/Turabian StyleGrech, Olivia, Susan P. Mollan, Benjamin R. Wakerley, Daniel Fulton, Gareth G. Lavery, and Alexandra J. Sinclair. 2021. "The Role of Metabolism in Migraine Pathophysiology and Susceptibility" Life 11, no. 5: 415. https://doi.org/10.3390/life11050415