
Evaluation of In Vitro Neuronal Protection by Postconditioning with Poloxamer 188 Following Simulated Traumatic Brain Injury


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- (1)
- Establishing a TBI model using compression and H/R to simulate I/R injury that leads to a moderate damage, still allowing P188 to attenuate cellular injury.
- (2)
- Determining a protective effect of P188 when administered at the start of reoxygenation and using the completely hydrophilic polymer PEG as an osmotic control to elucidate if any P188 effect would be based on its amphiphilic character.
2. Materials and Methods
2.1. Cell Culture
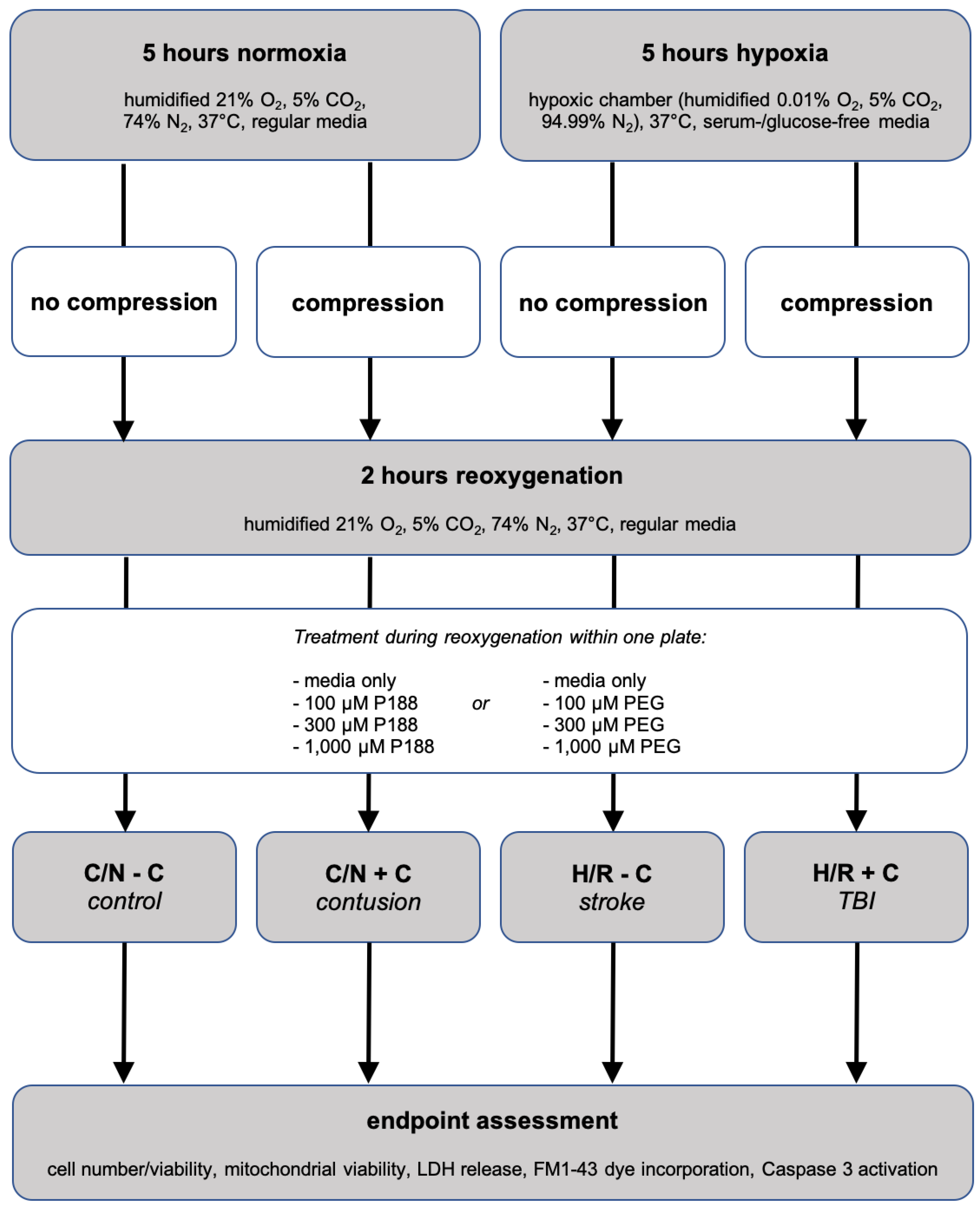
2.2. In Vitro Simulation of Traumatic Brain Injury
2.3. Model Development via Optimization of H/R ± Compression Duration
2.4. Treatment with P188 and PEG
2.5. Endpoint Assessments
2.6. Cellular Morphology Assessment
2.7. Statistics
3. Results
3.1. Model Development
3.2. Changes in Neuronal Morphology after Injury
3.3. Effects of P188 on Simulated Traumatic Brain Injury in Neurons
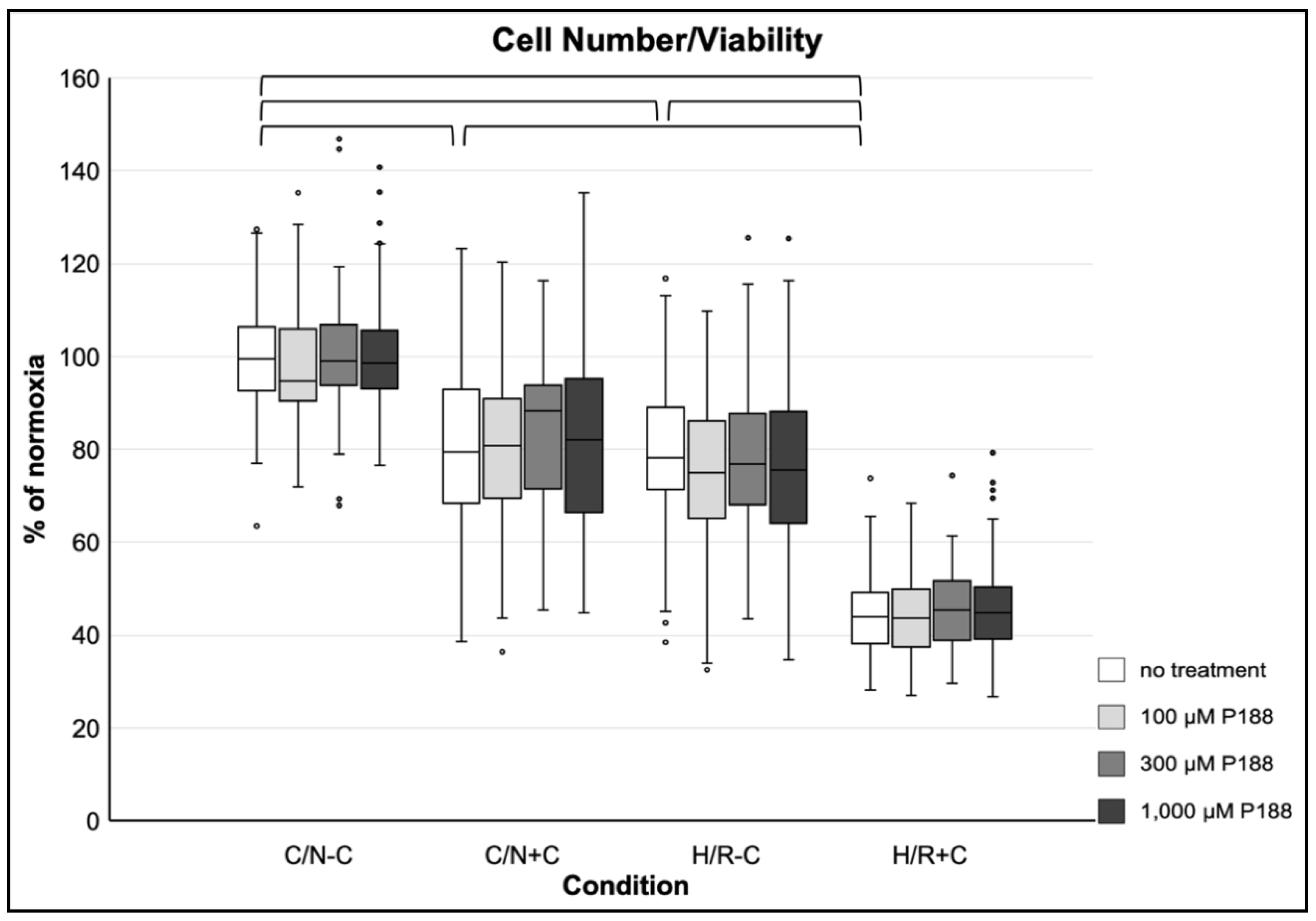
3.3.1. Cell Number and Viability
3.3.2. Mitochondrial Viability
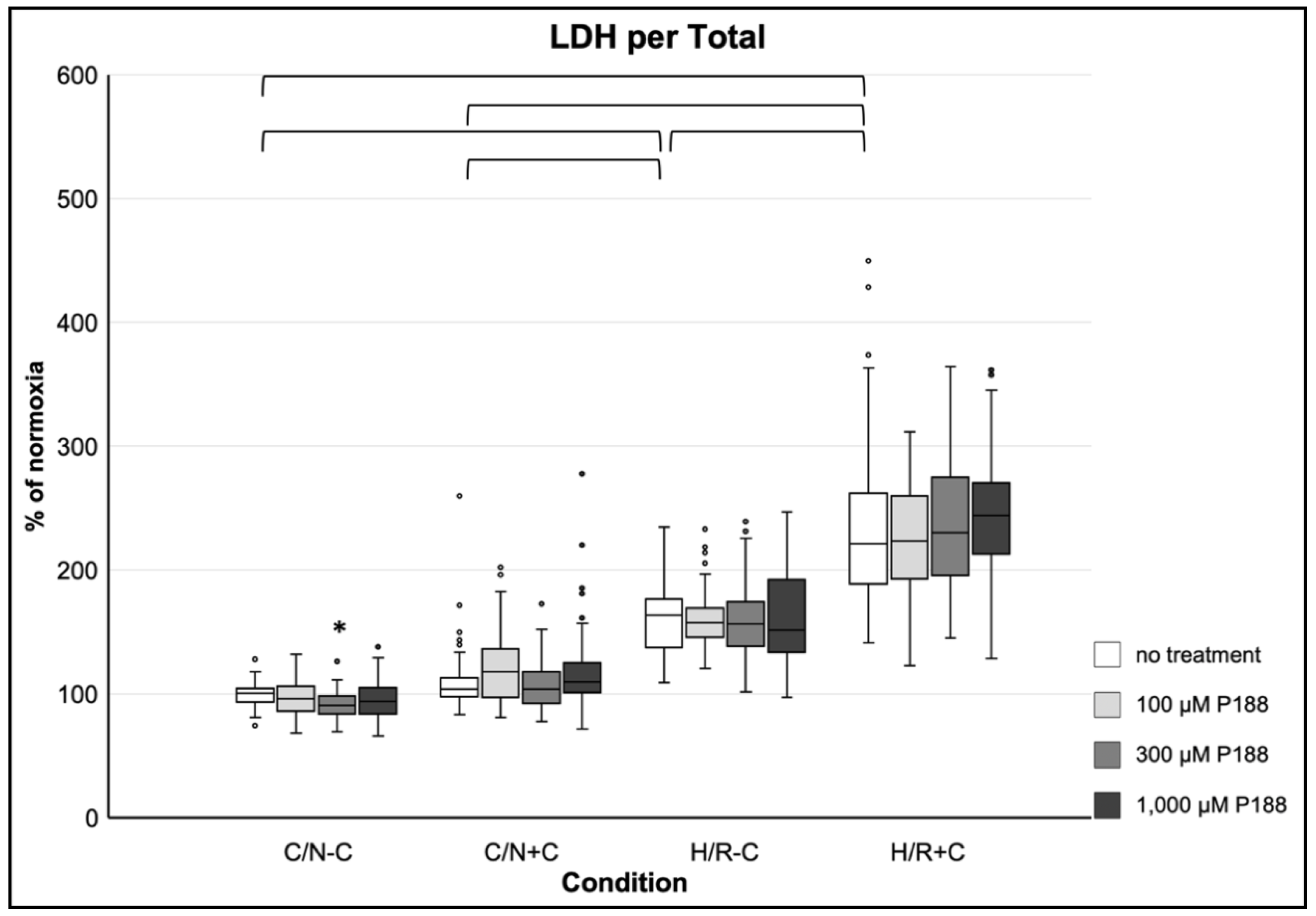
3.3.3. Membrane Damage as Assessed by LDH Release per Total
3.3.4. Membrane Damage as Assessed by FM1-43 Dye Incorporation
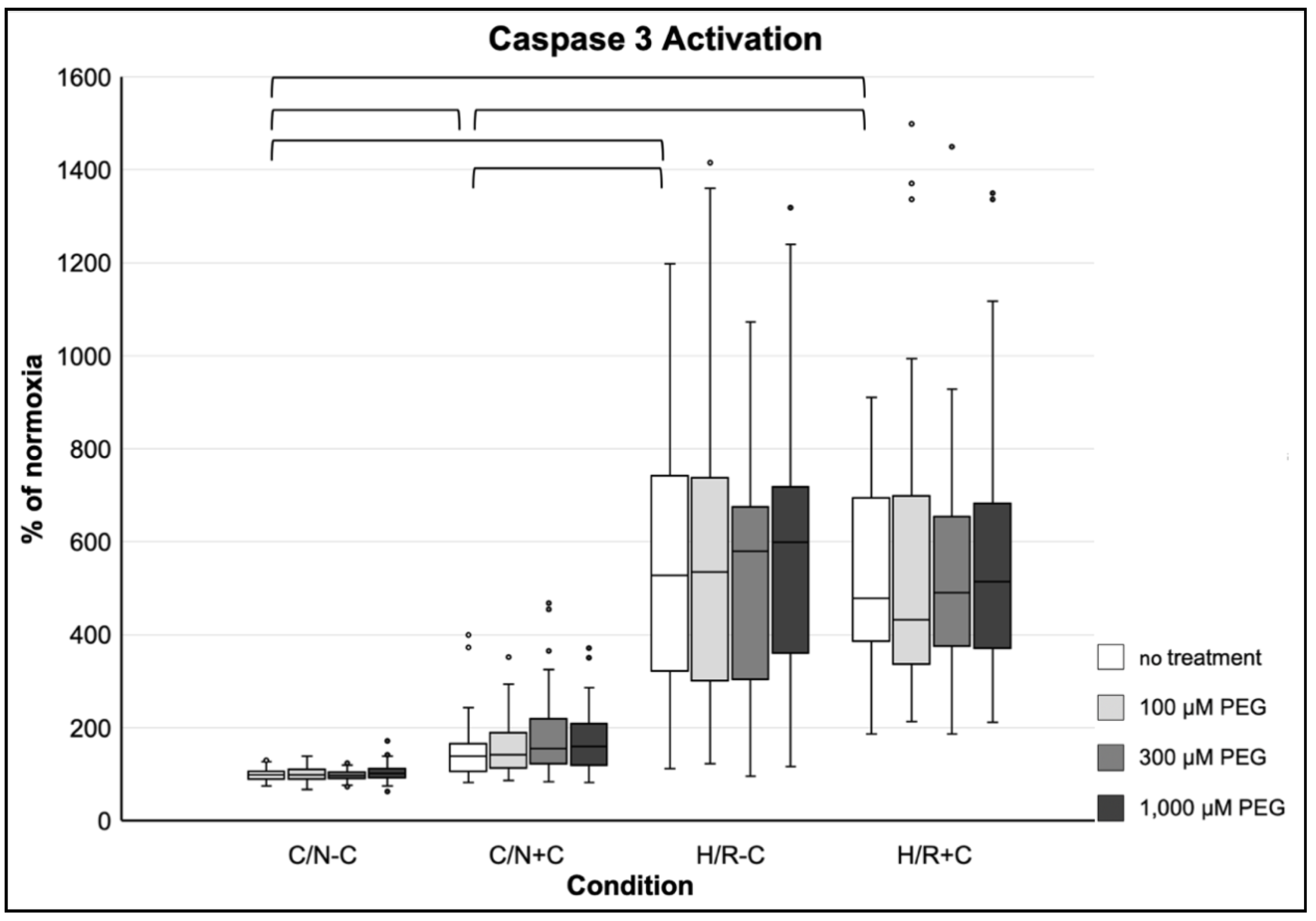
3.3.5. Induction of Apoptosis
3.4. Effects of PEG on Simulated Traumatic Brain Injury in Neurons
3.4.1. Cell Number and Viability
3.4.2. Mitochondrial Viability
3.4.3. Membrane Damage as Assessed by LDH per Total
3.4.4. Membrane Damage as Assessed by FM1-43 Dye Incorporation
3.4.5. Induction of Apoptosis
4. Discussion
4.1. Effect of P188 on Neuroprotection
4.2. Study Limitations
- The use of merely an in vitro TBI model of H/R ± compression injury represents both a strength and a limitation. In vitro experiments are indispensable in basic research as they provide a tool in the commencement of investigating novel therapeutic strategies and help identify pharmacological targets [56,57]. However, the use of an in vitro protocol generally implies that the applied mechanism to generate cellular damage might not represent the actual pathophysiological state of injury in a living body [56].
- The investigation of a primary culture—developed of immature tissue—might not appropriately simulate adult TBI. Primary neuronal cultures were harvested from newborn C57B/6 mouse tissue [58]. Thus, it could be argued that experiments on primary neurons might not be optimal for mimicking adult TBI but rather for investigations of the infantile brain. Indeed, neurons derived from embryonic tissue do not exhibit glutamate-mediated excitotoxicity because they do not express glutamatergic receptors. Excitotoxicity, however, plays an important role in adult neuronal injury. Moreover, for harvesting primary cultures mechanical and enzymatic dissociation processes are necessary, which can already lead to cellular disruption and injury [57].
- The compression with heavy aluminum lids and H/R injury used in our model might not be adequate for attenuation by P188. Different techniques to simulate TBI in vitro have been considered in other studies [4], some of which might be better-suited to study membrane resealing properties and, consequently possible positive effects of P188 on neuronal regeneration after TBI. TBI models with fluid shear stress have been observed to exert neuronal damage that can be abolished by P188 [16,17,18,20,21,22]. One might suggest that the injury induced by shear stress might be more adequate for investigating P188’s neuroprotective properties, possibly due to a shorter duration of damage and perhaps different cellular targets that might experience injury. Thus, the model used in the present investigation, while effective, might not have injured the neurons in a way that can be alleviated by P188.
- In the present model, cells in the center of a well suffered more mechanical injury than cells at the edge of a well due to the placement of the rods of the compression lids. Hence, it is possible that cells—especially in the center of a well—were excessively and irreversibly injured by compression, thus, making it possible that the injury was beyond what can be attenuated by potential sealing strategies. However, cells that did not undergo compression could also not be rescued by P188. Yet, it is additionally possible that the applied H/R injury was too excessive for being alleviated by P188.
- Neurons were only treated with P188 as a postconditioning agent after 5 h of injury. It is possible that an earlier beginning of P188 treatment could have allowed a better neuronal outcome. This hypothesis is supported by previous investigations: Serbest et al. observed a neuroprotective effect of P188 after injuring neuronal cells for a short duration (200 ms), and, consequently, an earlier P188 administration, already after 15 min; furthermore, cells were treated with P188 for 24 h [17,18]. Another study by Gu et al. achieved neuroprotection through a 43-h exposure to P188 [10]. Exposing the cells to P188 for no longer than 2 h, as in the present study, might not have been enough time for the neurons to regenerate. However, previous investigations of H/R and compression injury in different cells have revealed a protective effect of P188 after only 2 h of reoxygenation [29,32,60,61].
- No proven positive control/neuroprotectant was examined against this type of injury to show the general ability to protect neurons in this model. However, while P188 failed to attenuate neuronal cell injury, we have no evidence to suggest that our model would preclude other neuroprotective agents from being effective.
- Cell morphology changes have been described subjectively. Systematic quantitative assessment has been outside the scope of the present work. Yet, the observations made are concordant with the results of our objective endpoint assays (cell number/viability, mitochondrial viability, LDH, FM1-43 dye incorporation, Caspase 3 activation) and would not have changed our conclusions.
- Based on previous work of our study group, we have chosen 2 h for reoxygenation [32]. We did not further explore different reoxygenation times as this was outside the scope of our current work.
4.3. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A









References
- Dang, B.; Chen, W.; He, W.; Chen, G. Rehabilitation Treatment and Progress of Traumatic Brain Injury Dysfunction. Neural Plast. 2017, 2017, 1582182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackenberg, K.; Unterberg, A. Traumatic brain injury. Nervenarzt 2016, 87, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Dinet, V.; Petry, K.G.; Badaut, J. Brain–Immune Interactions and Neuroinflammation after Traumatic Brain Injury. Front. Neurosci. 2019, 13, 1178. [Google Scholar] [CrossRef] [Green Version]
- Morrison, B.; Elkin, B.S.; Dollé, J.-P.; Yarmush, M.L. In Vitro Models of Traumatic Brain Injury. Annu. Rev. Biomed. Eng. 2011, 13, 91–126. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, R.A. Cell Death Pathways in Acute Ischemia/Reperfusion Injury. J. Cardiovasc. Pharmacol. Ther. 2011, 16, 233–238. [Google Scholar] [CrossRef]
- Kalogeris, T.; Baines, C.P.; Krenz, M.; Korthuis, R.J. Cell Biology of Ischemia/Reperfusion Injury. Int. Rev. Cell Mol. Biol. 2012, 298, 229–317. [Google Scholar] [CrossRef] [Green Version]
- Bartos, J.A.; Matsuura, T.R.; Tsangaris, A.; Olson, M.; McKnite, S.H.; Rees, J.N.; Haman, K.; Shekar, K.C.; Riess, M.L.; Bates, F.S.; et al. Intracoronary Poloxamer 188 Prevents Reperfusion Injury in a Porcine Model of ST-Segment Elevation Myocardial Infarction. JACC Basic Transl. Sci. 2016, 1, 224–234. [Google Scholar] [CrossRef] [Green Version]
- Houang, E.M.; Bartos, J.; Hackel, B.J.; Lodge, T.P.; Yannopoulos, D.; Bates, F.S.; Metzger, J.M. Cardiac Muscle Membrane Stabilization in Myocardial Reperfusion Injury. JACC Basic Transl. Sci. 2019, 4, 275–287. [Google Scholar] [CrossRef]
- Martindale, J.J.; Metzger, J.M. Uncoupling of increased cellular oxidative stress and myocardial ischemia reperfusion injury by directed sarcolemma stabilization. J. Mol. Cell. Cardiol. 2014, 67, 26–37. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.-H.; Ge, J.-B.; Li, M.; Xu, H.-D.; Wu, F.; Qin, Z.-H. Poloxamer 188 Protects Neurons against Ischemia/Reperfusion Injury through Preserving Integrity of Cell Membranes and Blood Brain Barrier. PLoS ONE 2013, 8, e61641. [Google Scholar] [CrossRef]
- Luo, C.; Li, Q.; Gao, Y.; Shen, X.; Ma, L.; Wu, Q.; Wang, Z.; Zhang, M.; Zhao, Z.; Chen, X.; et al. Poloxamer 188 Attenuates Cerebral Hypoxia/Ischemia Injury in Parallel with Preventing Mitochondrial Membrane Permeabilization and Autophagic Activation. J. Mol. Neurosci. 2015, 56, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.C.; Bartos, J.A.; Matsuura, T.R.; Yannopoulos, D. The future is now: Neuroprotection during cardiopulmonary resuscitation. Curr. Opin. Crit. Care 2017, 23, 215–222. [Google Scholar] [CrossRef]
- Riess, M.L.; Salzman, M.M.; Cheng, Q.; Qi, A.; Afzal, A. Copolymer-Based Cell Membrane Stabilizers Attenuate Murine Cerebral Ischemia Reperfusion Injury [Abstract]. ASA Abstr. 2015, A3014. Available online: http://www.asaabstracts.com/strands/asaabstracts/abstractList.htm?year=2015&index=10 (accessed on 25 March 2021).
- Bao, H.; Yang, X.; Zhuang, Y.; Huang, Y.; Wang, T.; Zhang, M.; Dai, D.; Wang, S.; Xiao, H.; Huang, G.; et al. The effects of poloxamer 188 on the autophagy induced by traumatic brain injury. Neurosci. Lett. 2016, 634, 7–12. [Google Scholar] [CrossRef] [Green Version]
- Bao, H.-J.; Wang, T.; Zhang, M.-Y.; Liu, R.; Dai, D.-K.; Wang, Y.-Q.; Wang, L.; Zhang, L.; Gao, Y.-Z.; Qin, Z.-H. Poloxamer-188 attenuates TBI-induced blood–brain barrier damage leading to decreased brain edema and reduced cellular death. Neurochem. Res. 2012, 37, 2856–2867. [Google Scholar] [CrossRef]
- Luo, C.-L.; Chen, X.-P.; Li, L.-L.; Li, Q.-Q.; Li, B.-X.; Xue, A.-M.; Xu, H.-F.; Dai, D.-K.; Shen, Y.-W.; Tao, L.-Y.; et al. Poloxamer 188 Attenuates in vitro Traumatic Brain Injury-Induced Mitochondrial and Lysosomal Membrane Permeabilization Damage in Cultured Primary Neurons. J. Neurotrauma 2013, 30, 597–607. [Google Scholar] [CrossRef]
- Serbest, G.; Horwitz, J.; Barbee, K. The Effect of Poloxamer-188 on Neuronal Cell Recovery from Mechanical Injury. J. Neurotrauma 2005, 22, 119–132. [Google Scholar] [CrossRef]
- Serbest, G.; Horwitz, J.; Jost, M.; Barbee, K.A. Mechanisms of cell death and neuroprotection by poloxamer 188 after mechanical trauma. FASEB J. 2006, 20, 308–310. [Google Scholar] [CrossRef]
- Zhang, Y.; Chopp, M.; Emanuele, M.; Zhang, L.; Zhang, Z.G.; Lu, M.; Zhang, T.; Mahmood, A.; Xiong, Y. Treatment of Traumatic Brain Injury with Vepoloxamer (Purified Poloxamer 188). J. Neurotrauma 2018, 35, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Kilinc, D.; Gallo, G.; Barbee, K. Poloxamer 188 reduces axonal beading following mechanical trauma to cultured neurons. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2007, 2007, 5395–5398. [Google Scholar] [CrossRef]
- Kilinc, D.; Gallo, G.; Barbee, K.A. Mechanically-induced membrane poration causes axonal beading and localized cytoskeletal damage. Exp. Neurol. 2008, 212, 422–430. [Google Scholar] [CrossRef]
- Kilinc, D.; Gallo, G.; Barbee, K.A. Mechanical membrane injury induces axonal beading through localized activation of calpain. Exp. Neurol. 2009, 219, 553–561. [Google Scholar] [CrossRef] [Green Version]
- Marks, J.D.; Pan, C.Y.; Bushell, T.; Cromie, W.; Lee, R.C. Amphiphilic, tri-block copolymers provide potent membrane-targeted neuroprotection. FASEB J. 2001, 15, 1107–1109. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, U.; Goliaei, A.; Tsereteli, L.; Berkowitz, M.L. Properties of Poloxamer Molecules and Poloxamer Micelles Dissolved in Water and Next to Lipid Bilayers: Results from Computer Simulations. J. Phys. Chem. B 2016, 120, 5823–5830. [Google Scholar] [CrossRef]
- Poellmann, M.J.; Lee, R.C. Repair and Regeneration of the Wounded Cell Membrane. Regen. Eng. Transl. Med. 2017, 3, 111–132. [Google Scholar] [CrossRef]
- Glass, T.F.; Reeves, B.; Sharp, F.R. The impact of excitotoxic blockade on the evolution of injury following combined mechanical and hypoxic insults in primary rat neuronal culture. Neurobiol. Dis. 2004, 17, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Qin, Y.; Huang, Y.; Ji, D.; Wu, F. Poloxamer 188 rescues MPTP-induced lysosomal membrane integrity impairment in cellular and mouse models of Parkinson’s disease. Neurochem. Int. 2019, 126, 178–186. [Google Scholar] [CrossRef]
- Yıldırım, T.; Eylen, A.; Lule, S.; Erdener, S.E.; Vural, A.; Karatas, H.; Ozveren, M.F.; Dalkara, T.; Gursoy-Ozdemir, Y. Poloxamer-188 and citicoline provide neuronal membrane integrity and protect membrane stability in cortical spreading depression. Int. J. Neurosci. 2015, 125, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Lotze, F.P.; Salzman, M.M.; Pille, J.A.; Balzer, C.; Hees, J.E.; Cleveland, W.J.; Riess, M.L. Poloxamer 188 Protects Mouse Brain Microvascular Endothelial Cells in an in-vitro Traumatic Brain Injury Model [Abstract]. Circulation 2019, 140, A22. [Google Scholar]
- Lotze, F.P.; Salzman, M.M.; Pille, J.A.; Balzer, C.; Hees, J.E.; Cleveland, W.J.; Riess, M.L. In Vitro Traumatic Brain Injury Model of Mouse Brain Microvascular Endothelial Cells [Abstract]. FASEB J. 2019, 33, 527.4. [Google Scholar] [CrossRef]
- Meyer, L.J.; Salzman, M.M.; Eskaf, J.; Li, Z.; Riess, M.L. Traumatic Brain Injury Simulation in a Mouse Neuronal Cell Culture Model [Abstract]. Anesth Analg 2021. accepted for 2021 World Congress of Anaesthesiologists. Available online: https://www.wca2021.org/ (accessed on 25 March 2021).
- Salzman, M.M.; Bartos, J.A.; Yannopoulos, D.; Riess, M.L. Poloxamer 188 Protects Isolated Adult Mouse Cardiomyocytes from Reoxygenation Injury. Pharmacol. Res. Perspect. 2020, 8, 00639. [Google Scholar] [CrossRef]
- Hackel, B.J.; (University of Minnesota, Minneapolis, MN, USA). Personal communication, 2017.
- Kim, M.; Haman, K.J.; Houang, E.M.; Zhang, W.; Yannopoulos, D.; Metzger, J.M.; Bates, F.S.; Hackel, B.J. PEO–PPO Diblock Copolymers Protect Myoblasts from Hypo-Osmotic Stress In Vitro Dependent on Copolymer Size, Composition, and Architecture. Biomacromolecules 2017, 18, 2090–2101. [Google Scholar] [CrossRef]
- Yasuda, S.; Townsend, D.; Michele, D.E.; Favre, E.G.; Day, S.M.; Metzger, J.M. Dystrophic heart failure blocked by membrane sealant poloxamer. Nature 2005, 436, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Amaral, E.; Guatimosim, S.; Guatimosim, C. Using the fluorescent styryl dye FM1-43 to visualize synaptic vesicles exocytosis and endocytosis in motor nerve terminals. Methods Mol. Biol. 2011, 689, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Moloughney, J.G.; Weisleder, N. Poloxamer 188 (p188) as a membrane resealing reagent in biomedical applications. Recent Pat. Biotechnol. 2012, 6, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Poly(Ethylene Glycol) Average moL wt 8000, Powder. Sigma-Aldrich. Available online: https://www.sigmaaldrich.com/catalog/product/aldrich/p2139?lang=de®ion=DE (accessed on 12 May 2020).
- Kolliphor® P 188. Sigma-Aldrich. Available online: https://www.sigmaaldrich.com/catalog/substance/kolliphorp18812345900311611?lang=de®ion=DE (accessed on 12 May 2020).
- Borgens, R.B.; Bohnert, D.; Duerstock, B.; Spomar, D.; Lee, R.C. Subcutaneous tri-block copolymer produces recovery from spinal cord injury. J. Neurosci. Res. 2004, 76, 141–154. [Google Scholar] [CrossRef]
- Cadichon, S.B.; Le, H.M.; Wright, D.A.; Curry, D.J.; Kang, U.; Frim, D.M. Neuroprotective effect of the surfactant poloxamer 188 in a model of intracranial hemorrhage in rats. J. Neurosurg. Pediatr. 2007, 106, 36–40. [Google Scholar] [CrossRef]
- Chen, B.; Tjahja, J.; Malla, S.; Liebman, C.; Cho, M. Astrocyte Viability and Functionality in Spatially Confined Microcavitation Zone. ACS Appl. Mater. Interfaces 2019, 11, 4889–4899. [Google Scholar] [CrossRef]
- Curry, D.J.; Wright, D.A.; Lee, R.C.; Kang, U.J.; Frim, D.M. Poloxamer 188 Volumetrically Decreases Neuronal Loss in the Rat in a Time-dependent Manner. Neurosurgery 2004, 55, 943–949. [Google Scholar] [CrossRef]
- Curry, D.J.; Wright, D.A.; Lee, R.C.; Kang, U.J.; Frim, D.M. Surfactant poloxamer 188—Related decreases in inflammation and tissue damage after experimental brain injury in rats. J. Neurosurg. Pediatr. 2004, 101, 91–96. [Google Scholar] [CrossRef] [Green Version]
- Frim, D.M.; Wright, D.A.; Curry, D.J.; Cromie, W.; Lee, R.; Kang, U.J. The surfactant poloxamer-188 protects against glutamate toxicity in the rat brain. NeuroReport 2004, 15, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Inyang, E.; Abhyankar, V.; Chen, B.; Cho, M. Modulation of in vitro Brain Endothelium by Mechanical Trauma: Structural and Functional Restoration by Poloxamer 188. Sci. Rep. 2020, 10, 3054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanagaraj, J.; Chen, B.; Xiao, S.; Cho, M. Reparative Effects of Poloxamer P188 in Astrocytes Exposed to Controlled Microcavitation. Ann. Biomed. Eng. 2018, 46, 354–364. [Google Scholar] [CrossRef] [PubMed]
- Misra, A.; Kondaveeti, P.; Nissanov, J.; Barbee, K.; Shewokis, P.; Rioux, L.; Moxon, K.A. Preventing neuronal damage and inflammation in vivo during cortical microelectrode implantation through the use of Poloxamer P-188. J. Neural Eng. 2013, 10, 016011. [Google Scholar] [CrossRef] [Green Version]
- Quinn, M.; Mukhida, K.; Sadi, D.; Hong, M.; Méndez, I. Adjunctive use of the non-ionic surfactant Poloxamer 188 improves fetal dopaminergic cell survival and reinnervation in a neural transplantation strategy for Parkinson’s disease. Eur. J. Neurosci. 2007, 27, 43–52. [Google Scholar] [CrossRef]
- Wang, T.; Chen, X.; Wang, Z.; Zhang, M.; Meng, H.; Gao, Y.; Luo, B.; Tao, L.; Chen, Y. Poloxamer-188 Can Attenuate Blood–Brain Barrier Damage to Exert Neuroprotective Effect in Mice Intracerebral Hemorrhage Model. J. Mol. Neurosci. 2015, 55, 240–250. [Google Scholar] [CrossRef]
- Thal, S.C.; Neuhaus, W. The Blood–Brain Barrier as a Target in Traumatic Brain Injury Treatment. Arch. Med. Res. 2014, 45, 698–710. [Google Scholar] [CrossRef]
- Douglas, H.F.; Salzman, M.M.; Hackel, B.J.; Bartos, J.A.; Yannopoulos, D.; Riess, M.L. Dose Dependent Nitric Oxide Production by Poloxamer 188 in Rat Isolated Hearts [Abstract]. FASEB J. 2017, 31, 1006.3. [Google Scholar] [CrossRef]
- Salzman, M.M.; Cheng, Q.; Matsuura, T.; Yannopoulos, D.; Riess, M.L. Cardioprotection by Poloxamer 188 is Mediated by Nitric Oxide Synthase [Abstract]. FASEB J. 2015, 29, 1026.5. [Google Scholar] [CrossRef]
- Cyr, A.R.; Huckaby, L.V.; Shiva, S.S.; Zuckerbraun, B.S. Nitric Oxide and Endothelial Dysfunction. Crit. Care Clin. 2020, 36, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Lin, H.; Hong, X.; Ji, D.; Wu, F. Poloxamer 188-mediated anti-inflammatory effect rescues cognitive deficits in paraquat and maneb-induced mouse model of Parkinson’s disease. Toxicology 2020, 436, 152437. [Google Scholar] [CrossRef] [PubMed]
- Kumaria, A.; Tolias, C.M. In vitro models of neurotrauma. Br. J. Neurosurg. 2008, 22, 200–206. [Google Scholar] [CrossRef]
- Kumaria, A. In Vitro Models as a Platform to Investigate Traumatic Brain Injury. Altern. Lab. Anim. 2017, 45, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Sharma, J.; (CEO/CSO, Celprogen Inc., Torrance, CA, USA). Personal communication, 2020.
- Lüllmann-Rauch, R.; Asan, E. Taschenbuch Histologie. 5., Vollständig Überarbeitete Auflage; Georg Thieme Verlag: Stuttgart, Germany; New York, NY, USA, 2015. [Google Scholar]
- Salzman, M.M.; Hackel, B.J.; Bates, F.S.; Bartos, J.A.; Yannopoulos, D.; Riess, M.L. Poloxamer 188 Decreases Hypoxia/Reoxygenation-Induced LDH Release from Isolated Cardiomyocytes [Abstract]. FASEB J. 2017, 31, 1069.11. [Google Scholar] [CrossRef]
- Salzman, M.M.; Hackel, B.J.; Bates, F.S.; Bartos, J.A.; Yannopoulos, D.; Riess, M.L. Poloxamer 188 Protects Isolated Cardiomyocytes from Hypoxia/Reoxygenation Injury [Abstract]. FASEB J. 2017, 31, 1070.1. [Google Scholar] [CrossRef]
- Huh, D.; Hamilton, G.A.; Ingber, D.E. From 3D cell culture to organs-on-chips. Trends Cell Biol. 2011, 21, 745–754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, D.; Heo, I.; Clevers, H. Disease Modeling in Stem Cell-Derived 3D Organoid Systems. Trends Mol. Med. 2017, 23, 393–410. [Google Scholar] [CrossRef]
- Houang, E.M.; Sham, Y.Y.; Bates, F.S.; Metzger, J.M. Muscle membrane integrity in Duchenne muscular dystrophy: Recent advances in copolymer-based muscle membrane stabilizers. Skelet. Muscle 2018, 8, 31. [Google Scholar] [CrossRef] [Green Version]
- Houang, E.M.; Haman, K.J.; Kim, M.; Zhang, W.; Lowe, D.A.; Sham, Y.Y.; Lodge, T.P.; Hackel, B.J.; Bates, F.S.; Metzger, J.M. Chemical End Group Modified Diblock Copolymers Elucidate Anchor and Chain Mechanism of Membrane Stabilization. Mol. Pharm. 2017, 14, 2333–2339. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meyer, L.J.; Riess, M.L. Evaluation of In Vitro Neuronal Protection by Postconditioning with Poloxamer 188 Following Simulated Traumatic Brain Injury. Life 2021, 11, 316. https://doi.org/10.3390/life11040316
Meyer LJ, Riess ML. Evaluation of In Vitro Neuronal Protection by Postconditioning with Poloxamer 188 Following Simulated Traumatic Brain Injury. Life. 2021; 11(4):316. https://doi.org/10.3390/life11040316
Chicago/Turabian StyleMeyer, Luise J., and Matthias L. Riess. 2021. "Evaluation of In Vitro Neuronal Protection by Postconditioning with Poloxamer 188 Following Simulated Traumatic Brain Injury" Life 11, no. 4: 316. https://doi.org/10.3390/life11040316