
The Role of the CREB Protein Family Members and the Related Transcription Factors in Radioresistance Mechanisms

 ,
,
Abstract
:1. Introduction
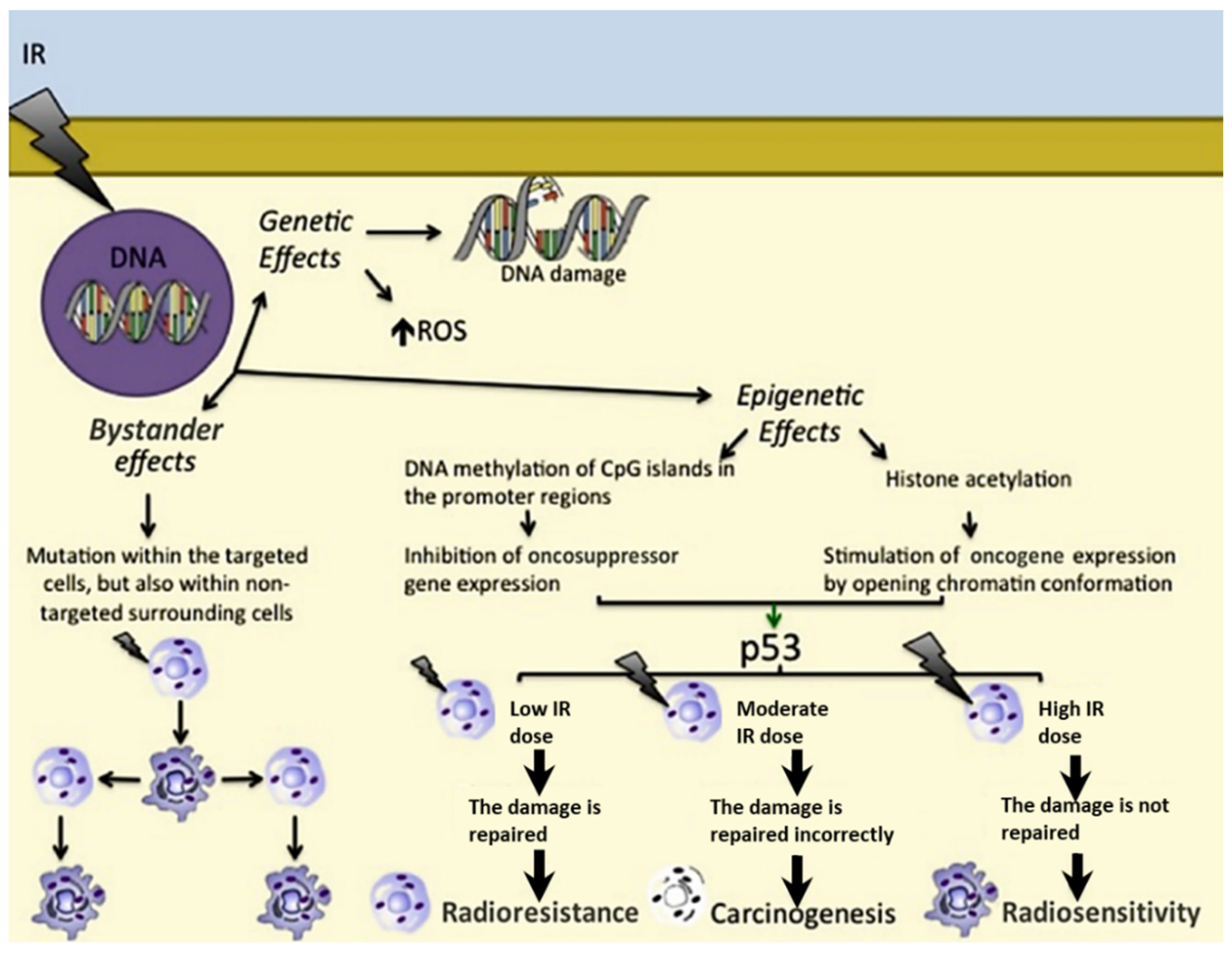
2. Ionising Radiation: Radioresistance and Radiosensitivity
2.1. Genetic Effects
2.2. Epigenetic Effects
2.3. Bystander Effects
3. CREB Family Members and CREB-Binding Proteins
4. NF-κB and p53: Two Crucial CREB-Related Transcription Factors
5. CREB Family Members: An Active Role in Tumours
6. CREB Family Members and Related Transcription Factors in Radiotherapy of Solid Tumours and Leukaemia
6.1. Cell Responses to Ionising Radiation
6.2. CREB and Other Factors in Radioresistance and Radiosensitivity
6.3. CREB and Related Transcription Factors as Possible Targets in the Treatment of Tumours
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- El-Jaby, S.; Lewis, B.J.; Tomi, L. A commentary on the impact of modelling results to inform mission planning and shield design. Life Sci. Sp. Res. 2020, 25, 148–150. [Google Scholar] [CrossRef]
- Li, M.; Gonon, G.; Buonanno, M.; Autsavapromporn, N.; De Toledo, S.M.; Pain, D.; Azzam, E.I. Health Risks of Space Exploration: Targeted and Nontargeted Oxidative Injury by High-Charge and High-Energy Particles. Antioxid. Redox Signal. 2014, 20, 1501. [Google Scholar] [CrossRef] [Green Version]
- Romero, E.; Francisco, D. The NASA human system risk mitigation process for space exploration. Acta Astronaut. 2020, 175, 606–615. [Google Scholar] [CrossRef]
- Sridharan, D.M.; Asaithamby, A.; Bailey, S.M.; Costes, S.V.; Doetsch, P.W.; Dynan, W.S.; Kronenberg, A.; Rithidech, K.N.; Saha, J.; Snijders, A.M.; et al. Understanding cancer development processes after HZE-particle exposure: Roles of ROS, DNA damage repair and inflammation. Radiat. Res. 2015, 183, 1–26. [Google Scholar] [CrossRef]
- Prasad, B.; Grimm, D.; Strauch, S.M.; Erzinger, G.S.; Corydon, T.J.; Lebert, M.; Magnusson, N.E.; Infanger, M.; Richter, P.; Krüger, M. Influence of Microgravity on Apoptosis in Cells, Tissues, and Other Systems In Vivo and In Vitro. Int. J. Mol. Sci. 2020, 21, 9373. [Google Scholar] [CrossRef]
- Simonsen, L.C.; Slaba, T.C.; Guida, P.; Rusek, A. NASA’s first ground-based Galactic Cosmic Ray Simulator: Enabling a new era in space radiobiology research. PLoS Biol. 2020, 18, e3000669. [Google Scholar] [CrossRef]
- Barcellos-Hoff, M.H.; Blakely, E.A.; Burma, S.; Fornace, A.J.; Gerson, S.; Hlatky, L.; Kirsch, D.G.; Luderer, U.; Shay, J.; Wang, Y.; et al. Concepts and challenges in cancer risk prediction for the space radiation environment. Life Sci. Sp. Res. 2015, 6, 92–103. [Google Scholar] [CrossRef] [Green Version]
- Montminy, M. Transcriptionl regulation by ciclic amp. Annu. Rev. Biochem. 2003, 66, 807–822. [Google Scholar] [CrossRef]
- Papavassiliou, A.G. The CREB/ATF family of transcription factors: Modulation by reversible phosphorylation. Anticancer Res. 1994, 14, 1801–1805. [Google Scholar]
- Niu, M.; Tabari, E.; Ni, P.; Su, Z. Towards a map of cis-regulatory sequences in the human genome. Nucleic Acids Res. 2018, 46, 5395–5409. [Google Scholar] [CrossRef] [Green Version]
- D’Auria, F.; Centurione, L.; Centurione, M.A.; Angelini, A.; Di Pietro, R. Regulation of Cancer Cell Responsiveness to Ionizing Radiation Treatment by Cyclic AMP Response Element Binding Nuclear Transcription Factor. Front. Oncol. 2017, 7, 76. [Google Scholar] [CrossRef] [Green Version]
- Lavrovsky, Y.; Schwartzman, M.L.; Levere, R.D.; Kappas, A.; Abraham, N.G. Identification of binding sites for transcription factors NF-kappa B and AP-2 in the promoter region of the human heme oxygenase 1 gene. Proc. Natl. Acad. Sci. USA 1994, 91, 5987–5991. [Google Scholar] [CrossRef] [Green Version]
- Kogure, Y.; Kataoka, K. Genetic alterations in adult T-cell leukemia/lymphoma. Cancer Sci. 2017, 108, 1719–1725. [Google Scholar] [CrossRef] [Green Version]
- Cataldi, A.; Rapino, M.; Centurione, L.; Sabatini, N.; Grifone, G.; Garaci, F.; Rana, R. NF-kB activation plays an antiapoptotic role in human leukemic K562 cells exposed to ionizing radiation. J. Cell. Biochem. 2003, 89, 956–963. [Google Scholar] [CrossRef]
- Yu, H.; Aravindan, N.; Xu, J.; Natarajan, M. Inter- and intra-cellular mechanism of NF-kB-dependent survival advantage and clonal expansion of radio-resistant cancer cells. Cell. Signal. 2017, 31, 105–111. [Google Scholar] [CrossRef]
- Mortezaee, K.; Najafi, M.; Farhood, B.; Ahmadi, A.; Shabeeb, D.; Musa, A.E. NF-κB targeting for overcoming tumor resistance and normal tissues toxicity. J. Cell. Physiol. 2019, 234, 17187–17204. [Google Scholar] [CrossRef]
- Obrador, E.; Salvador, R.; Villaescusa, J.I.; Soriano, J.M.; Estrela, J.M.; Montoro, A. Radioprotection and Radiomitigation: From the Bench to Clinical Practice. Biomedicines 2020, 8, 461. [Google Scholar] [CrossRef]
- Tang, F.R.; Loke, W.K.; Khoo, B.C. Low-dose or low-dose-rate ionizing radiation–induced bioeffects in animal models. J. Radiat. Res. 2017, 58, 165–182. [Google Scholar] [CrossRef]
- Spitz, D.R.; Azzam, E.I.; Jian Li, J.; Gius, D. Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: A unifying concept in stress response biology. Cancer Metastasis Rev. 2004, 23, 311–322. [Google Scholar] [CrossRef]
- Chaudhry, M.A. Bystander effect: Biological endpoints and microarray analysis. Mutat. Res. Mol. Mech. Mutagen. 2006, 597, 98–112. [Google Scholar] [CrossRef]
- Fleming, A.M.; Ding, Y.; Burrows, C.J. Oxidative DNA damage is epigenetic by regulating gene transcription via base excision repair. Proc. Natl. Acad. Sci. USA 2017, 114, 2604–2609. [Google Scholar] [CrossRef]
- Tudek, B.; Winczura, A.; Janik, J.; Siomek, A.; Foksinski, M.; Oliński, R. Involvement of oxidatively damaged DNA and repair in cancer development and aging. Am. J. Transl. Res. 2010, 2, 254–284. [Google Scholar] [PubMed]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Tubiana, M. Can we reduce the incidence of second primary malignancies occurring after radiotherapy? A critical review. Radiother. Oncol. 2009, 91, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.V.; Hora, S.; Pal, A.; Jha, S.; Taneja, R. Stressing the (Epi)Genome: Dealing with Reactive Oxygen Species in Cancer. Antioxid. Redox Signal. 2018, 29, 1273–1292. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, N.; Burns, D.M.; Blau, H.M. DNA Demethylation Dynamics. Cell 2011, 146, 866–872. [Google Scholar] [CrossRef] [Green Version]
- Church, M.C.; Fleming, A.B. A role for histone acetylation in regulating transcription elongation. Transcription 2018, 9, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef]
- Mladenov, E.; Li, F.; Zhang, L.; Klammer, H.; Iliakis, G. Intercellular communication of DNA damage and oxidative status underpin bystander effects. Int. J. Radiat. Biol. 2018, 94, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Ilnytskyy, Y.; Kovalchuk, O. Non-targeted radiation effects—An epigenetic connection. Mutat. Res. Mol. Mech. Mutagen. 2011, 714, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Albanese, J.; Dainiak, N. Ionizing radiation alters Fas antigen ligand at the cell surface and on exfoliated plasma membrane-derived vesicles: Implications for apoptosis and intercellular signaling. Radiat. Res. 2000, 153, 49–61. [Google Scholar] [CrossRef]
- Azzam, E.I.; de Toledo, S.M.; Gooding, T.; Little, J.B. Intercellular communication is involved in the bystander regulation of gene expression in human cells exposed to very low fluences of alpha particles. Radiat. Res. 1998, 150, 497–504. [Google Scholar] [CrossRef]
- Barcellos-Hoff, M.H.; Brooks, A.L. Extracellular signaling through the microenvironment: A hypothesis relating carcinogenesis, bystander effects, and genomic instability. Radiat. Res. 2001, 156, 618–627. [Google Scholar] [CrossRef]
- Nakamura, N. A hypothesis: Radiation carcinogenesis may result from tissue injuries and subsequent recovery processes which can act as tumor promoters and lead to an earlier onset of cancer. Br. J. Radiol. 2020, 93, 1–7. [Google Scholar] [CrossRef]
- Ishikawa, K.; Ishii, H.; Saito, T. DNA Damage-Dependent Cell Cycle Checkpoints and Genomic Stability. DNA Cell Biol. 2006, 25, 406–411. [Google Scholar] [CrossRef]
- Nakano, H.; Yonekawa, H.; Shinohara, K. Threshold Level of p53 Required for the Induction of Apoptosis in X-Irradiated MOLT-4 Cells. Int. J. Radiat. Oncol. 2007, 68, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Heylmann, D.; Ponath, V.; Kindler, T.; Kaina, B. Comparison of DNA repair and radiosensitivity of different blood cell populations. Sci. Rep. 2021, 11, 2478. [Google Scholar] [CrossRef]
- Portella, L.; Scala, S. Ionizing radiation effects on the tumor microenvironment. Semin. Oncol. 2019, 46, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, H.; Hayashi, S.; Hatashita, M.; Ohnishi, K.; Shioura, H.; Ohtsubo, T.; Kitai, R.; Ohnishi, T.; Kano, E. Induction of radioresistance by a nitric oxide-mediated bystander effect. Radiat. Res. 2001, 153, 387–396. [Google Scholar] [CrossRef]
- González, R.; Molina-Ruiz, F.J.; Bárcena, J.A.; Padilla, C.A.; Muntané, J. Regulation of Cell Survival, Apoptosis, and Epithelial-to-Mesenchymal Transition by Nitric Oxide-Dependent Post-Translational Modifications. Antioxid. Redox Signal. 2018, 29, 1312–1332. [Google Scholar] [CrossRef]
- Coleman, C.N.; Eke, I.; Makinde, A.Y.; Chopra, S.; Demaria, S.; Formenti, S.C.; Martello, S.; Bylicky, M.; Mitchell, J.B.; Aryankalayil, M.J. Radiation-induced Adaptive Response: New Potential for Cancer Treatment. Clin. Cancer Res. 2020, 26, 5781–5790. [Google Scholar] [CrossRef]
- Chevalier, F.; Hamdi, D.H.; Saintigny, Y.; Lefaix, J.-L. Proteomic overview and perspectives of the radiation-induced bystander effects. Mutat. Res. Mutat. Res. 2015, 763, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Yamashita, S. Radiation-Induced Bystander Response: Mechanism and Clinical Implications. Adv. Wound Care 2014, 3, 16–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tazawa, H.; Kagawa, S.; Fujiwara, T. p53 Replacement Therapy for Cancer. Recent Results Cancer Res. 2016, 209, 1–15. [Google Scholar] [CrossRef]
- Acharya, A.; Rishi, V.; Moll, J.; Vinson, C. Experimental identification of homodimerizing B-ZIP families in Homo sapiens. J. Struct. Biol. 2006, 155, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yu, H.; Wang, P.; Ding, Q.; Wang, Z. Screening of transcription factors with transcriptional initiation activity. Gene 2013, 531, 64–70. [Google Scholar] [CrossRef]
- Sapio, L.; Salzillo, A.; Ragone, A.; Illiano, M.; Spina, A.; Naviglio, S. Targeting CREB in Cancer Therapy: A Key Candidate or One of Many? An Update. Cancers 2020, 12, 3166. [Google Scholar] [CrossRef] [PubMed]
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2001, 2, 599–609. [Google Scholar] [CrossRef]
- Naqvi, S.; Martin, K.J.; Arthur, J.S.C. CREB phosphorylation at Ser133 regulates transcription via distinct mechanisms downstream of cAMP and MAPK signalling. Biochem. J. 2014, 458, 469–479. [Google Scholar] [CrossRef]
- Pregi, N.; Belluscio, L.M.; Berardino, B.G.; Castillo, D.S.; Cánepa, E.T. Oxidative stress-induced CREB upregulation promotes DNA damage repair prior to neuronal cell death protection. Mol. Cell. Biochem. 2017, 425, 9–24. [Google Scholar] [CrossRef]
- Kehat, I.; Hasin, T.; Aronheim, A. The Role of Basic Leucine Zipper Protein-Mediated Transcription in Physiological and Pathological Myocardial Hypertrophy. Ann. N. Y. Acad. Sci. 2006, 1080, 97–109. [Google Scholar] [CrossRef]
- Medzhitov, R.; Horng, T. Transcriptional control of the inflammatory response. Nat. Rev. Immunol. 2009, 9, 692–703. [Google Scholar] [CrossRef]
- Cai, D.H.; Wang, D.; Keefer, J.; Yeamans, C.; Hensley, K.; Friedman, A.D. C/EBPα:AP-1 leucine zipper heterodimers bind novel DNA elements, activate the PU.1 promoter and direct monocyte lineage commitment more potently than C/EBPα homodimers or AP-1. Oncogene 2008, 27, 2772–2779. [Google Scholar] [CrossRef] [Green Version]
- Hai, T.; Curran, T. Cross-family dimerization of transcription factors Fos/Jun and ATF/CREB alters DNA binding specificity. Proc. Natl. Acad. Sci. USA 1991, 88, 3720–3724. [Google Scholar] [CrossRef] [Green Version]
- Wek, R.C.; Anthony, T.G. EXtENDINg β cell survival by UPRegulating ATF4 translation. Cell Metab. 2006, 4, 333–334. [Google Scholar] [CrossRef] [Green Version]
- Hai, T.; Hartman, M.G. The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: Activating transcription factor proteins and homeostasis. Gene 2001, 273, 1–11. [Google Scholar] [CrossRef]
- Gao, S.; Gao, L.; Wang, S.; Shi, X.; Yue, C.; Wei, S.; Zuo, L.; Zhang, L.; Qin, X. ATF3 Suppresses Growth and Metastasis of Clear Cell Renal Cell Carcinoma by Deactivating EGFR/AKT/GSK3β/β-Catenin Signaling Pathway. Front. Cell Dev. Biol. 2021, 9, 618987. [Google Scholar] [CrossRef] [PubMed]
- Hasim, M.S.; Nessim, C.; Villeneuve, P.J.; Vanderhyden, B.C.; Dimitroulakos, J. Activating Transcription Factor 3 as a Novel Regulator of Chemotherapy Response in Breast Cancer. Transl. Oncol. 2018, 11, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.G.; Haynes, K.A.; Hegde, A.N. Degradation of Transcriptional Repressor ATF4 during Long-Term Synaptic Plasticity. Int. J. Mol. Sci. 2020, 21, 8543. [Google Scholar] [CrossRef] [PubMed]
- Holmqvist, P.-H.; Mannervik, M. Genomic occupancy of the transcriptional co-activators p300 and CBP. Transcription 2013, 4, 18–23. [Google Scholar] [CrossRef] [Green Version]
- Attar, N.; Kurdistani, S.K. Exploitation of EP300 and CREBBP Lysine Acetyltransferases by Cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a026534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mink, S.; Haenig, B.; Klempnauer, K.H. Interaction and functional collaboration of p300 and C/EBPbeta. Mol. Cell. Biol. 1997, 17, 6609–6617. [Google Scholar] [CrossRef] [Green Version]
- Burda, P.; Laslo, P.; Stopka, T. The role of PU.1 and GATA-1 transcription factors during normal and leukemogenic hematopoiesis. Leukemia 2010, 24, 1249–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johannessen, M.; Delghandi, M.P.; Seternes, O.M.; Johansen, B.; Moens, U. Synergistic activation of CREB-mediated transcription by forskolin and phorbol ester requires PKC and depends on the glutamine-rich Q2 transactivation domain. Cell. Signal. 2004, 16, 1187–1199. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [Green Version]
- Qin, F.; Fan, Q.; Yu, P.K.N.; Almahi, W.A.; Kong, P.; Yang, M.; Cao, W.; Nie, L.; Chen, G.; Han, W. Properties and gene expression profiling of acquired radioresistance in mouse breast cancer cells. Ann. Transl. Med. 2021, 9, 628. [Google Scholar] [CrossRef]
- Sokolova, O.; Naumann, M. NF-κB Signaling in Gastric Cancer. Toxins 2017, 9, 119. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Karin, M. Regulation and Function of NF-κB Transcription Factors in the Immune System. Ann. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef]
- Shoji, S.; Hanada, K.; Takahashi, M.; Watanabe, K.; Yonemochi, M.; Tomabechi, Y.; Shirouzu, M. The NF-κB regulator IκBβ exhibits different molecular interactivity and phosphorylation status from IκBα in an IKK2-catalysed reaction. FEBS Lett. 2020, 594, 1532–1549. [Google Scholar] [CrossRef]
- Schmitz, M.L.; Bacher, S.; Kracht, M. IκB-independent control of NF-κB activity by modulatory phosphorylations. Trends Biochem. Sci. 2001, 26, 186–190. [Google Scholar] [CrossRef]
- Mukherjee, S.P.; Behar, M.; Birnbaum, H.A.; Hoffmann, A.; Wright, P.E.; Ghosh, G. Analysis of the RelA:CBP/p300 Interaction Reveals Its Involvement in NF-κB-Driven Transcription. PLoS Biol. 2013, 11, e1001647. [Google Scholar] [CrossRef] [Green Version]
- Ono, H.; Basson, M.D.; Ito, H. P300 inhibition enhances gemcitabine-induced apoptosis of pancreatic cancer. Oncotarget 2016, 7, 51301–51310. [Google Scholar] [CrossRef] [Green Version]
- Dyson, H.J.; Wright, P.E. Role of Intrinsic Protein Disorder in the Function and Interactions of the Transcriptional Coactivators CREB-binding Protein (CBP) and p300. J. Biol. Chem. 2016, 291, 6714–6722. [Google Scholar] [CrossRef] [Green Version]
- Huante-Mendoza, A.; Silva-García, O.; Oviedo-Boyso, J.; Hancock, R.E.W.; Baizabal-Aguirre, V.M. Peptide IDR-1002 Inhibits NF-κB Nuclear Translocation by Inhibition of IκBα Degradation and Activates p38/ERK1/2–MSK1-Dependent CREB Phosphorylation in Macrophages Stimulated with Lipopolysaccharide. Front. Immunol. 2016, 7, 533. [Google Scholar] [CrossRef] [Green Version]
- Wen, A.Y.; Sakamoto, K.M.; Miller, L.S. The Role of the Transcription Factor CREB in Immune Function. J. Immunol. 2010, 185, 6413–6419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossman, S.R. p300/CBP/p53 interaction and regulation of the p53 response. Eur. J. Biochem. 2001, 268, 2773–2778. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Deng, X.; Shyu, Y.J.; Li, J.J.; Taparowsky, E.J.; Hu, C.-D. Mutual regulation of c-Jun and ATF2 by transcriptional activation and subcellular localization. EMBO J. 2006, 25, 1058–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, S.; Murphy, M.E. Genetic Modifiers of the p53 Pathway. Cold Spring Harb. Perspect. Med. 2016, 6, a026302. [Google Scholar] [CrossRef]
- Vaseva, A.V.; Moll, U.M. The mitochondrial p53 pathway. Biochim. Biophys. Acta-Bioenerg. 2009, 1787, 414–420. [Google Scholar] [CrossRef] [Green Version]
- Reinhardt, H.C.; Schumacher, B. The p53 network: Cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012, 28, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Pflaum, J.; Schlosser, S.; Müller, M. p53 Family and Cellular Stress Responses in Cancer. Front. Oncol. 2014, 4, 285. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F.; Gueven, N. The complexity of p53 stabilization and activation. Cell Death Differ. 2006, 13, 941–950. [Google Scholar] [CrossRef]
- Joruiz, S.M.; Bourdon, J.-C. p53 Isoforms: Key Regulators of the Cell Fate Decision. Cold Spring Harb. Perspect. Med. 2016, 6, a026039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef] [Green Version]
- Chasov, V.; Mirgayazova, R.; Zmievskaya, E.; Khadiullina, R.; Valiullina, A.; Stephenson Clarke, J.; Rizvanov, A.; Baud, M.G.J.; Bulatov, E. Key Players in the Mutant p53 Team: Small Molecules, Gene Editing, Immunotherapy. Front. Oncol. 2020, 10, 1460. [Google Scholar] [CrossRef]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef]
- Mogensen, T.H. IRF and STAT Transcription Factors—From Basic Biology to Roles in Infection, Protective Immunity, and Primary Immunodeficiencies. Front. Immunol. 2019, 9, 3047. [Google Scholar] [CrossRef] [PubMed]
- Steven, A.; Friedrich, M.; Jank, P.; Heimer, N.; Budczies, J.; Denkert, C.; Seliger, B. What turns CREB on? And off? And why does it matter? Cell. Mol. Life Sci. 2020, 77, 4049–4067. [Google Scholar] [CrossRef]
- Siu, Y.-T.; Jin, D.-Y. CREB—A real culprit in oncogenesis. FEBS J. 2007, 274, 3224–3232. [Google Scholar] [CrossRef]
- D’Auria, F.; Di Pietro, R. Role of CREB Protein Family Members in Human Haematological Malignancies. In Cancer Treatment—Conventional and Innovative Approaches; InTechOpen: London, UK, 2013; ISBN 978-953-51-1098-9. [Google Scholar] [CrossRef] [Green Version]
- Cho, E.-C.; Mitton, B.; Sakamoto, K. CREB and Leukemogenesis. Crit. Rev. Oncog. 2011, 16, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Cataldi, A.; Di Giacomo, V.; Rapino, M.; Genovesi, D.; Rana, R.A. Cyclic Nucleotide Response Element Binding Protein (CREB) Activation Promotes Survival Signal in Human K562 Erythroleukemia Cells Exposed to Ionising Radiation/Etoposide Combined Treatment. J. Radiat. Res. 2006, 47, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Di Nisio, C.; Sancilio, S.; Di Giacomo, V.; Rapino, M.; Sancillo, L.; Genovesi, D.; Di Siena, A.; Rana, R.A.; Cataldi, A.; Di Pietro, R. Involvement of cyclic-nucleotide response element-binding family members in the radiation response of Ramos B lymphoma cells. Int. J. Oncol. 2016, 48, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Steven, A.; Seliger, B. Control of CREB expression in tumors: From molecular mechanisms and signal transduction pathways to therapeutic target. Oncotarget 2016, 7, 35454–35465. [Google Scholar] [CrossRef] [Green Version]
- Jean, D.; Menashe, B.-E.; Bar-Eli, M. Regulation of tumor growth and metastasis of human melanoma by the CREB transcription factor family. Mol. Cell. Biochem. 2000, 212, 19–28. [Google Scholar] [CrossRef]
- Sarkar, D.; Leung, E.Y.; Baguley, B.C.; Finlay, G.J.; Askarian-Amiri, M.E. Epigenetic regulation in human melanoma: Past and future. Epigenetics 2015, 10, 103–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braeuer, R.R.; Zigler, M.; Villares, G.J.; Dobroff, A.S.; Bar-Eli, M. Transcriptional control of melanoma metastasis: The importance of the tumor microenvironment. Semin. Cancer Biol. 2011, 21, 83–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.J.; Shin, K.J.; Park, S.-A.; Park, K.S.; Park, S.; Heo, K.; Seo, Y.-K.; Noh, D.-Y.; Ryu, S.H.; Suh, P.-G. G-protein-coupled receptor 81 promotes a malignant phenotype in breast cancer through angiogenic factor secretion. Oncotarget 2016, 7, 70898–70911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caravatta, L.; Sancilio, S.; di Giacomo, V.; Rana, R.; Cataldi, A.; Di Pietro, R. PI3-K/Akt-dependent activation of cAMP-response element-binding (CREB) protein in Jurkat T leukemia cells treated with TRAIL. J. Cell. Physiol. 2008, 214, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Shankar, D.B.; Cheng, J.C.; Kinjo, K.; Federman, N.; Moore, T.B.; Gill, A.; Rao, N.P.; Landaw, E.M.; Sakamoto, K.M. The role of CREB as a proto-oncogene in hematopoiesis and in acute myeloid leukemia. Cancer Cell 2005, 7, 351–362. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.W.; Chen, X.; Gao, J.W.; Zhang, H.; Ma, R.R.; Gao, Z.H.; Gao, P. High expression of cAMP responsive element binding protein 1 (CREB1) is associated with metastasis, tumor stage and poor outcome in gastric cancer. Oncotarget 2015, 6, 10646–10657. [Google Scholar] [CrossRef]
- Abramovitch, R.; Tavor, E.; Jacob-Hirsch, J.; Zeira, E.; Amariglio, N.; Pappo, O.; Rechavi, G.; Galun, E.; Honigman, A. A Pivotal Role of Cyclic AMP-Responsive Element Binding Protein in Tumor Progression. Cancer Res. 2004, 64, 1338–1346. [Google Scholar] [CrossRef] [Green Version]
- Ye, C.-G.; Yeung, J.H.-K.; Huang, G.-L.; Cui, P.; Wang, J.; Zou, Y.; Zhang, X.-N.; He, Z.-W.; Cho, C.-H. Increased glutathione and mitogen-activated protein kinase phosphorylation are involved in the induction of doxorubicin resistance in hepatocellular carcinoma cells. Hepatol. Res. 2013, 43, 289–299. [Google Scholar] [CrossRef]
- Belkhiri, A.; Dar, A.A.; Zaika, A.; Kelley, M.; El-Rifai, W. t-Darpp Promotes Cancer Cell Survival by Up-regulation of Bcl2 through Akt-Dependent Mechanism. Cancer Res. 2008, 68, 395–403. [Google Scholar] [CrossRef] [Green Version]
- Sang, M.; Hulsurkar, M.; Zhang, X.; Song, H.; Zheng, D.; Zhang, Y.; Li, M.; Xu, J.; Zhang, S.; Ittmann, M.; et al. GRK3 is a direct target of CREB activation and regulates neuroendocrine differentiation of prostate cancer cells. Oncotarget 2016, 7, 45171–45185. [Google Scholar] [CrossRef] [Green Version]
- Darrington, R.S.; Campa, V.M.; Walker, M.M.; Bengoa-Vergniory, N.; Gorrono-Etxebarria, I.; Uysal-Onganer, P.; Kawano, Y.; Waxman, J.; Kypta, R.M. Distinct expression and activity of GSK-3α and GSK-3β in prostate cancer. Int. J. Cancer 2012, 131, E872–E883. [Google Scholar] [CrossRef]
- Banerji, V.; Frumm, S.M.; Ross, K.N.; Li, L.S.; Schinzel, A.C.; Hahn, C.K.; Kakoza, R.M.; Chow, K.T.; Ross, L.; Alexe, G.; et al. The intersection of genetic and chemical genomic screens identifies GSK-3α as a target in human acute myeloid leukemia. J. Clin. Investig. 2012, 122, 935–947. [Google Scholar] [CrossRef] [Green Version]
- Bang, D.; Wilson, W.; Ryan, M.; Yeh, J.J.; Baldwin, A.S. GSK-3α Promotes Oncogenic KRAS Function in Pancreatic Cancer via TAK1–TAB Stabilization and Regulation of Noncanonical NF-κB. Cancer Discov. 2013, 3, 690–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.-A.; Lee, J.W.; Herbst, R.S.; Koo, J.S. GSK-3α Is a Novel Target of CREB and CREB-GSK-3α Signaling Participates in Cell Viability in Lung Cancer. PLoS ONE 2016, 11, e0153075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cataldi, A.; Di Giacomo, V.; Rapino, M.; Zara, S.; Rana, R.A. Ionizing Radiation Induces Apoptotic Signal Through Protein Kinase Cδ (delta) and Survival Signal Through Akt and Cyclic-Nucleotide Response Element-Binding Protein (CREB) in Jurkat T Cells. Biol. Bull. 2009, 217, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Katanyutanon, S.; Wu, R.; Wang, P. The effect of whole-body radiation on blood levels of gastrointestinal peptides in the rat. Int. J. Clin. Exp. Med. 2008, 1, 332–337. [Google Scholar] [PubMed]
- Chen, X.; Shen, B.; Xia, L.; Khaletzkiy, A.; Wong, J.Y.C.; Li, J.J.; Chu, D. Activation of nuclear factor κB in radioresistance of TP53-inactive human keratinocytes. Cancer Res. 2002, 62, 1213–1221. [Google Scholar]
- Chua, J.S.; Liew, H.P.; Guo, L.; Lane, D.P. Tumor-specific signaling to p53 is mimicked by Mdm2 inactivation in zebrafish: Insights from mdm2 and mdm4 mutant zebrafish. Oncogene 2015, 34, 5933–5941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart-Ornstein, J.; Iwamoto, Y.; Miller, M.A.; Prytyskach, M.A.; Ferretti, S.; Holzer, P.; Kallen, J.; Furet, P.; Jambhekar, A.; Forrester, W.C.; et al. p53 dynamics vary between tissues and are linked with radiation sensitivity. Nat. Commun. 2021, 12, 898. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.-P.; Shao, S.-H.; Li, D.-D.; Zhou, G.-P. A dynamic model for the p53 stress response networks under ion radiation. Amino Acids 2007, 33, 75–83. [Google Scholar] [CrossRef]
- Qi, J.; Shao, S.; Shen, Y. Cellular responding DNA damage: An improved modeling of P53 gene regulatory networks under ion radiation (IR). Appl. Math. Comput. 2008, 205, 73–83. [Google Scholar] [CrossRef]
- Macaluso, M.; Montanari, M.; Cinti, C.; Giordano, A. Modulation of Cell Cycle Components by Epigenetic and Genetic Events. Semin. Oncol. 2005, 32, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Teufel, D.P.; Bycroft, M.; Fersht, A.R. Regulation by phosphorylation of the relative affinities of the N-terminal transactivation domains of p53 for p300 domains and Mdm2. Oncogene 2009, 28, 2112–2118. [Google Scholar] [CrossRef] [Green Version]
- Sokolov, M.; Neumann, R. Changes in gene expression as one of the key mechanisms involved in radiation-induced bystander effect (Review). Biomed. Rep. 2018, 9, 99–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeVries-Seimon, T.A.; Ohm, A.M.; Humphries, M.J.; Reyland, M.E. Induction of Apoptosis Is Driven by Nuclear Retention of Protein Kinase Cδ. J. Biol. Chem. 2007, 282, 22307–22314. [Google Scholar] [CrossRef] [Green Version]
- Bluwstein, A.; Kumar, N.; Léger, K.; Traenkle, J.; van Oostrum, J.; Rehrauer, H.; Baudis, M.; Hottiger, M.O. PKC signaling prevents irradiation-induced apoptosis of primary human fibroblasts. Cell Death Dis. 2013, 4, e498. [Google Scholar] [CrossRef] [Green Version]
- Martelli, A.M.; Evangelisti, C.; Nyakern, M.; Manzoli, F.A. Nuclear protein kinase C. Biochim. Biophys. Acta 2006, 1761, 542–551. [Google Scholar] [CrossRef]
- Xue, Y.; Ren, J.; Gao, X.; Jin, C.; Wen, L.; Yao, X. GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell. Proteomics 2008, 7, 1598–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickoloff, J.A.; Sharma, N.; Taylor, L. Clustered DNA Double-Strand Breaks: Biological Effects and Relevance to Cancer Radiotherapy. Genes 2020, 11, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, M.; Kashyap, T.; Pramanik, K.K.; Singh, A.K.; Nagini, S.; Mishra, R. The elevated activation of NFκB and AP-1 is correlated with differential regulation of Bcl-2 and associated with oral squamous cell carcinoma progression and resistance. Clin. Oral Investig. 2017, 21, 2721–2731. [Google Scholar] [CrossRef] [PubMed]
- Voboril, R.; Rychterova, V.; Voborilova, J.; Kubecova, M.; Fanta, J.; Dvorak, J. NF-κB/p65 expression before and after treatment in rectal cancer patients undergoing neoadjuvant (chemo)radiotherapy and surgery: Prognostic marker for disease progression and survival. Neoplasma 2016, 63, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Rosenzweig, K.R.; Youmell, M.; Price, B.D. The DNA-Dependent Protein Kinase Participates in the Activation of NFκB Following DNA Damage. Biochem. Biophys. Res. Commun. 1998, 247, 79–83. [Google Scholar] [CrossRef]
- Madhusoodhanan, R.; Natarajan, M.N.; Veeraraghavan, J.; Herman, T.S.; Aravindan, N. NFκB activity and transcriptional responses in human breast adenocarcinoma cells after single and fractionated irradiation. Cancer Biol. Ther. 2009, 8, 765–773. [Google Scholar] [CrossRef] [Green Version]
- Ramos, P.M.M.; Pezuk, J.A.; Castro-Gamero, A.M.; Oliveira, H.F.; Scrideli, C.A.; Umezawa, K.; Tone, L.G. Antineoplastic Effects of NF-κB Inhibition by DHMEQ (Dehydroxymethylepoxyquinomicin) Alone and in Co-treatment with Radio-and Chemotherapy in Medulloblastoma Cell Lines. Anticancer. Agents Med. Chem. 2018, 18, 541–549. [Google Scholar] [CrossRef]
- Samuel, T.; Fadlalla, K.; Gales, D.N.; Putcha, B.D.; Manne, U. Variable NF-κB pathway responses in colon cancer cells treated with chemotherapeutic drugs. BMC Cancer 2014, 14, 599. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Singh, A.K.; Repasky, E.A. Highlighting the Potential for Chronic Stress to Minimize Therapeutic Responses to Radiotherapy through Increased Immunosuppression and Radiation Resistance. Cancers 2020, 12, 3853. [Google Scholar] [CrossRef]
- Nelson, E.C.; Cambio, A.J.; Yang, J.C.; Ok, J.-H.; Lara, P.N.; Evans, C.P. Clinical implications of neuroendocrine differentiation in prostate cancer. Prostate Cancer Prostatic 2007, 10, 6–14. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.-H.; Zhang, Y.-Q.; Huang, J.-T. Neuroendocrine cells of prostate cancer: Biologic functions and molecular mechanisms. Asian J. Androl. 2019, 21, 291. [Google Scholar] [CrossRef]
- Liu, M.; Guyot-Sionnest, P. Mechanism of silver(I)-assisted growth of gold nanorods and bipyramids. J. Phys. Chem. B 2005, 109, 22192–22200. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Liu, H.; Huang, J.; Cheng, L.; Keller, E.T.; Parsons, S.J.; Hu, C.-D. Ionizing Radiation Induces Prostate Cancer Neuroendocrine Differentiation through Interplay of CREB and ATF2: Implications for Disease Progression. Cancer Res. 2008, 68, 9663–9670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Pietro, R.; Fang, H.; Fields, K.; Miller, S.; Flora, M.; Petricoin, E.C.; Dveksler, G.; Rana, R.A.; Grimley, P.M. Peroxiredoxin Genes are Not Induced in Myeloid Leukemia Cells Exposed to Ionizing Radiation. Int. J. Immunopathol. Pharmacol. 2006, 19, 517–524. [Google Scholar] [CrossRef] [Green Version]
- Rhee, S.G. Overview on Peroxiredoxin. Mol. Cells 2016, 39, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Diaz, A.J.G.; Yen, Y. The role of peroxiredoxin II in chemoresistance of breast cancer cells. Breast Cancer Targ. Ther. 2014, 6, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alper, O.; Bergmann-Leitner, E.S.; Abrams, S.; Cho-Chung, Y.S. Apoptosis, growth arrest and suppression of invasiveness by CRE-decoy oligonucleotide in ovarian cancer cells: Protein kinase A downregulation and cytoplasmic export of CRE-binding proteins. Mol. Cell. Biochem. 2001, 218, 55–63. [Google Scholar] [CrossRef]
- Steven, A.; Leisz, S.; Massa, C.; Iezzi, M.; Lattanzio, R.; Lamolinara, A.; Bukur, J.; Müller, A.; Hiebl, B.; Holzhausen, H.-J.; et al. HER-2/neu Mediates Oncogenic Transformation via Altered CREB Expression and Function. Mol. Cancer Res. 2013, 11, 1462–1477. [Google Scholar] [CrossRef] [Green Version]
- Xie, F.; Li, B.X.; Kassenbrock, A.; Xue, C.; Wang, X.; Qian, D.Z.; Sears, R.C.; Xiao, X. Identification of a Potent Inhibitor of CREB-Mediated Gene Transcription with Efficacious in Vivo Anticancer Activity. J. Med. Chem. 2015, 58, 5075–5087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsolou, A.; Liousia, M.; Kalamida, D.; Pouliliou, S.; Giatromanolaki, A.; Koukourakis, M. Inhibition of IKK-NFκB pathway sensitizes lung cancer cell lines to radiation. Cancer Biol. Med. 2017, 14, 293–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molavi Pordanjani, S.; Jalal Hosseinimehr, S. The Role of NF-κB Inhibitors in Cell Response to Radiation. Curr. Med. Chem. 2016, 23, 3951–3963. [Google Scholar] [CrossRef] [PubMed]
- Hara, K.; Takeda, A.; Tsurugai, Y.; Saigusa, Y.; Sanuki, N.; Eriguchi, T.; Maeda, S.; Tanaka, K.; Numata, K. Radiotherapy for Hepatocellular Carcinoma Results in Comparable Survival to Radiofrequency Ablation: A Propensity Score Analysis. Hepatology 2019, 69, 2533–2545. [Google Scholar] [CrossRef]
- Fuchs-Tarlovsky, V. Role of antioxidants in cancer therapy. Nutrition 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Impicciatore, G.; Sancilio, S.; Miscia, S.; Di Pietro, R. Nutlins and Ionizing Radiation in Cancer Therapy. Curr. Pharm. Des. 2010, 16, 1427–1442. [Google Scholar] [CrossRef]
- Nayak, S.K.; Khatik, G.L.; Narang, R.; Monga, V.; Chopra, H.K. p53-Mdm2 Interaction Inhibitors as Novel Nongenotoxic Anticancer Agents. Curr. Cancer Drug Targ. 2017, 18, 749–772. [Google Scholar] [CrossRef]
- Zauli, G.; Celeghini, C.; Melloni, E.; Voltan, R.; Ongari, M.; Tiribelli, M.; di Iasio, M.G.; Lanza, F.; Secchiero, P. The sorafenib plus nutlin-3 combination promotes synergistic cytotoxicity in acute myeloid leukemic cells irrespectively of FLT3 and p53 status. Haematologica 2012, 97, 1722–1730. [Google Scholar] [CrossRef] [Green Version]
- Gu, L.; Zhu, N.; Findley, H.W.; Zhou, M. MDM2 antagonist nutlin-3 is a potent inducer of apoptosis in pediatric acute lymphoblastic leukemia cells with wild-type p53 and overexpression of MDM2. Leukemia 2008, 22, 730–739. [Google Scholar] [CrossRef] [Green Version]
- Zauli, G.; Voltan, R.; Bosco, R.; Melloni, E.; Marmiroli, S.; Rigolin, G.M.; Cuneo, A.; Secchiero, P. Dasatinib Plus Nutlin-3 Shows Synergistic Antileukemic Activity in Both p53 wild-type and p53 mutated B Chronic Lymphocytic Leukemias by Inhibiting the Akt Pathway. Clin. Cancer Res. 2011, 17, 762–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roh, J.L.; Ko, J.H.; Moon, S.J.; Ryu, C.H.; Choi, J.Y.; Koch, W.M. The p53-reactivating small-molecule RITA enhances cisplatin-induced cytotoxicity and apoptosis in head and neck cancer. Cancer Lett. 2012, 325, 35–41. [Google Scholar] [CrossRef]
- Wiman, K.G. Pharmacological reactivation of mutant p53: From protein structure to the cancer patient. Oncogene 2010, 29, 4245–4252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hientz, K.; Mohr, A.; Bhakta-Guha, D.; Efferth, T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2017, 8, 8921–8946. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| ↑ CREB/ATF | Cofactors | Type of Cancer |
|---|---|---|
| CREB | ↑ GM-CSF | Acute myeloid leukaemia (AML) |
| CREB | ↑ GM-CSF | Acute lymphoblastic leukaemia (ALL) |
| CREB/ATF-1 | ↓ AP-2α, ↑ Bcl-2 | Human melanoma |
| CREB | ↑ GPR-81 | Breast cancer |
| CREB-1 | ↓ miR-1297 | Gastric cancer |
| CREB | ↑ Bcl-2, P-gp | Hepatocellular carcinoma (HCC) |
| CREB | ↑ GRK-3α | Pancreatic cancer |
| CREB | ↑ GRK-3α | Lung cancer |
| CREB | ↑ GRK-3α | Neuroendocrine prostate cancer (NEPC) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stati, G.; Passaretta, F.; Gindraux, F.; Centurione, L.; Di Pietro, R. The Role of the CREB Protein Family Members and the Related Transcription Factors in Radioresistance Mechanisms. Life 2021, 11, 1437. https://doi.org/10.3390/life11121437
Stati G, Passaretta F, Gindraux F, Centurione L, Di Pietro R. The Role of the CREB Protein Family Members and the Related Transcription Factors in Radioresistance Mechanisms. Life. 2021; 11(12):1437. https://doi.org/10.3390/life11121437
Chicago/Turabian StyleStati, Gianmarco, Francesca Passaretta, Florelle Gindraux, Lucia Centurione, and Roberta Di Pietro. 2021. "The Role of the CREB Protein Family Members and the Related Transcription Factors in Radioresistance Mechanisms" Life 11, no. 12: 1437. https://doi.org/10.3390/life11121437