Mitochondrial Dysfunction in Pancreatic Alpha and Beta Cells Associated with Type 2 Diabetes Mellitus






{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Model
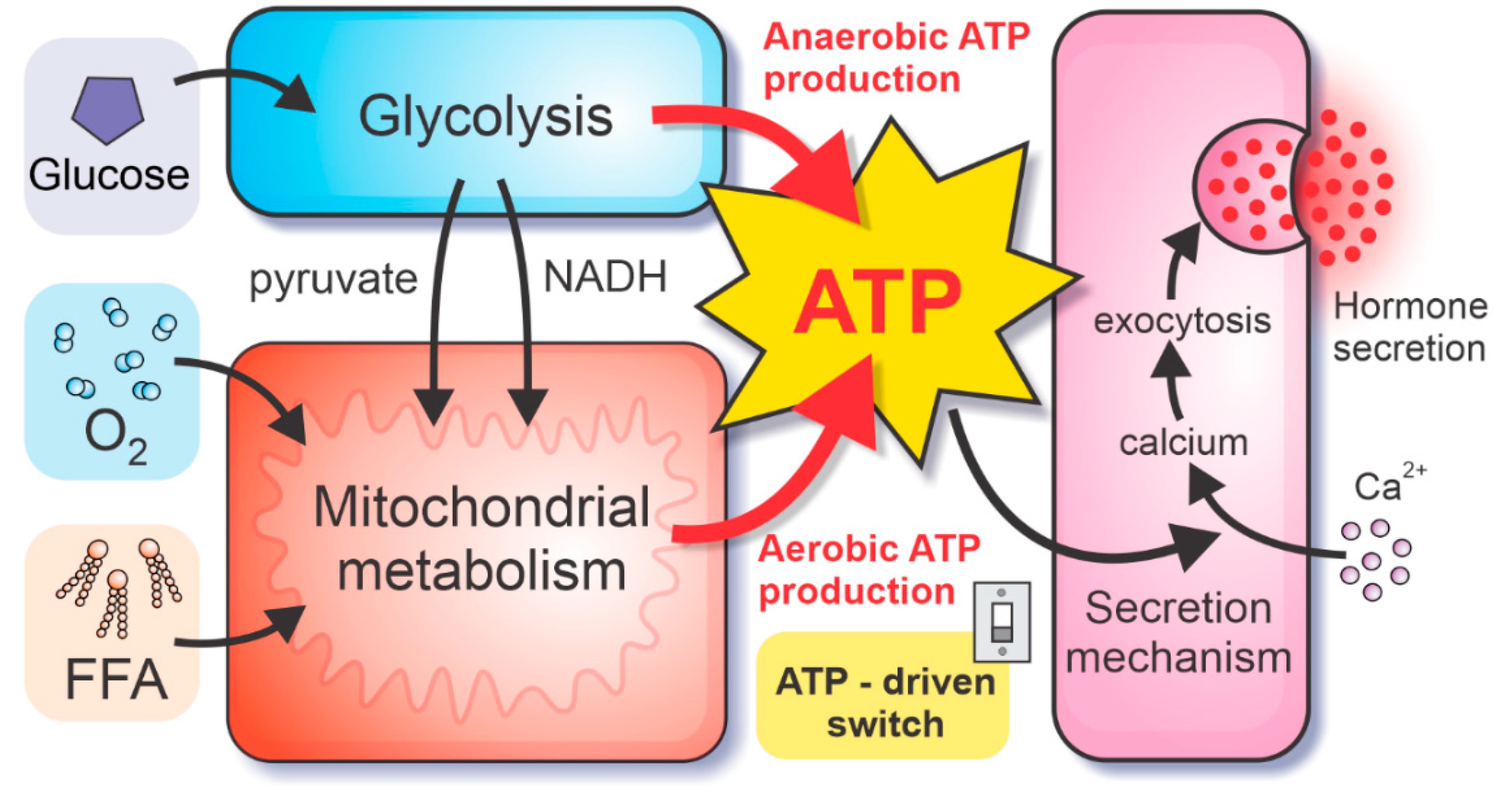
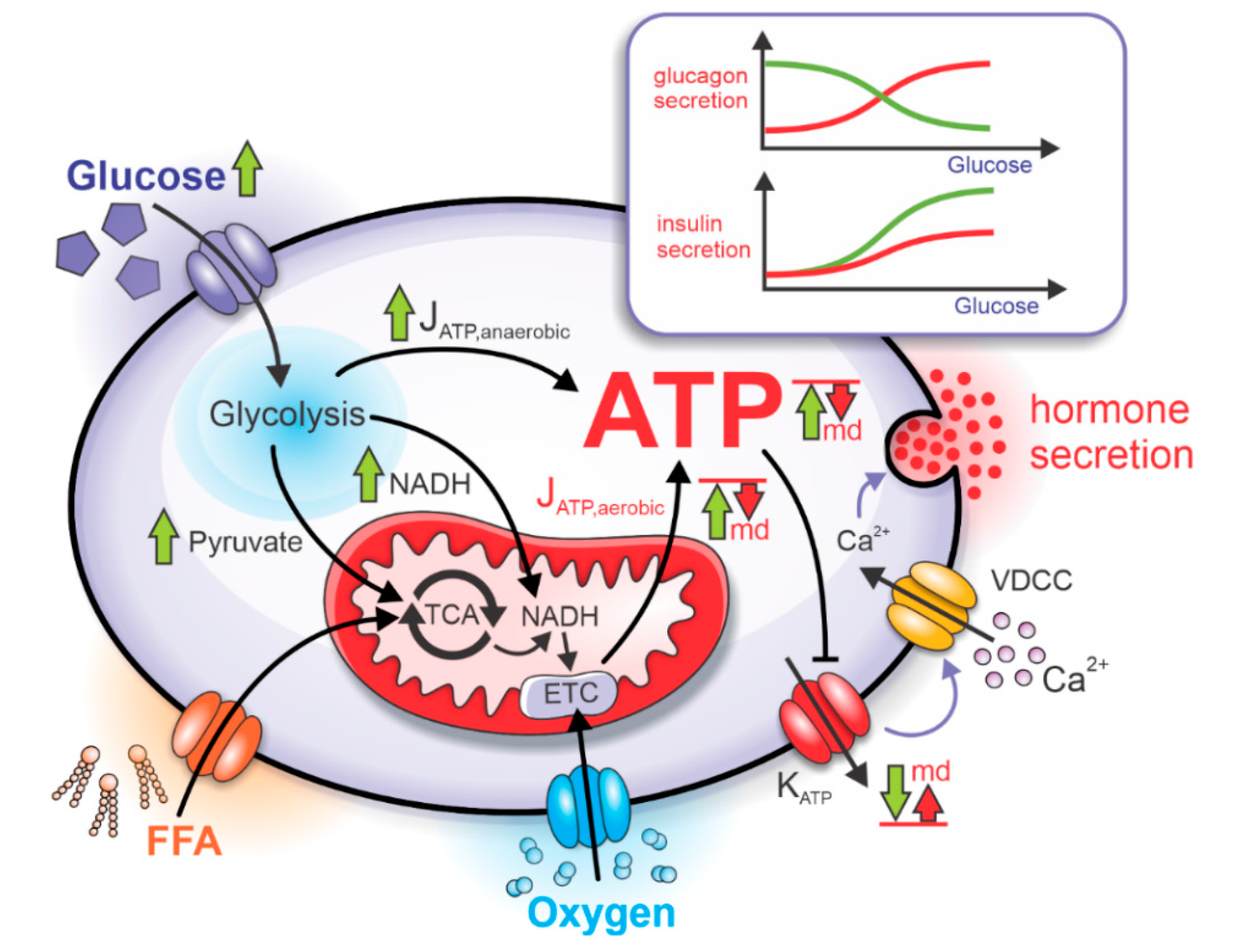
2.1. Bioenergetics and ATP-Driven Switch for Hormone Secretion
2.2. Mitochondrial Dysfunction and Pathophysiological Hormone Secretion
3. Results
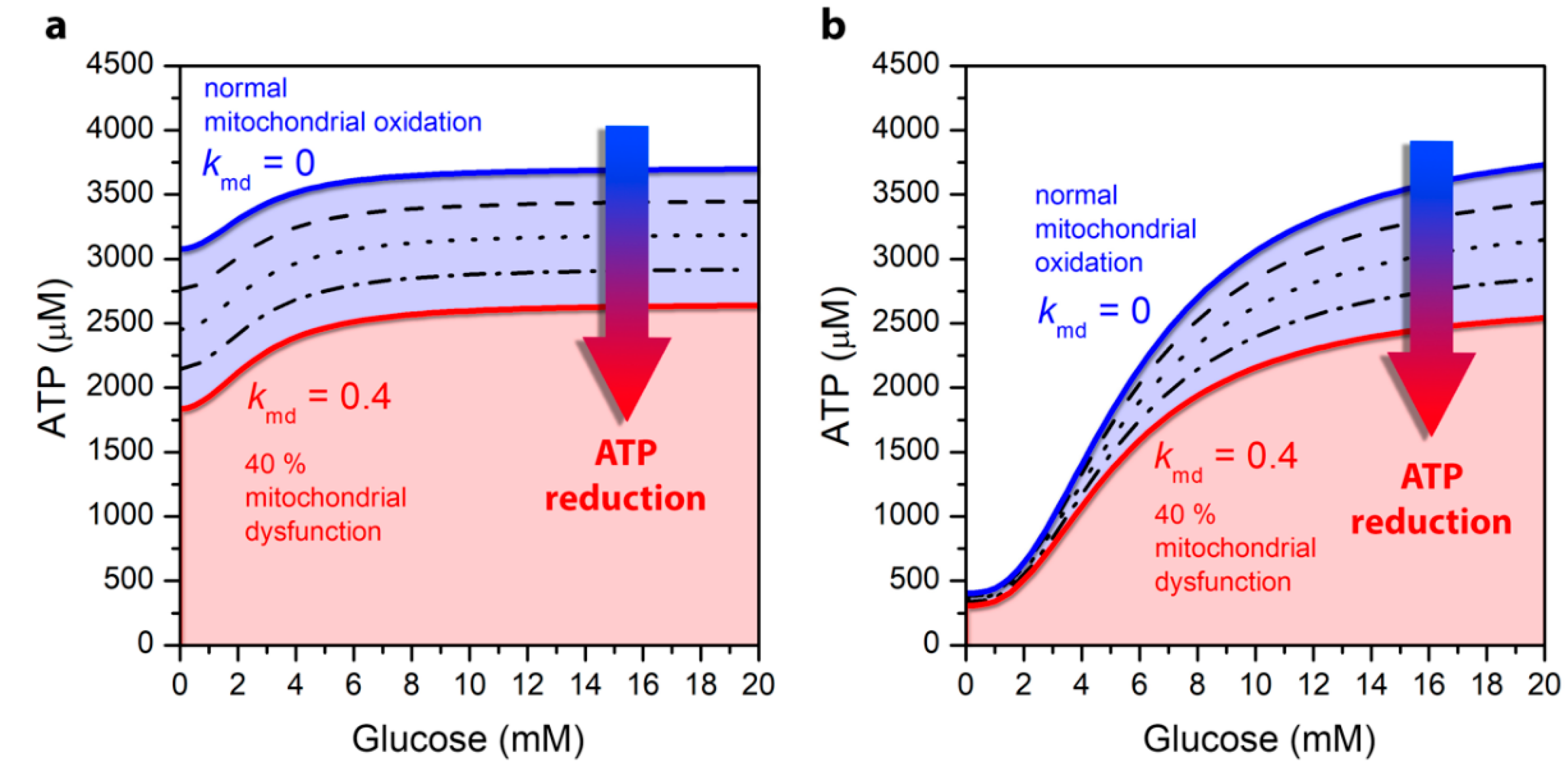
3.1. Mitochondrial Function Affects ATP Production
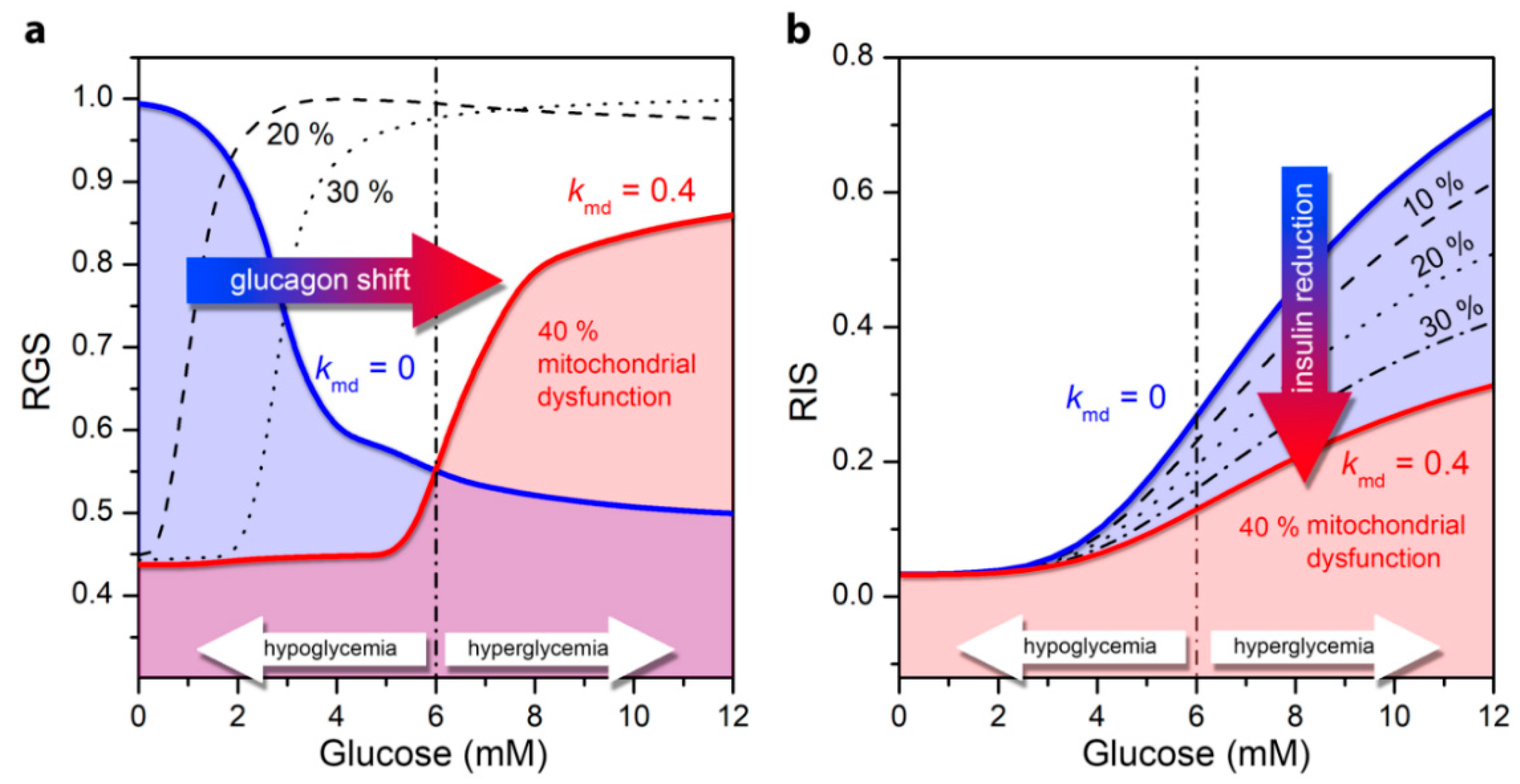
3.2. Impaired Glucagon and Insulin Secretion in the Pathogenesis of T2DM
4. Discussion
Author Contributions
Funding
Conflicts of Interest
Appendix A
References
- Legault, J.T.; Strittmatter, L.; Tardif, J.; Sharma, R.; Tremblay-Vaillancourt, V.; Aubut, C.; Boucher, G.; Clish, C.B.; Cyr, D.; Daneault, C.; et al. A Metabolic Signature of Mitochondrial Dysfunction Revealed through a Monogenic Form of Leigh Syndrome. Cell Rep. 2015, 13, 981–989. [Google Scholar] [CrossRef] [Green Version]
- Maechler, P.; Wollheim, C.B. Mitochondrial function in normal and diabetic β-cells. Nature 2001, 414, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Haythorne, E.; Rohm, M.; van de Bunt, M.; Brereton, M.F.; Tarasov, A.I.; Blacker, T.S.; Sachse, G.; dos Santos, M.S.; Exposito, R.T.; Davis, S.; et al. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β-cells. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Madreiter-Sokolowski, C.T.; Malli, R.M.; Graier, W.F. Mitochondrial–Endoplasmic Reticulum Interplay: A Lifelong On–Off Relationship? Contact 2019, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Zheng, X.; Ansari, Z.; Bunnell, M.C.; Herdy, J.R.; Traxler, L.; Lee, H.; Paquola, A.C.M.; Blithikioti, C.; Ku, M.; et al. Mitochondrial Aging Defects Emerge in Directly Reprogrammed Human Neurons due to Their Metabolic Profile. Cell Rep. 2018, 23, 2550–2558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabral-Costa, J.V.; Kowaltowski, A.J. Neurological disorders and mitochondria. Mol. Asp. Med. 2020, 71, 100826. [Google Scholar] [CrossRef]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.S.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Groschner, L.N.; Waldeck-Weiermair, M.; Malli, R.; Graier, W.F. Endothelial mitochondria-less respiration, more integration. Pflug. Arch. Eur. J. Physiol. 2012, 464, 63–76. [Google Scholar] [CrossRef] [Green Version]
- Bozi, L.H.M.; Campos, J.C.; Zambelli, V.O.; Ferreira, N.D.; Ferreira, J.C.B. Mitochondrially-targeted treatment strategies. Mol. Asp. Med. 2020, 71, 100836. [Google Scholar] [CrossRef]
- Madreiter-Sokolowski, C.T.; Sokolowski, A.A.; Waldeck-Weiermair, M.; Malli, R.; Graier, W.F. Targeting mitochondria to counteract age-related cellular dysfunction. Genes 2018, 9, 165. [Google Scholar] [CrossRef] [Green Version]
- Kowaltowski, A.J.; Oliveira, M.F. Mitochondria: New developments in pathophysiology. Mol. Asp. Med. 2020, 71. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.E.; He, J.; Menshikova, E.V.; Ritov, V.B. Dysfunction of mitochondria in human skeletal muscle in type 2 diabetes. Diabetes 2002, 51, 2944–2950. [Google Scholar] [CrossRef] [Green Version]
- Patti, M.E.; Butte, A.J.; Crunkhorn, S.; Cusi, K.; Berria, R.; Kashyap, S.; Miyazaki, Y.; Kohane, I.; Costello, M.; Saccone, R.; et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003, 100, 8466–8471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu Bakar, M.H.; Sarmidi, M.R. Association of cultured myotubes and fasting plasma metabolite profiles with mitochondrial dysfunction in type 2 diabetes subjects. Mol. Biosyst. 2017, 13, 1838–1853. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K. Mitochondria in Diabetes Mellitus. In Mitochondria in Health and Disease; Berdanier, C.D., Ed.; CRC Press: Boca Raton, FL, USA, 2005; pp. 377–454. [Google Scholar]
- Patti, M.E.; Corvera, S. The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr. Rev. 2010, 31, 364–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunemaker, C.S.; Zhang, M.; Satin, L.S. Insulin feedback alters mitochondrial activity through an ATP-sensitive K+ channel-dependent pathway in mouse islets and β-cells. Diabetes 2004, 53, 1765–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anello, M.; Lupi, R.; Spampinato, D.; Piro, S.; Masini, M.; Boggi, U.; Del Prato, S.; Rabuazzo, A.M.; Purrello, F.; Marchetti, P. Functional and morphological alterations of mitochondria in pancreatic beta cells from type 2 diabetic patients. Diabetologia 2005, 48, 282–289. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Koshkin, V.; Allister, E.M.; Gyulkhandanyan, A.V.; Wheeler, M.B. Molecular and metabolic evidence for mitochondrial defects associated with β-cell dysfunction in a mouse model of type 2 diabetes. Diabetes 2010, 59, 448–459. [Google Scholar] [CrossRef] [Green Version]
- Sivitz, W.I.; Yorek, M.A. Mitochondrial dysfunction in diabetes: From molecular mechanisms to functional significance and therapeutic opportunities. Antioxid. Redox Signal. 2010, 12, 537–577. [Google Scholar] [CrossRef] [Green Version]
- Del Guerra, S.; Lupi, R.; Marselli, L.; Masini, M.; Bugliani, M.; Sbrana, S.; Torri, S.; Pollera, M.; Boggi, U.; Mosca, F.; et al. Functional and molecular defects of pancreatic islets in human type 2 diabetes. Diabetes 2005, 54, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Petersen, K.F.; Befroy, D.; Dufour, S.; Dziura, J.; Ariyan, C.; Rothman, D.L.; DiPietro, L.; Cline, G.W.; Shulman, G.I. Mitochondrial dysfunction in the elderly: Possible role in insulin resistance. Science 2003, 300, 1140–1142. [Google Scholar] [CrossRef] [Green Version]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Garcia, R.; Shulman, G.I. Impaired Mitochondrial Activity in the Insulin-Resistant Offspring of Patients with Type 2 Diabetes. N. Engl. J. Med. 2004, 350, 664–671. [Google Scholar] [CrossRef] [Green Version]
- Assali, E.A.; Shlomo, D.; Zeng, J.; Taddeo, E.P.; Trudeau, K.M.; Erion, K.A.; Colby, A.H.; Grinstaff, M.W.; Liesa, M.; Las, G.; et al. Nanoparticle-mediated lysosomal reacidification restores mitochondrial turnover and function in β cells under lipotoxicity. FASEB J. 2019, 33, 4154–4165. [Google Scholar] [CrossRef] [PubMed]
- Rutter, G.A.; Georgiadou, E.; Martinez-Sanchez, A.; Pullen, T.J. Metabolic and functional specialisations of the pancreatic beta cell: Gene disallowance, mitochondrial metabolism and intercellular connectivity. Diabetologia 2020, 63, 1990–1998. [Google Scholar] [CrossRef] [PubMed]
- Grubelnik, V.; Markovič, R.; Lipovšek, S.; Leitinger, G.; Gosak, M.; Dolenšek, J.; Valladolid-Acebes, I.; Berggren, P.O.; Stožer, A.; Perc, M.; et al. Modelling of dysregulated glucagon secretion in type 2 diabetes by considering mitochondrial alterations in pancreatic α-cells. R. Soc. Open Sci. 2020, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubelnik, V.; Zmazek, J.; Markovič, R.; Gosak, M.; Marhl, M. Modelling of energy-driven switch for glucagon and insulin secretion. J. Theor. Biol. 2020, 493. [Google Scholar] [CrossRef]
- Wilson, D.F.; Cember, A.T.J.; Matschinsky, F.M. The thermodynamic basis of glucose-stimulated insulin release: A model of the core mechanism. Physiol. Rep. 2017, 5, 1–13. [Google Scholar] [CrossRef]
- Rorsman, P.; Braun, M.; Zhang, Q. Regulation of calcium in pancreatic α- and β-cells in health and disease. Cell Calcium 2012, 51, 300–308. [Google Scholar] [CrossRef] [Green Version]
- Rorsman, P.; Ashcroft, F.M. Pancreatic β-cell electrical activity and insulin secretion: Of mice and men. Physiol. Rev. 2018, 98, 117–214. [Google Scholar] [CrossRef]
- Ramracheya, R.; Ward, C.; Shigeto, M.; Walker, J.N.; Amisten, S.; Zhang, Q.; Johnson, P.R.; Rorsman, P.; Braun, M. Membrane potential-dependent inactivation of voltage-gated ion channels in α-cells inhibits glucagon secretion from human islets. Diabetes 2010, 59, 2198–2208. [Google Scholar] [CrossRef] [Green Version]
- Montefusco, F.; Pedersen, M.G. Mathematical modelling of local calcium and regulated exocytosis during inhibition and stimulation of glucagon secretion from pancreatic alpha-cells. J. Physiol. 2015, 593, 4519–4530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, M.G.; Bertram, R.; Sherman, A. Intra- and inter-islet synchronization of metabolically driven insulin secretion. Biophys. J. 2005, 89, 107–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doliba, N.M.; Qin, W.; Najafi, H.; Liu, C.; Buettger, C.W.; Sotiris, J.; Collins, H.W.; Li, C.; Stanley, C.A.; Wilson, D.F.; et al. Glucokinase activation repairs defective bioenergetics of islets of Langerhans isolated from type 2 diabetics. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E87–E102. [Google Scholar] [CrossRef] [Green Version]
- Knudsen, J.G.; Hamilton, A.; Ramracheya, R.; Tarasov, A.I.; Brereton, M.; Haythorne, E.; Chibalina, M.V.; Spégel, P.; Mulder, H.; Zhang, Q.; et al. Dysregulation of Glucagon Secretion by Hyperglycemia-Induced Sodium-Dependent Reduction of ATP Production. Cell Metab. 2019, 29, 430–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, E.D.; Wollheim, C.B. Role of mitochondrial calcium in metabolism-secretion coupling in nutrient-stimulated insulin release. Diabetes Metab. 1998, 24, 15–24. [Google Scholar] [PubMed]
- Quesada, I.; Todorova, M.G.; Alonso-Magdalena, P.; Beltrá, M.; Carneiro, E.M.; Martin, F.; Nadal, A.; Soria, B. Glucose induces opposite intracellular Ca2+ concentration oscillatory patterns in identified {alpha}- and β-cells within intact human islets of Langerhans. Diabetes 2006, 55, 2463–2469. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Ramracheya, R.; Lahmann, C.; Tarasov, A.; Bengtsson, M.; Braha, O.; Braun, M.; Brereton, M.; Collins, S.; Galvanovskis, J.; et al. Role of KATPchannels in glucose-regulated glucagon secretion and impaired counterregulation in type 2 diabetes. Cell Metab. 2013, 18, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Yu, Q.; Ahooghalandari, P.; Gribble, F.M.; Reimann, F.; Tengholm, A.; Gylfe, E. Submembrane ATP and Ca2+ kinetics in α-cells: Unexpected signaling for glucagon secretion. FASEB J. 2015, 29, 3379–3388. [Google Scholar] [CrossRef] [Green Version]
- Kusminski, C.M.; Chen, S.; Ye, R.; Sun, K.; Wang, Q.A.; Spurgin, S.B.; Sanders, P.E.; Brozinick, J.T.; Geldenhuys, W.J.; Li, W.H.; et al. MitoNEET-Parkin effects in pancreatic α- and β-cells, cellular survival, and intrainsular cross talk. Diabetes 2016, 65, 1534–1555. [Google Scholar] [CrossRef] [Green Version]
- Hosker, J.P.; Rudenski, A.S.; Burnett, M.A.; Matthews, D.R.; Turner, R.C. Similar reduction of first- and second-phase B-cell responses at three different glucose levels in type II diabetes and the effect of gliclazide therapy. Metabolism 1989, 38, 767–772. [Google Scholar] [CrossRef]
- Silva, J.P.; Köhler, M.; Graff, C.; Oldfors, A.; Magnuson, M.A.; Berggren, P.O.; Larsson, N.G. Impaired insulin secretion and β-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nat. Genet. 2000, 26, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G.; Lombardi, A.; Sorriento, D.; Anastasio, A.; Del Giudice, C.; Formisano, P.; Béguinot, F.; Trimarco, B.; Miele, C.; Iaccarino, G. Age-related impairment in insulin release: The essential role of β 2-adrenergic receptor. Diabetes 2012, 61, 692–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grespan, E.; Giorgino, T.; Arslanian, S.; Natali, A.; Ferrannini, E.; Mari, A. Defective amplifying pathway of β-cell secretory response to glucose in type 2 diabetes: Integrated modeling of in vitro and in vivo evidence. Diabetes 2018, 67, 496–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, E.D.; Maechler, P.; Wollheim, C.B. Effects of depletion of mitochondrial DNA in metabolism secretion coupling in INS-1 cells. Diabetes 1998, 47, 374–380. [Google Scholar] [CrossRef] [PubMed]
- Quesada, I.; Todorova, M.G.; Soria, B. Different metabolic responses in α-, β-, and δ-cells of the islet of langerhans monitored by redox confocal microscopy. Biophys. J. 2006, 90, 2641–2650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantley, J.; Ashcroft, F.M. Q & A: Insulin secretion and type 2 diabetes: Why do β-cells fail? BMC Biol. 2015, 13, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Rahier, J.; Guiot, Y.; Goebbels, R.M.; Sempoux, C.; Henquin, J.C. Pancreatic β-cell mass in European subjects with type 2 diabetes. Diabetes Obes. Metab. 2008, 10, 32–42. [Google Scholar] [CrossRef]
- Weir, G.C.; Bonner-Weir, S. Islet β cell mass in diabetes and how it relates to function, birth, and death. Ann. N. Y. Acad. Sci. 2013, 1281, 92–105. [Google Scholar] [CrossRef]
- Olsen, H.L.; Theander, S.; Bokvist, K.; Buschard, K.; Wollheim, C.B.; Gromada, J. Glucose stimulates glucagon release in single rat α-cells by mechanisms that mirror the stimulus-secretion coupling in β-cells. Endocrinology 2005, 146, 4861–4870. [Google Scholar] [CrossRef] [Green Version]
- Briant, L.J.B.; Dodd, M.S.; Chibalina, M.V.; Rorsman, N.J.G.; Johnson, P.R.V.; Carmeliet, P.; Rorsman, P.; Knudsen, J.G. CPT1a-Dependent Long-Chain Fatty Acid Oxidation Contributes to Maintaining Glucagon Secretion from Pancreatic Islets. Cell Rep. 2018, 23, 3300–3311. [Google Scholar] [CrossRef]
- Janah, L.; Kjeldsen, S.; Galsgaard, K.D.; Winther-Sørensen, M.; Stojanovska, E.; Pedersen, J.; Knop, F.K.; Holst, J.J.; Albrechtsen, N.J.W. Glucagon Receptor Signaling and Glucagon Resistance. Int. J. Mol. Sci. 2019, 20, 3314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unger, R.H.; Ohneda, A.; Aguilar-Parada, E.; Eisentraut, A.M. The role of aminogenic glucagon secretion in blood glucose homeostasis. J. Clin. Investig. 1969, 48, 810–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basco, D.; Zhang, Q.; Salehi, A.; Tarasov, A.; Dolci, W.; Herrera, P.; Spiliotis, I.; Berney, X.; Tarussio, D.; Rorsman, P.; et al. A-Cell Glucokinase Suppresses Glucose-Regulated Glucagon Secretion. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holst, J.J.; Albrechtsen, N.J.W.; Pedersen, J.; Knop, F.K. Glucagon and Amino Acids Are Linked in a Mutual Feedback Cycle: The Liver-α-Cell Axis. Diabetes 2017, 66, 235–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, E.D. A primary role for α-cells as amino acid sensors. Diabetes 2020, 69, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Dean, E.D.; Li, M.; Prasad, N.; Wisniewski, S.N.; Von Deylen, A.; Spaeth, J.; Maddison, L.; Botros, A.; Sedgeman, L.R.; Bozadjieva, N.; et al. Interrupted Glucagon Signaling Reveals Hepatic α Cell Axis and Role for L-Glutamine in α Cell Proliferation. Cell Metab. 2017, 25, 1362–1373. [Google Scholar] [CrossRef] [Green Version]
- Newsholme, P.; Brennan, L.; Bender, K. Amino acid metabolism, β-cell function, and diabetes. Diabetes 2006, 55, 39–47. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Li, C. Mechanisms of amino acid-stimulated insulin secretion in congenital hyperinsulinism. Acta Biochim. Biophys. Sin. 2013, 45, 36–43. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.A.; Sakura, H.; Coles, B.; Gummerson, N.; Proks, P.; Ashcroft, F.M. Electrogenic arginine transport mediates stimulus-secretion coupling in mouse pancreatic beta-cells. J. Physiol. 1997, 499, 625–635. [Google Scholar] [CrossRef] [Green Version]
- Newsholme, P.; Krause, M. Nutritional Regulation of Insulin Secretion: Implications for Diabetes. Clin. Biochem. Rev. 2012, 33, 35–47. [Google Scholar]
- Tengholm, A. Cyclic AMP dynamics in the pancreatic β-cell. Ups. J. Med. Sci. 2012, 117, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Seino, S. Cell signalling in insulin secretion: The molecular targets of ATP, cAMP and sulfonylurea. Diabetologia 2012, 55, 2096–2108. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.; Sandler, S.; Gylfe, E.; Tengholm, A. Glucose- and hormone-induced cAMP oscillations in α- and β-cells within intact pancreatic islets. Diabetes 2011, 60, 1535–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tengholm, A.; Gylfe, E. cAMP signalling in insulin and glucagon secretion. Diabetes Obes. Metab. 2017, 19, 42–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Q.; Shuai, H.; Ahooghalandari, P.; Gylfe, E.; Tengholm, A. Glucose controls glucagon secretion by directly modulating cAMP in alpha cells. Diabetologia 2019, 62, 1212–1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Y.E.; Chun, J.N.; Kwon, M.J.; Ji, Y.S.; Jeong, M.H.; Kim, H.H.; Park, S.H.; Rah, J.C.; Kang, J.S.; Lee, S.H.; et al. Endocytosis of KATP Channels Drives Glucose-Stimulated Excitation of Pancreatic β Cells. Cell Rep. 2018, 22, 471–481. [Google Scholar] [CrossRef] [Green Version]
- Onyango, A.N. Mechanisms of the Regulation and Dysregulation of Glucagon Secretion. Oxid. Med. Cell. Longev. 2020, 2020. [Google Scholar] [CrossRef]
- Hughes, J.W.; Ustione, A.; Lavagnino, Z.; Piston, D.W. Regulation of islet glucagon secretion: Beyond calcium. Diabetes Obes. Metab. 2018, 20, 127–136. [Google Scholar] [CrossRef] [Green Version]
- Rackham, C.L.; Hubber, E.L.; Czajka, A.; Malik, A.N.; King, A.J.F.; Jones, P.M. Optimizing beta cell function through mesenchymal stromal cell-mediated mitochondria transfer. Stem Cells 2020, 38, 574–584. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Li, H.; Yao, Y.; Zhao, T.; Chen, Y.Y.; Shen, Y.L.; Wang, L.L.; Zhu, Y. Stem cell-derived mitochondria transplantation: A novel strategy and the challenges for the treatment of tissue injury. Stem Cell Res. Ther. 2018, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Ji, K.; Guo, L.; Wu, W.; Lu, H.; Shan, P.; Yan, C. Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc. Res. 2014, 92, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Vasic, V.; Barth, K.; Schmidt, M.H.H. Neurodegeneration and neuro-regeneration—Alzheimer’s disease and stem cell therapy. Int. J. Mol. Sci. 2019, 20, 4272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raoof, R.; van der Vlist, M.; Willemen, H.L.D.M.; Prado, J.; Versteeg, S.; Vos, M.; Lockhorst, R.; Pasterkamp, J.; Khoury-Hanold, W.; Meyaard, L.; et al. Macrophages transfer mitochondria to sensory neurons to resolve inflammatory pain. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Leng, Z.; Zhu, R.; Hou, W.; Feng, Y.; Yang, Y.; Han, Q.; Shan, G.; Meng, F.; Du, D.; Wang, S.; et al. Transplantation of ACE2-Mesenchymal stem cells improves the outcome of patients with covid-19 pneumonia. Aging Dis. 2020, 11, 216–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorlund, K.; Dron, L.; Park, J.; Hsu, G.; Forrest, J.I.; Mills, E.J. A real-time dashboard of clinical trials for COVID-19. Lancet Digit. Health 2020, 2, e286–e287. [Google Scholar] [CrossRef]
- Bavamian, S.; Klee, P.; Britan, A.; Populaire, C.; Caille, D.; Cancela, J.; Charollais, A.; Meda, P. Islet-cell-to-cell communication as basis for normal insulin secretion. Diabetes Obes. Metab. 2007, 9, 118–132. [Google Scholar] [CrossRef]
- Jain, R.; Lammert, E. Cell-cell interactions in the endocrine pancreas. Diabetes Obes. Metab. 2009, 11, 159–167. [Google Scholar] [CrossRef]
- Briant, L.J.B.; Reinbothe, T.M.; Spiliotis, I.; Miranda, C.; Rodriguez, B.; Rorsman, P. Δ-Cells and Β-Cells Are Electrically Coupled and Regulate A-Cell Activity Via Somatostatin. J. Physiol. 2018, 596, 197–215. [Google Scholar] [CrossRef]
- Salem, V.; Silva, L.D.; Suba, K.; Georgiadou, E.; Gharavy, S.N.M.; Akhtar, N.; Martin-Alonso, A.; Gaboriau, D.C.A.; Rothery, S.M.; Stylianides, T.; et al. Leader β-cells coordinate Ca2+ dynamics across pancreatic islets in vivo. Nat. Metab. 2019, 1, 615–629. [Google Scholar] [CrossRef] [Green Version]
- Zavala, E.; Wedgwood, K.C.A.; Voliotis, M.; Tabak, J.; Spiga, F.; Lightman, S.L.; Tsaneva-Atanasova, K. Mathematical Modelling of Endocrine Systems. Trends Endocrinol. Metab. 2019, 30, 244–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klemen, M.S.; Dolenšek, J.; Rupnik, M.S.; Stožer, A. The triggering pathway to insulin secretion: Functional similarities and differences between the human and the mouse β cells and their translational relevance. Islets 2017, 9, 109–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosco, D.; Haefliger, J.A.; Meda, P. Connexins: Key mediators of endocrine function. Physiol. Rev. 2011, 91, 1393–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benninger, R.K.P.; Piston, D.W. Cellular communication and heterogeneity in pancreatic islet insulin secretion dynamics. Trends Endocrinol. Metab. 2014, 25, 399–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šterk, M.; Dolenšek, J.; Bombek, L.K.; Markovič, R.; Zakelšek, D.; Perc, M.; Pohorec, V.; Stožer, A.; Gosak, M. Assessing the origin and velocity of Ca2+ waves in three-dimensional tissue: Insights from a mathematical model and confocal imaging in mouse pancreas tissue slices. Commun. Nonlinear Sci. Numer. Simul. 2021, 93, 105495. [Google Scholar] [CrossRef]
- Hauge-Evans, A.C.; King, A.J.; Fairhall, K.; Persaud, S.J.; Jones, P.M. A role for islet somatostatin in mediating sympathetic regulation of glucagon secretion. Islets 2010, 2, 341–344. [Google Scholar] [CrossRef] [Green Version]
- Vergari, E.; Knudsen, J.G.; Ramracheya, R.; Salehi, A.; Zhang, Q.; Adam, J.; Asterholm, I.W.; Benrick, A.; Briant, L.J.B.; Chibalina, M.V.; et al. Insulin inhibits glucagon release by SGLT2-induced stimulation of somatostatin secretion. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Kellard, J.A.; Rorsman, N.J.G.; Hill, T.G.; Armour, S.L.; van de Bunt, M.; Rorsman, P.; Knudsen, J.G.; Briant, L.J.B. Reduced somatostatin signalling leads to hypersecretion of glucagon in mice fed a high-fat diet. Mol. Metab. 2020, 40, 101021. [Google Scholar] [CrossRef]
- Xavier, G.D.S.; Rutter, G.A. Metabolic and Functional Heterogeneity in Pancreatic β Cells. J. Mol. Biol. 2020, 432, 1395–1406. [Google Scholar] [CrossRef]
- Dwulet, J.A.M.; Ludin, N.W.F.; Piscopio, R.A.; Schleicher, W.E.; Moua, O.; Westacott, M.J.; Benninger, R.K.P. How Heterogeneity in Glucokinase and Gap-Junction Coupling Determines the Islet [Ca2+] Response. Biophys. J. 2019, 117, 2188–2203. [Google Scholar] [CrossRef]
- Stožer, A.; Markovič, R.; Dolenšek, J.; Perc, M.; Marhl, M.; Rupnik, M.S.; Gosak, M. Heterogeneity and delayed activation as hallmarks of self-organization and criticality in excitable tissue. Front. Physiol. 2019, 10, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Stožer, A.; Gosak, M.; Dolenšek, J.; Perc, M.; Marhl, M.; Rupnik, M.S.; Korošak, D. Functional Connectivity in Islets of Langerhans from Mouse Pancreas Tissue Slices. PLoS Comput. Biol. 2013, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, N.R.; Mitchell, R.K.; Haythorne, E.; Pessoa, M.P.; Semplici, F.; Ferrer, J.; Piemonti, L.; Marchetti, P.; Bugliani, M.; Bosco, D.; et al. Beta Cell Hubs Dictate Pancreatic Islet Responses to Glucose. Cell Metab. 2016, 24, 389–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gosak, M.; Markovič, R.; Dolenšek, J.; Rupnik, M.S.; Marhl, M.; Stožer, A.; Perc, M. Network science of biological systems at different scales: A review. Phys. Life Rev. 2018, 24, 118–135. [Google Scholar] [CrossRef] [PubMed]
- Gosak, M.; Stožer, A.; Markovič, R.; Dolenšek, J.; Marhl, M.; Rupnik, M.S.; Perc, M. The relationship between node degree and dissipation rate in networks of diffusively coupled oscillators and its significance for pancreatic beta cells. Chaos 2015, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benninger, R.K.P.; Hodson, D.J. New understanding of β-cell heterogeneity and in situ islet function. Diabetes 2018, 67, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Westacott, M.J.; Farnsworth, N.L.; Clair, J.R.S.; Poffenberger, G.; Heintz, A.; Ludin, N.W.; Hart, N.J.; Powers, A.C.; Benninger, R.K.P. Age-dependent decline in the coordinated [Ca2+] and insulin secretory dynamics in human pancreatic islets. Diabetes 2017, 66, 2436–2445. [Google Scholar] [CrossRef] [Green Version]
- Kahn, S.E.; Hull, R.L.; Utzschneider, K.M. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 2006, 444, 840–846. [Google Scholar] [CrossRef]
- Estrada, E. Network robustness to targeted attacks. the interplay of expansibility and degree distribution. Eur. Phys. J. B 2006, 52, 563–574. [Google Scholar] [CrossRef]
- Huang, Y.C.; Rupnik, M.; Gaisano, H.Y. Unperturbed islet α-cell function examined in mouse pancreas tissue slices. J. Physiol. 2011, 589, 395–408. [Google Scholar] [CrossRef]
- Briant, L.J.B.; Zhang, Q.; Vergari, E.; Kellard, J.A.; Rodriguez, B.; Ashcroft, F.M.; Rorsman, P. Functional identification of islet cell types by electrophysiological fingerprinting. J. R. Soc. Interface 2017, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montefusco, F.; Cortese, G.; Pedersen, M.G. Heterogeneous alpha-cell population modeling of glucose-induced inhibition of electrical activity. J. Theor. Biol. 2020, 485. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grubelnik, V.; Zmazek, J.; Markovič, R.; Gosak, M.; Marhl, M. Mitochondrial Dysfunction in Pancreatic Alpha and Beta Cells Associated with Type 2 Diabetes Mellitus. Life 2020, 10, 348. https://doi.org/10.3390/life10120348
Grubelnik V, Zmazek J, Markovič R, Gosak M, Marhl M. Mitochondrial Dysfunction in Pancreatic Alpha and Beta Cells Associated with Type 2 Diabetes Mellitus. Life. 2020; 10(12):348. https://doi.org/10.3390/life10120348
Chicago/Turabian StyleGrubelnik, Vladimir, Jan Zmazek, Rene Markovič, Marko Gosak, and Marko Marhl. 2020. "Mitochondrial Dysfunction in Pancreatic Alpha and Beta Cells Associated with Type 2 Diabetes Mellitus" Life 10, no. 12: 348. https://doi.org/10.3390/life10120348