Measurement of Process Conditions Present in Pilot Scale Iron Ore Sintering


Abstract
:1. Introduction
2. Methodology
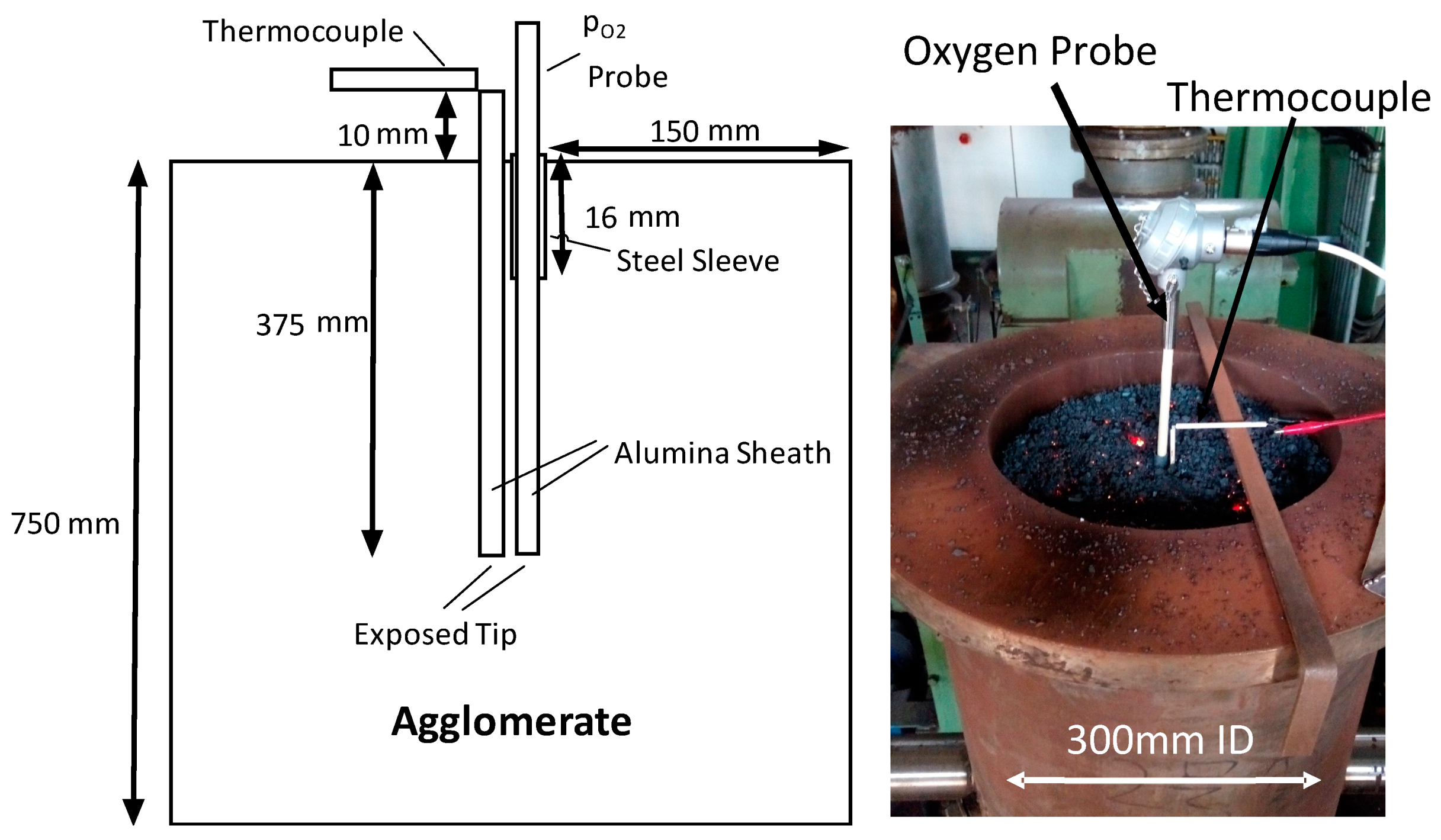
2.1. Pilot Scale Sinter Pot Tests
- Local differences in phase and particle distribution. These are directly related to the materials used in the feed, particle size distribution and methods of granulation. The differences in local chemical composition and phase assemblage result in differences in enthalpy of reaction and enthalpy of fusion. The combustion of coke provides a local heat source.
- Partial Melting. Whilst the initial sinter charge resembles a randomly packed bed of solids during sintering, partial melting of the bed takes place resulting in channelling of the gas and changes to bed porosity. These changes result in changes to the local rates of heat transfer from gas to condensed phases.
- Bed shrinkage. The partial melting of the burden leads to an increase in bulk density and shrinkage of the bed. The interaction of the sinter bed and the probes during sintering can lead to the creation of voids and changes to the local voidage around the probe. This can be minimised by inserting the measuring devices from the top of the bed rather than from the side, enabling the probe to move with the bed and reduce the hold-up of the bed by the probe. However, it is recognised that the bed has moved relative to the thermocouple during the measurement. The extent of the movement depends on the initial position of the thermocouple and the overall change in bed height during sintering, typically 13–15 vol. %.
- Thermocouple type. To protect the metal thermocouples, prevent the thermocouple wires from breaking during bed shrinkage, provide stiffness for insertion into the bed and prevent a junction forming from crossed thermocouple wires, the thermocouple wires are inserted into an aluminium oxide sheath. The material and mass of the oxide sheath has the potential to influence the local thermal profile and response time, particularly if the thermocouple tip is enclosed by the sheath.
- Probe type. A range of materials and designs can be used in solid state oxygen probes. The materials and dimensions of the sensor tip can influence the response time and sensitivity of the probe to changes in temperature and oxygen potential in the gas phase with time. To determine the oxygen partial pressure, separate or combined measurement of temperature and electromotive force (EMF), ∆E across a solid electrolyte may be undertaken. The calculated oxygen partial pressure, (∆E = 2.303 RT/nF. ln(pO2, unknown./pO2, reference.)) is a function of both the measured temperature (T) and the measured electrical potential difference (∆E). As such, the difference, if any, between the measured temperature and that at the tip of the solid electrolyte is required in order to accurately determine the actual pO2.
- The rate of response of the oxygen probe to changes in oxygen pressure is dependent on temperature. At high temperatures the response times are rapid and the EMF measurements provide accurate measurement of the pO2. At low temperatures in rapidly changing systems, the EMF measured is limited by the rates of ionic transfer in the solid state probes, and measurements should be treated with caution as these may not accurately reflect the pO2 at the time of measurement.
- The rate of response to changes in the oxygen partial pressure of the system is dependent on mass transfer from the system to the outer surface of the probe. The mass transfer is fastest when the oxygen probe is in contact with the gas phase. If, during the sinter process, the sensor tip is partially or fully covered with liquid and/or solids, again the EMF recorded may not accurately reflect the gas partial pressure at the time of measurement.
2.2. Laboratory Scale Tests
3. Results
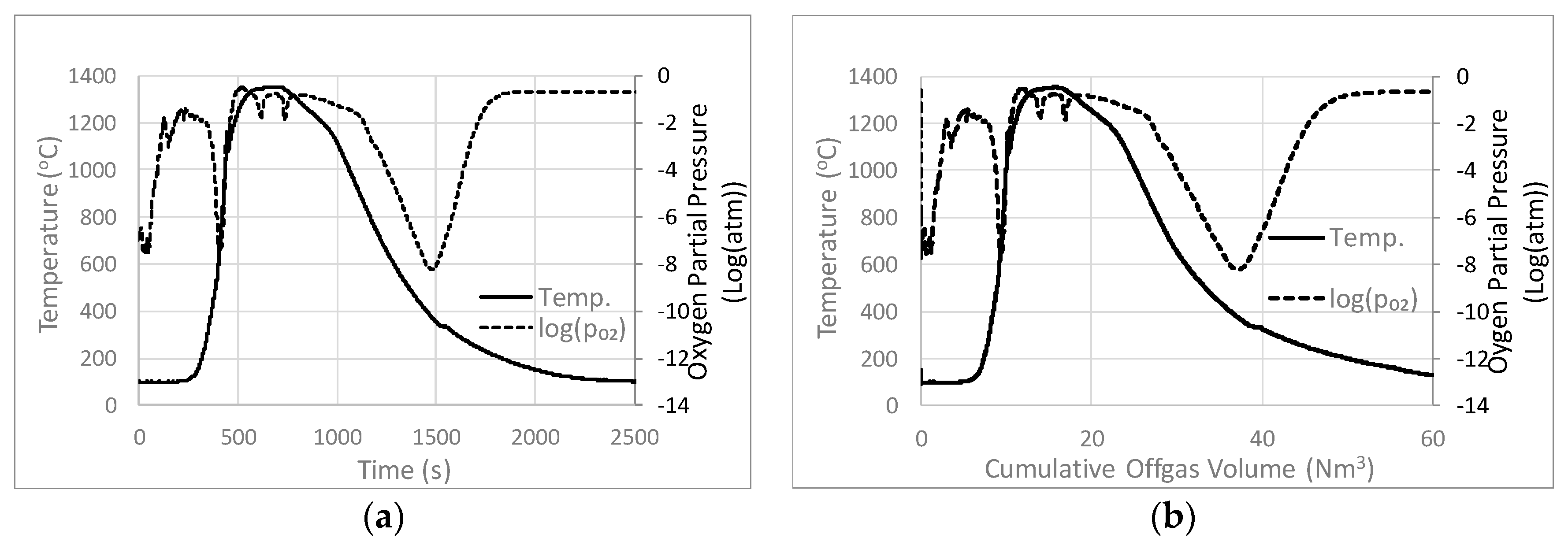
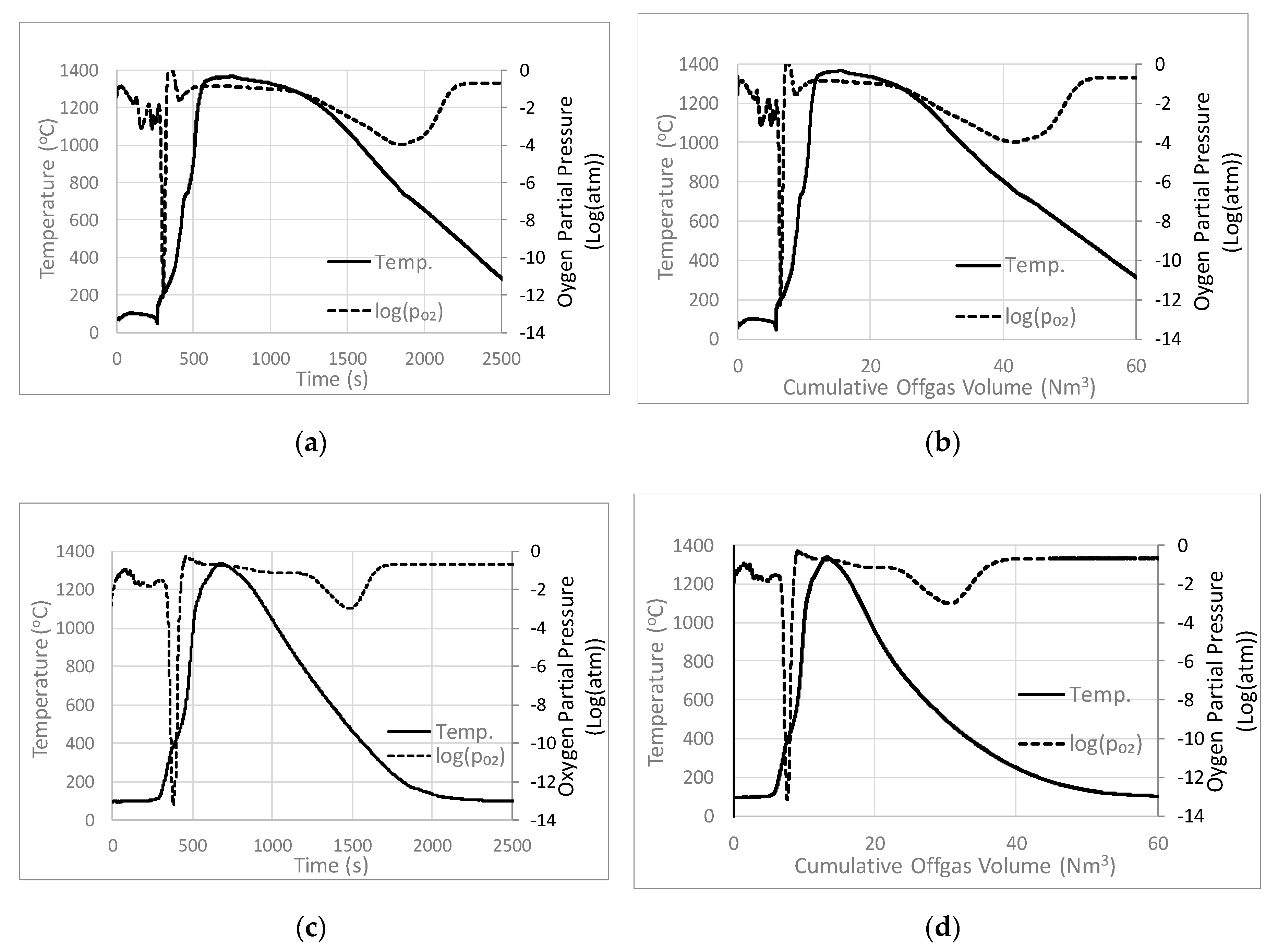
3.1. Base Case Temperature and pO2
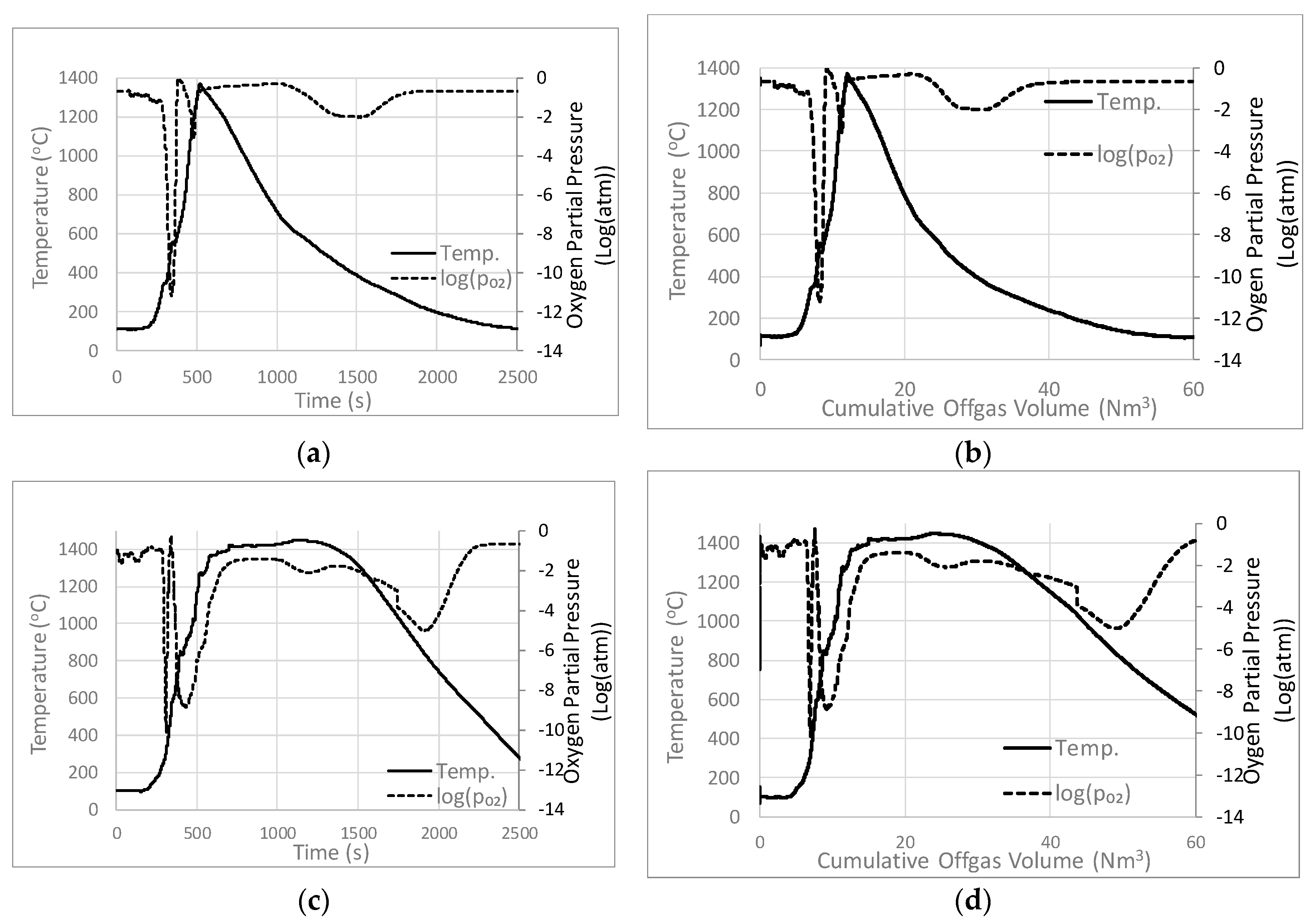
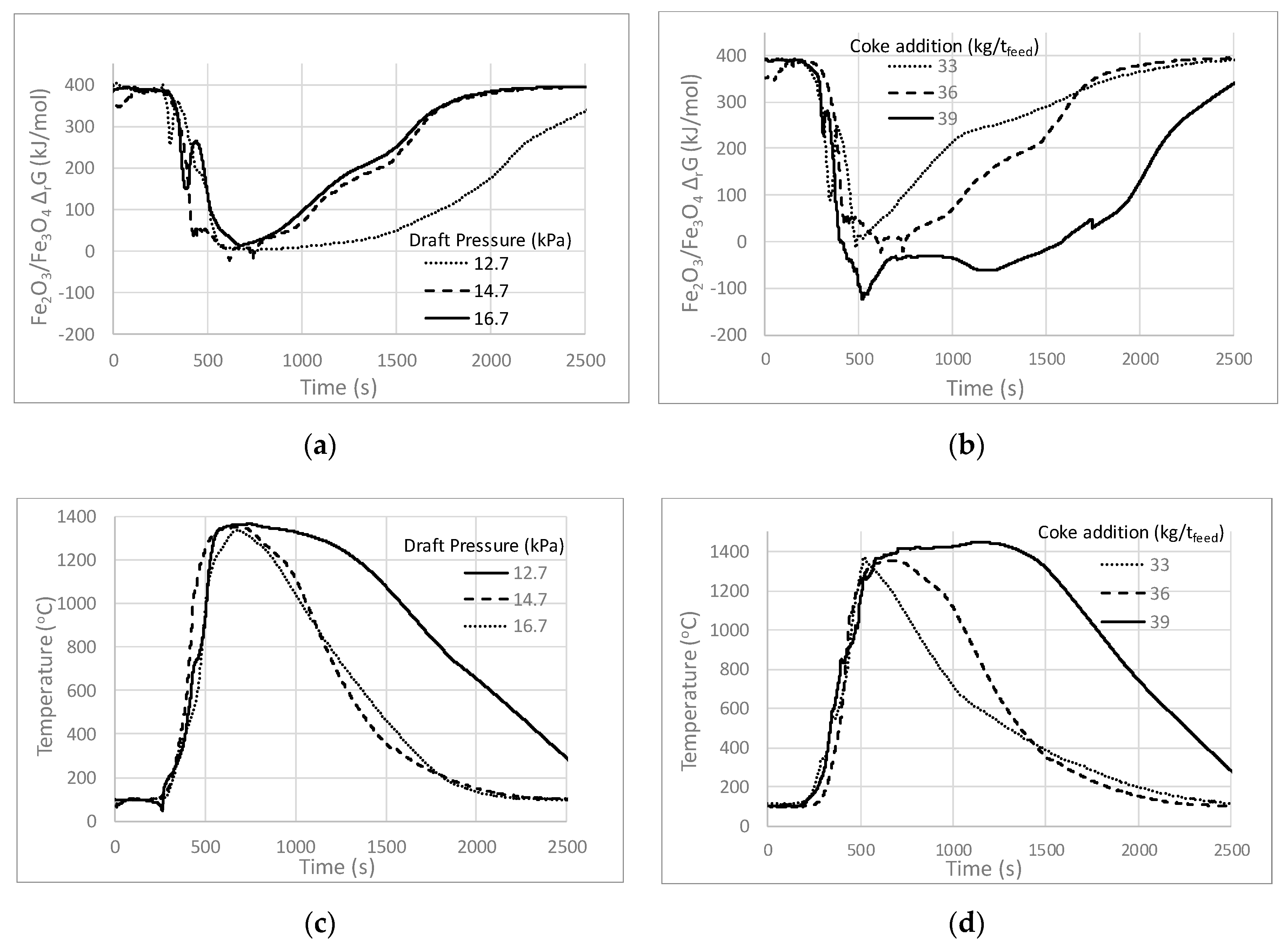
3.2. Effects of Coke Addition on Temperature and pO2
3.3. Effects of Draft Pressure on Temperature and pO2
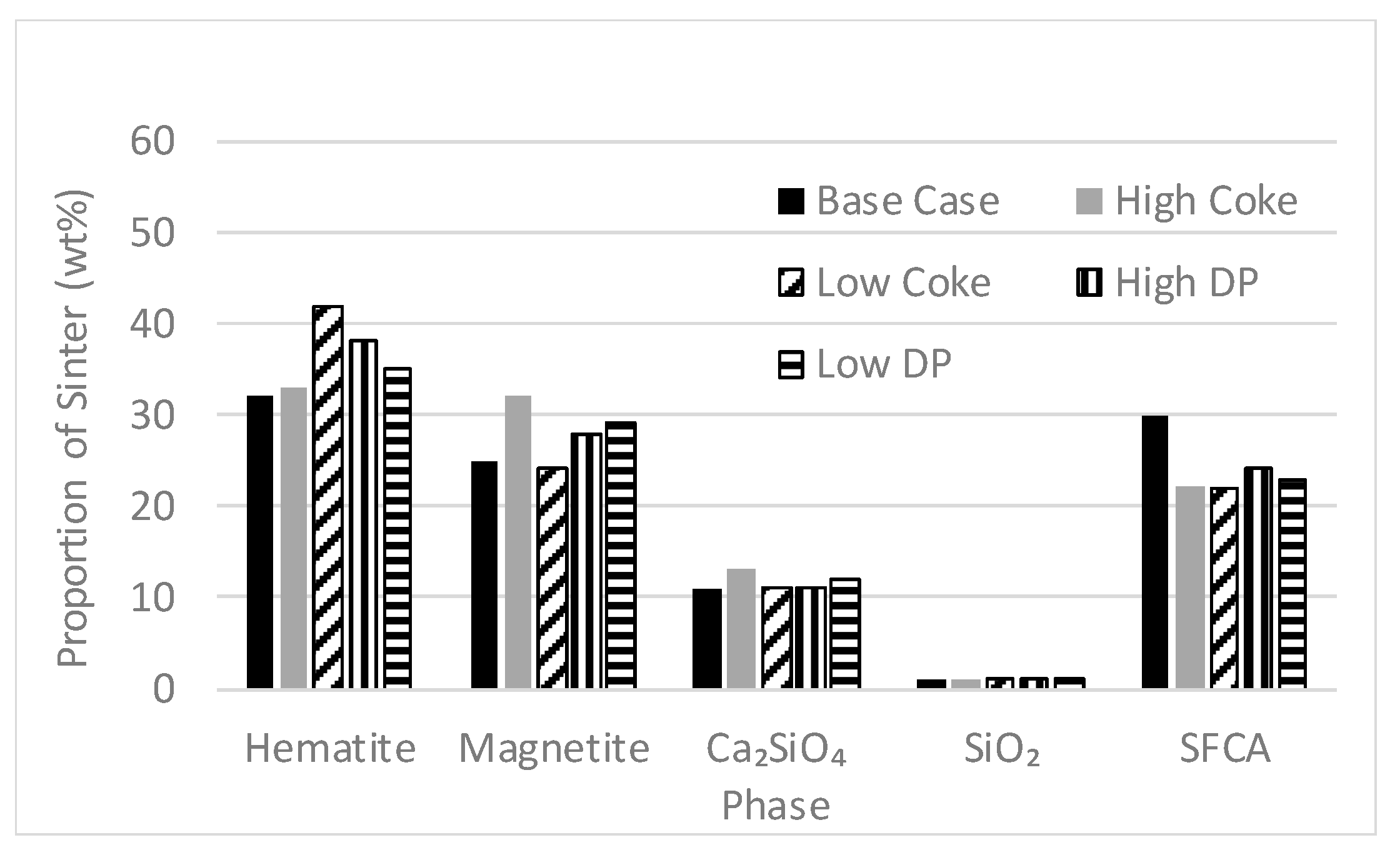
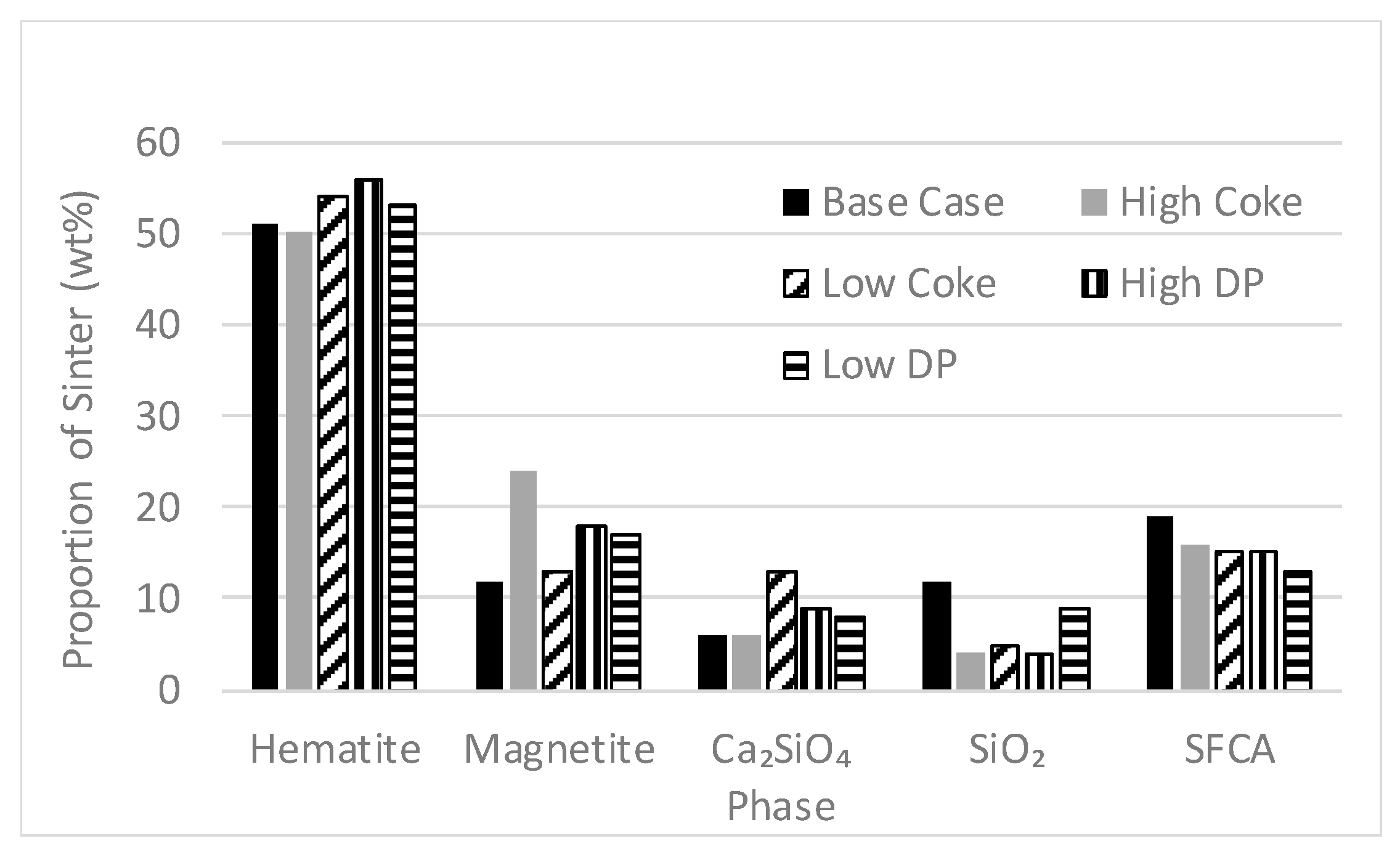
3.4. Proportions of Phases in Sinter Products and Fines
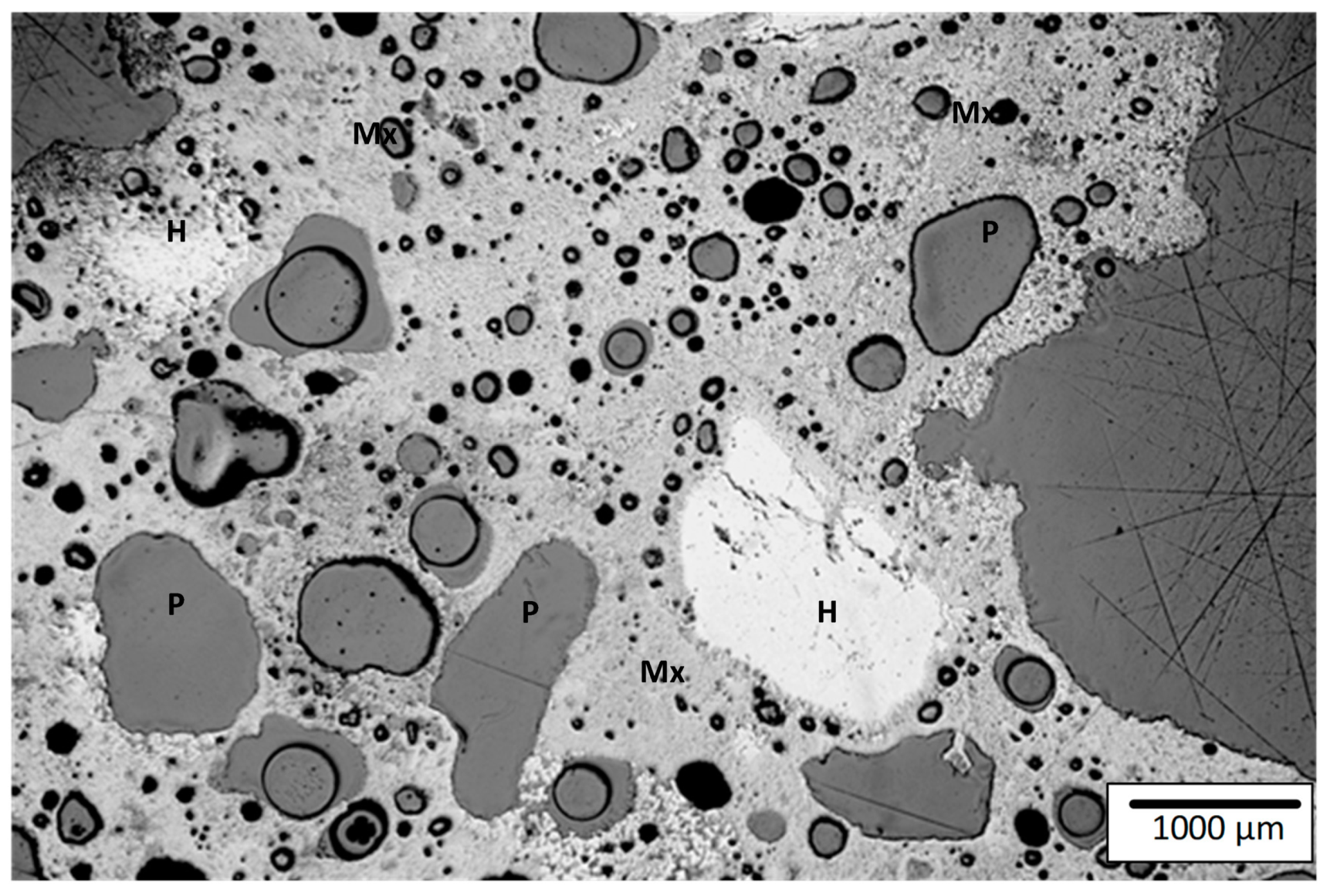
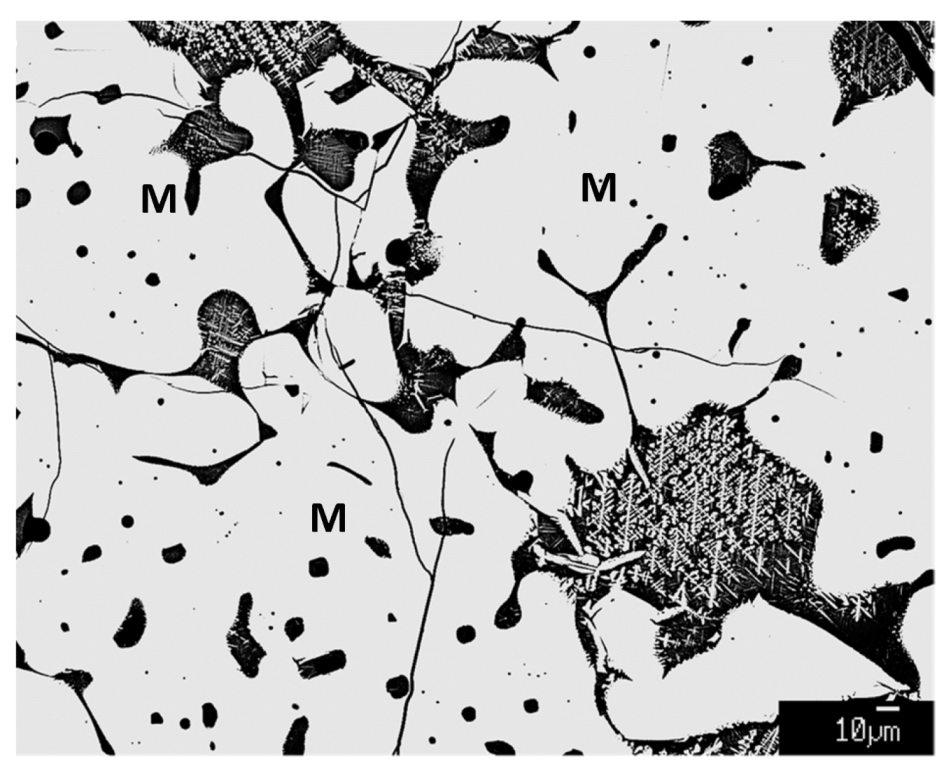
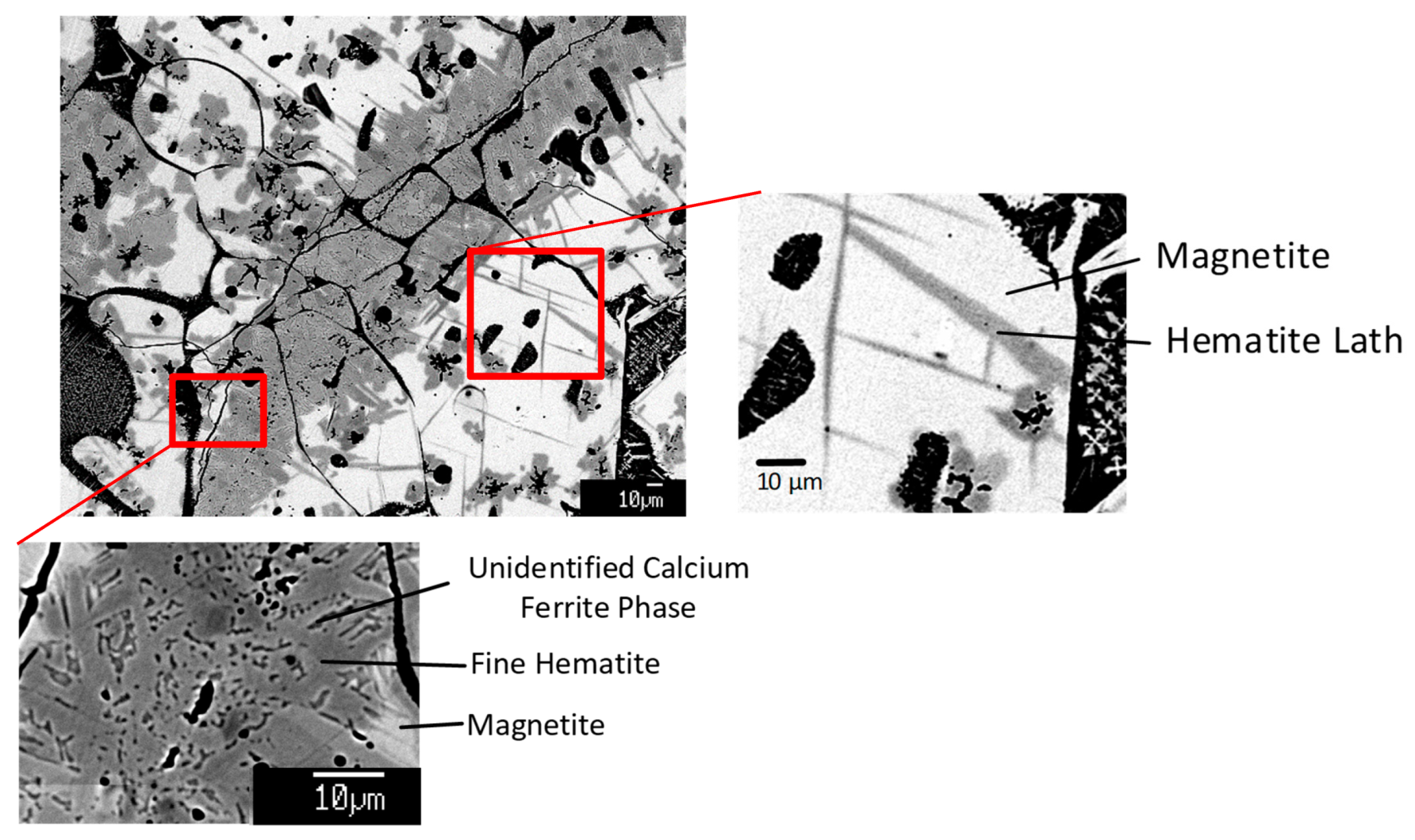
3.5. Microstructures formed in Sinter Products
4. Discussion
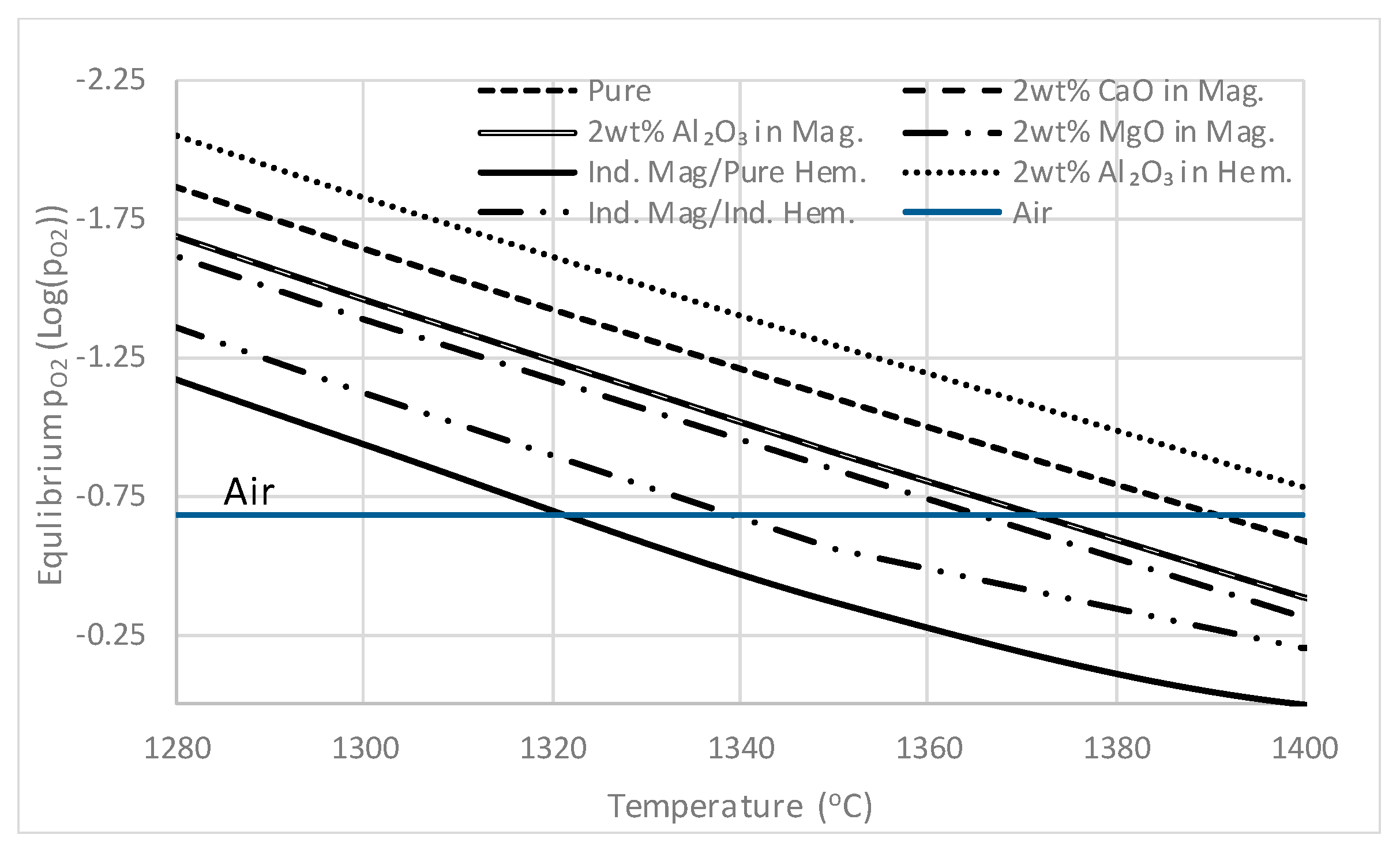
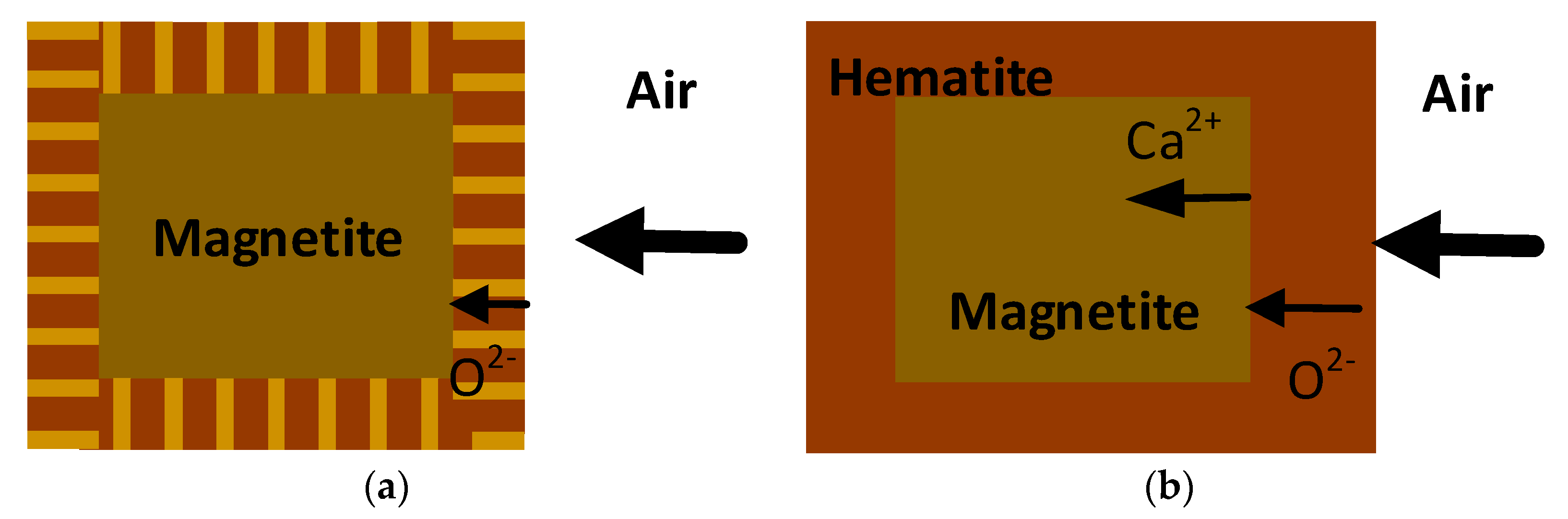
4.1. Hematite and Magnetite in Sinter
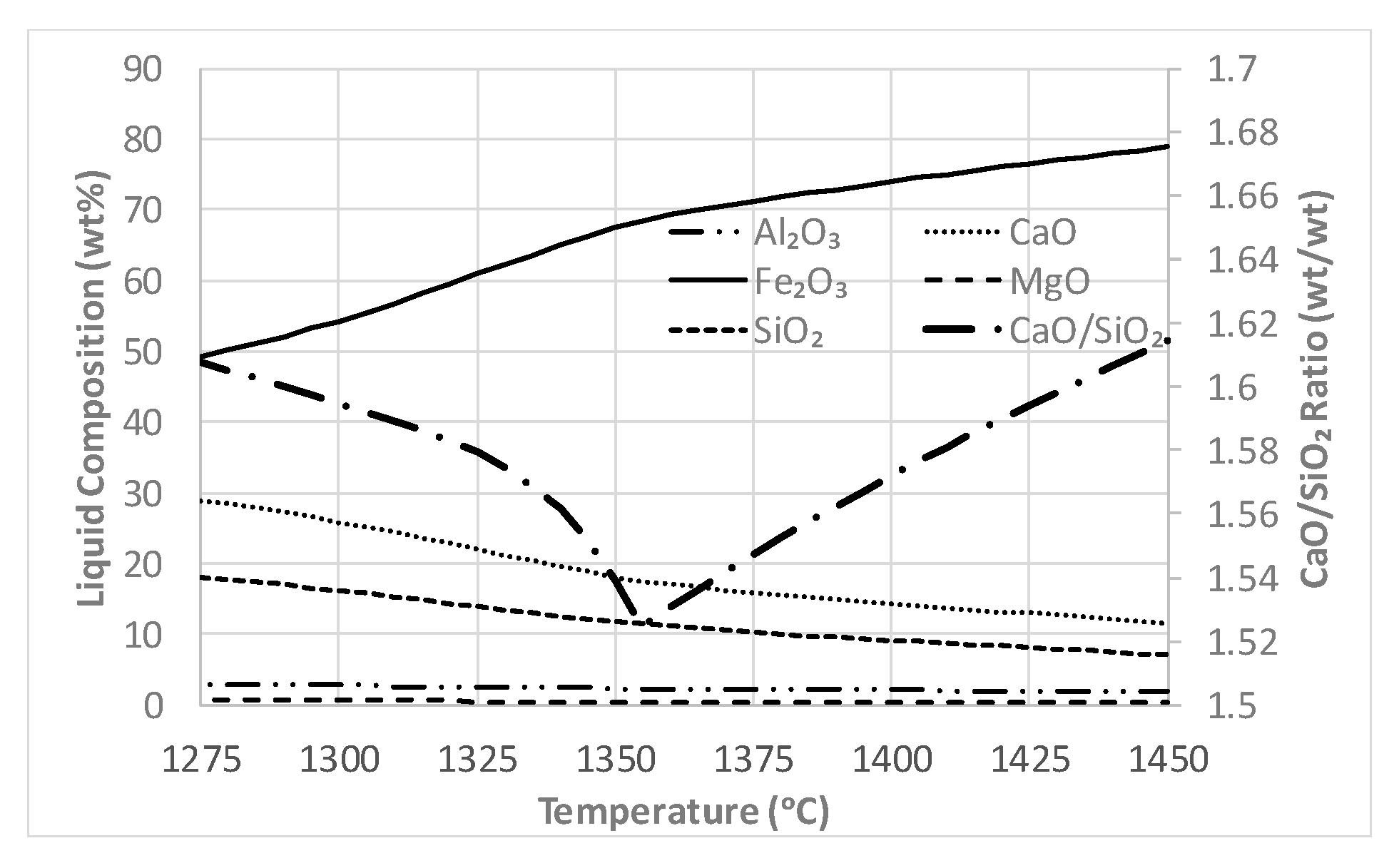
4.1.1. Hematite and Magnetite Stability
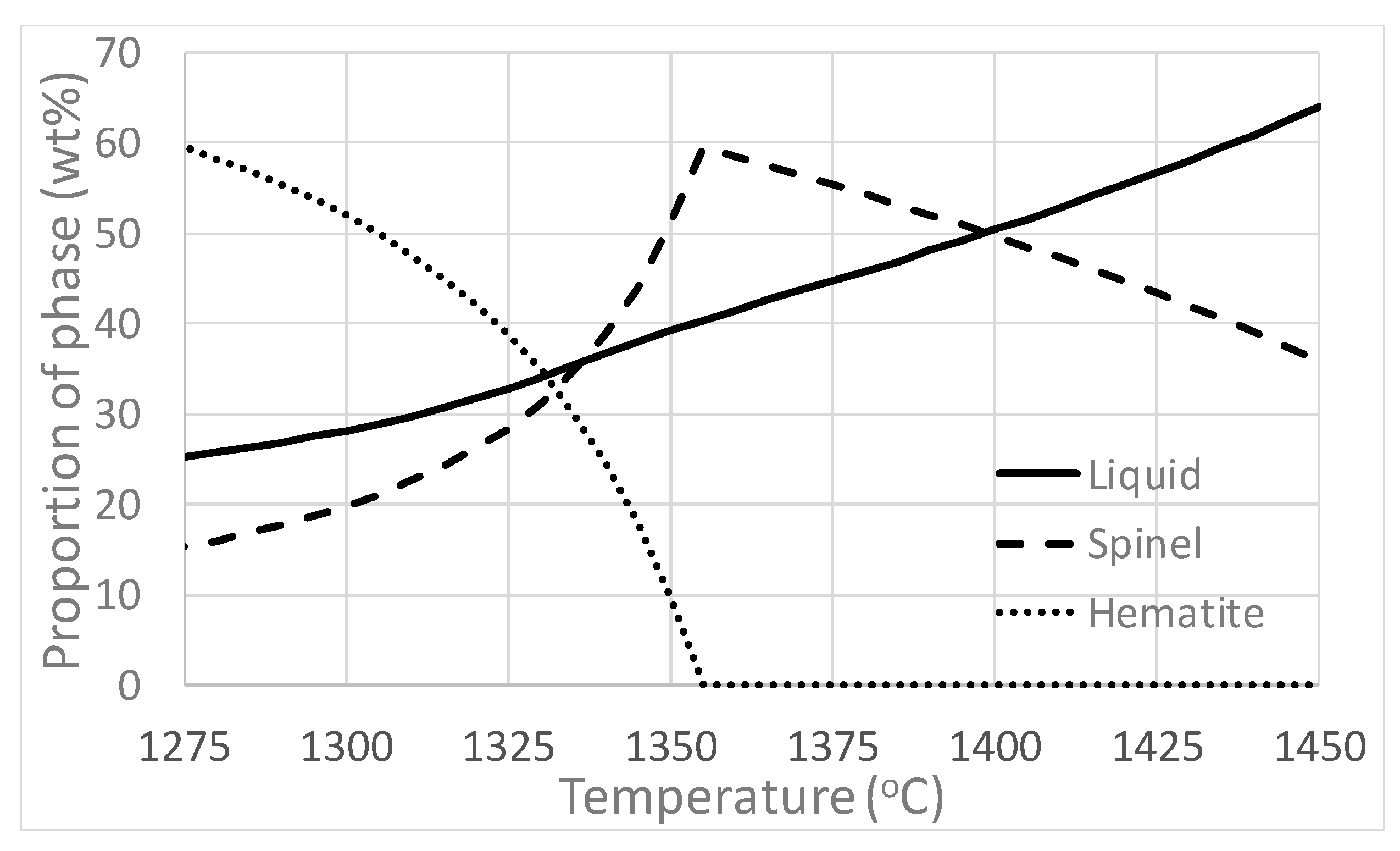
4.1.2. Targeted Laboratory Scale Experiments
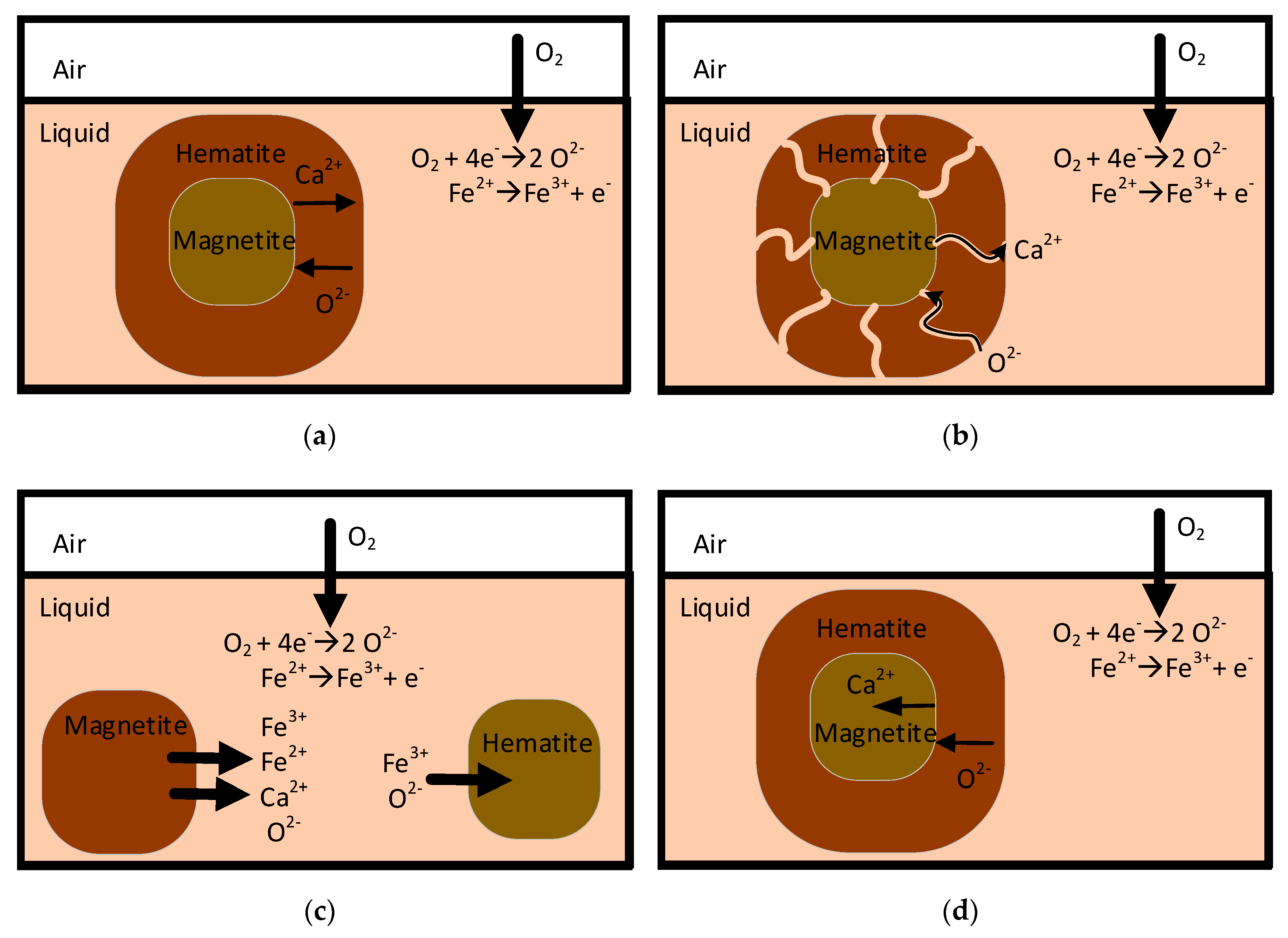
4.1.3. Mechanisms of Formation of Hematite and Magnetite
4.2. Analysis of Process Conditions
Relationship between Process Conditions and Microstructure
Comparison with Previous Studies
4.3. Processes Taking Place during Sintering
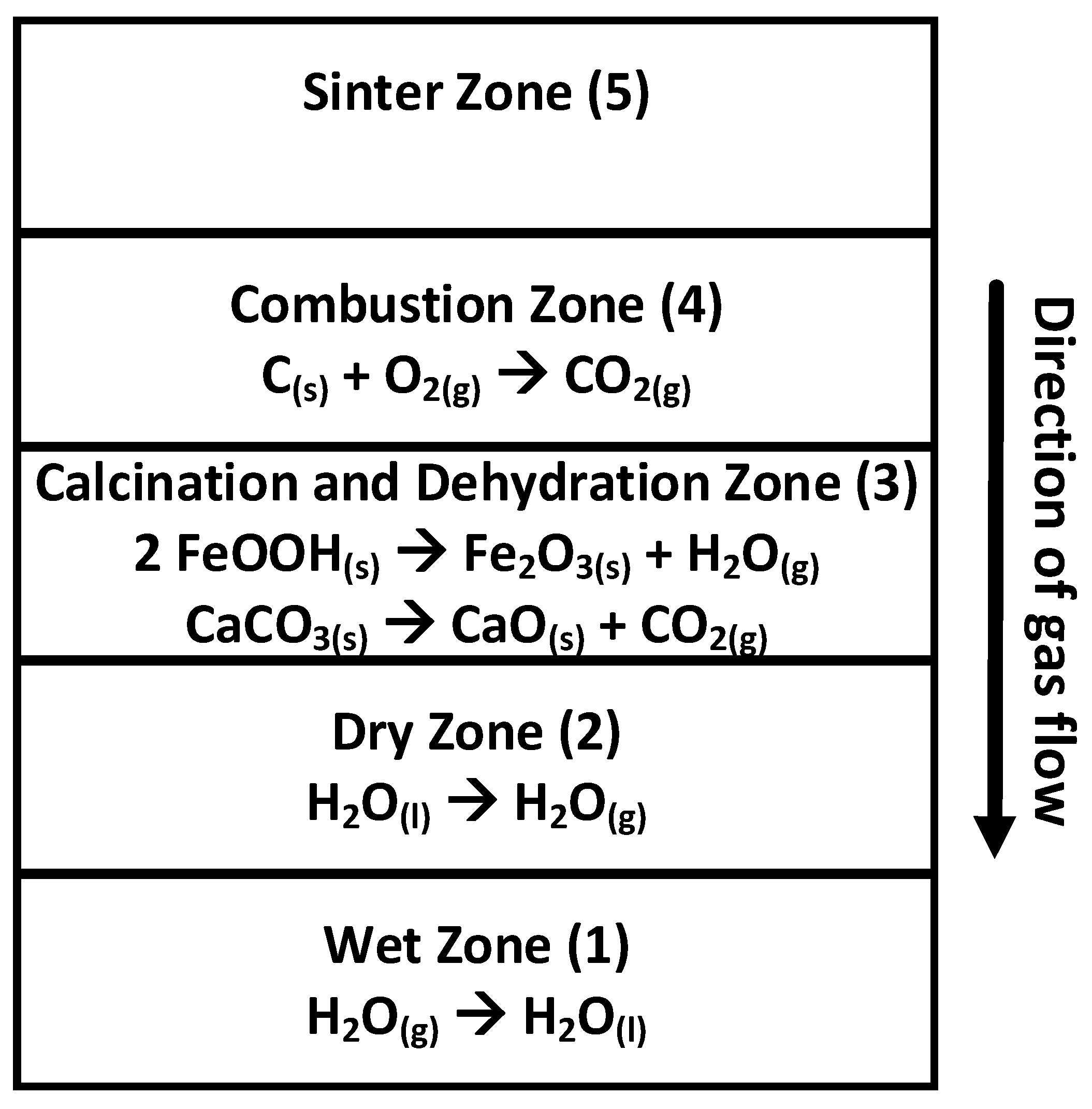
4.3.1. Heating: Calcination, Dehydration and Combustion
4.3.2. Peak Bed Temperature
4.3.3. Cooling
4.4. Stages of Sintering
- 1a.
- Heating solids under high/low oxygen pressures
- 1b.
- Heating solid/liquid mixtures under high/low oxygen pressures
- 2.
- Peak bed temperature under high/low oxygen pressures with solid/liquid mixtures
- 3.
- Cooling in solid/liquid mixtures under high oxygen pressures
- 4.
- Cooling solid mixtures under high oxygen pressures
5. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Ball, D. Agglomeration of Iron Ores; Heinemann Educational: London, UK, 1973. [Google Scholar]
- McBriar, E.; Johnson, W.; Andrews, K.W.; Davies, W. Nature of Ironstone Sinter. JISI 1954, 177, 316–323. [Google Scholar]
- Burlingame, R.; Bitsianes, G.; Joseph, T. Reaction Zones in the Iron Ore Sintering Process. JOM 1956, 8, 853–861. [Google Scholar] [CrossRef]
- Van den Berg, T.; de Villiers, J.; Cromarty, R. Variation of the redox conditions and the resultant phase assemblages during iron ore sintering. Int. J. Miner. Process. 2016, 150, 47–53. [Google Scholar] [CrossRef] [Green Version]
- The 94th Lecture Meeting of the Japan Iron and Steel Institute Lecture, Ironmaking/Environment. 1977. Available online: https://www.jstage.jst.go.jp/article/tetsutohagane1955/63/11/63_11_S512/_pdf/-char/ (accessed on 20 June 2019).
- Muller, J.; De Vries, T.; Dippenaar, B.; Vreugdenburg, J. A finite difference model of the iron ore sinter process. J. S. Afr. Inst. Min. Metall. 2015, 115, 409–417. [Google Scholar] [CrossRef]
- Yang, W.; Ryu, C.; Choi, S.; Choi, E.; Lee, D.; Huh, W. Modeling of Combustion and Heat Transfer in an Iron Ore Sintering Bed with Considerations of Multiple Solid Phases. ISIJ Int. 2004, 44, 492–499. [Google Scholar] [CrossRef]
- Zhou, H.; Zhao, J.P.; Loo, C.E.; Ellis, B.G.; Cen, K.F. Model Predictions of Important Bed and Gas Properties during Iron Ore Sintering. ISIJ Int. 2012, 52, 12–2168. [Google Scholar] [CrossRef]
- Zhou, H.; Zhao, J.P.; Loo, C.E.; Ellis, B.G.; Cen, K.F. Numerical Modeling of the Iron Ore Sintering Process. ISIJ Int. 2012, 52, 9–1550. [Google Scholar] [CrossRef]
- Zhao, J.; Loo, C. Dependence of flame front speed on iron ore sintering conditions. Miner. Process. Extr. Metall. Rev. 2016, 126, 3–165. [Google Scholar] [CrossRef]
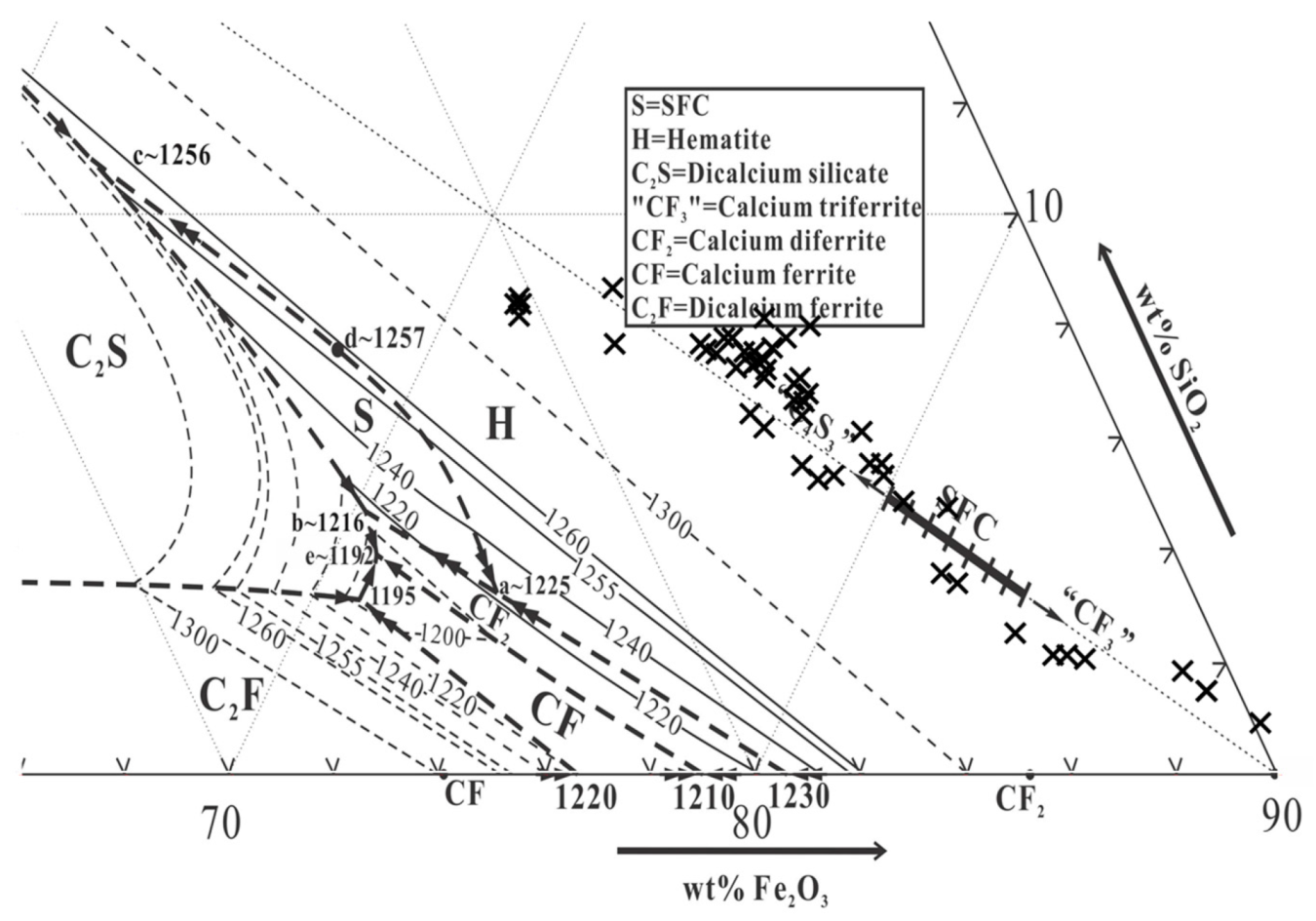
- Nicol, S.; Jak, E.; Hayes, P. Microstructure evolution during controlled solidification of “Fe2O3”-CaO-SiO2 liquids in air. Metall. Mater. Trans. B 2019, in press. [Google Scholar]
- Chen, J.; Shevchenko, M.; Hayes, P.C.; Jak, E. A phase equilibrium study of the iron-rich corner of the CaO-FeO–Fe2O3–SiO2 systems in air and the determination of the SFC primary phase field. ISIJ Int. 2019, 59, 795–804. [Google Scholar] [CrossRef]
- Pownceby, M.; Clout, J.; Fisher-White, M. Phase equilibria for the Fe2O3-rich part of the system Fe2O3–CaO–SiO2 in air at 1240–1300 °C. Trans. Inst. Min. Metall. Sect. C 1998, 107, C1–C10. [Google Scholar]
- Muan, A.; Osborn, E. Phase Equilibria among Oxides in Steelmaking; Reading Mass: Addison-Wesley Pub. Co.: Reading, MA, USA, 1965. [Google Scholar]
- Nicol, S.; Jak, E.; Hayes, P. Controlled solidification of liquids within the SFC primary phase field of the “Fe2O3”-CaO-SiO2 system in air. Metall. Mater. Trans. B 2019, in press. [Google Scholar]
- Simmonds, T.; Hayes, P. Isothermal Oxidation of Magnetite to Hematite in Air and Cyclic Reduction/Oxidation Under Carbon Looping Combustion Conditions. Metall. Mater. Trans. E 2017, 4, 2–114. [Google Scholar] [CrossRef]
- Földvári, M. Handbook of Thermogravimetric System of Minerals and Its Use in Geological Practice; Geological Institute of Hungary: Budapest, Hungary, 2011; Volume 213. [Google Scholar]
- Wall, T.; Gururajan, V.; Lucas, J.; Gupta, R.; Dong-Ke, Z.; Smith, I.; Young, B.; Wall, T. The ignition, burning rate and reactivity of petroleum coke. Symp. (Int.) Combust. 1991, 23, 1177–1184. [Google Scholar] [CrossRef]
- Sebastian, J.; Mayers, M. Coke Reactivity Determination by a Modified Ignition Point Method. Ind. Eng. Chem. 1937, 29, 10–1118. [Google Scholar] [CrossRef]
- Umadevi, T.; Brahmacharyulu, A.; Sah, R.; Mahapatra, P.C. Influence of sinter grate suction pressure (flame front speed) on microstructure, productivity and quality of iron ore sinter. Ironmak. Steelmak. 2014, 41, 6–410. [Google Scholar] [CrossRef]


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sintering Parameter | Base Case | Low Coke | High Coke | Low Draft | High Draft |
|---|---|---|---|---|---|
| Draft Pressure drop (kPa) | 14.7 | 14.7 | 14.7 | 12.7 | 16.7 |
| Coke (kg/t feed) | 36 | 33 | 39 | 36 | 33 |
| Coke breeze size (mm) | 1.5–1.8 | 1.5–1.8 | 1.5–1.8 | 1.5–1.8 | 1.5–1.8 |
| Sinter Returns (wt %) | 18.0 | 18.0 | 18.0 | 18.0 | 18.0 |
| Moisture (wt %) | 7.2 | 7.2 | 7.2 | 7.1 | 7.2 |
| Sinter Feed Size (mm) | 2.6 | 2.6 | 2.8 | 2.7 | 2.6 |
| Goethite in feed (wt %) | 20 | 20 | 20 | 20 | 20 |
| Hematite in feed (wt %) | 40 | 40 | 40 | 40 | 40 |
| Magnetite in feed (wt %) | 6–8 | 6–8 | 6–8 | 6–8 | 6–8 |
| Flame Front Speed (mm/min) | 23.9 | 18.5 | 22.8 | 19.7 | 24.2 |
| Sinter Yield (wt %) | 80.1 | 78.3 | 82.4 | 80.1 | 80.2 |
| Mean Sinter Lump Size (mm) | 24.2 | 26.1 | 24.6 | 26.5 | 23.5 |
| Sinter Composition | |||||
| SiO2 (wt %) | 4.6 | 4.6 | 4.8 | 4.7 | 4.5 |
| Al2O3 (wt %) | 1.7 | 1.7 | 1.8 | 1.6 | 1.6 |
| CaO (wt %) | 7.7 | 7.9 | 7.9 | 8.2 | 7.9 |
| MgO (wt %) | 1.2 | 1.3 | 1.3 | 1.3 | 1.3 |
| FeO (wt %) | 9.0 | 8.1 | 10.3 | 9.6 | 9.2 |
| Total Fe (wt %) | 59.8 | 59.6 | 59.6 | 59.5 | 59.9 |
| CaO/SiO2 (wt/wt) | 1.66 | 1.70 | 1.65 | 1.75 | 1.75 |
| Gas composition | |||||
| O2 (vol. %) | 13.54 | 11.51 | 11.55 | 13.70 | 13.28 |
| CO (vol. %) | 0.25 | 0.51 | 0.59 | 0.35 | 0.30 |
| CO2 (vol. %) | 2.42 | 4.51 | 4.50 | 3.59 | 2.47 |
| H2O (vol. %) | 19.73 | 20.00 | 20.00 | 17.13 | 20.00 |
| CO/CO2 (vol./vol.) | 0.10 | 0.11 | 0.13 | 0.10 | 0.12 |
| Phase | Composition (wt %) | ||||||
|---|---|---|---|---|---|---|---|
| “Fe2O3” | SiO2 | CaO | Al2O3 | MgO | MnO | ||
| Hematite | 1 | 97.8 | 0.0 | 0.2 | 1.9 | 0.1 | 0.0 |
| 2 | 98.6 | 0.2 | 0.7 | 0.5 | 0.0 | 0.0 | |
| 3 | 98.8 | 0.0 | 0.0 | 1.1 | 0.1 | 0.0 | |
| 4 | 98.9 | 0.0 | 0.2 | 0.8 | 0.0 | 0.1 | |
| 5 | 98.9 | 0.0 | 0.0 | 1.1 | 0.0 | 0.0 | |
| 6 | 99.0 | 0.0 | 0.1 | 0.9 | 0.0 | 0.0 | |
| Average | 98.7 | 0.0 | 0.2 | 1.1 | 0.0 | 0.0 | |
| Magnetite | 1 | 90.6 | 0.0 | 0.8 | 1.7 | 6.5 | 0.4 |
| 2 | 92.2 | 0.0 | 1.1 | 1.0 | 5.1 | 0.6 | |
| 3 | 92.3 | 0.0 | 1.3 | 1.1 | 4.8 | 0.5 | |
| 4 | 92.4 | 0.0 | 2.8 | 1.5 | 2.9 | 0.4 | |
| 5 | 93.0 | 0.0 | 1.6 | 2.0 | 3.1 | 0.3 | |
| 6 | 93.0 | 0.0 | 1.4 | 1.0 | 4.4 | 0.2 | |
| 7 | 93.6 | 0.0 | 2.3 | 1.0 | 2.9 | 0.2 | |
| 8 | 94.1 | 0.0 | 1.9 | 1.1 | 2.6 | 0.3 | |
| 9 | 94.1 | 0.0 | 1.8 | 0.8 | 3.0 | 0.3 | |
| 10 | 94.8 | 0.0 | 2.0 | 0.9 | 2.0 | 0.3 | |
| 11 | 95.7 | 0.0 | 0.9 | 0.6 | 2.5 | 0.3 | |
| Average | 93.3 | 0.0 | 1.6 | 1.2 | 3.6 | 0.3 | |
| SFCA | 1 | 65.5 | 8.8 | 14.6 | 7.8 | 3.0 | 0.2 |
| 2 | 65.6 | 9.0 | 14.9 | 7.6 | 2.8 | 0.1 | |
| 3 | 66.6 | 8.2 | 14.3 | 8.3 | 2.4 | 0.1 | |
| 4 | 66.9 | 8.4 | 16.5 | 8.1 | 0.0 | 0.0 | |
| 5 | 67.0 | 8.3 | 15.7 | 8.9 | 0.0 | 0.0 | |
| 6 | 67.2 | 8.1 | 16.1 | 8.6 | 0.1 | 0.0 | |
| 7 | 67.3 | 8.9 | 14.4 | 6.0 | 3.0 | 0.4 | |
| 8 | 67.5 | 9.1 | 14.7 | 5.8 | 2.8 | 0.1 | |
| 9 | 67.5 | 9.1 | 14.6 | 6.6 | 2.0 | 0.2 | |
| 10 | 70.9 | 7.9 | 15.4 | 4.6 | 1.0 | 0.2 | |
| 11 | 71.4 | 7.3 | 14.4 | 6.1 | 0.8 | 0.0 | |
| 12 | 71.6 | 7.7 | 15.2 | 4.3 | 1.0 | 0.2 | |
| 13 | 71.9 | 7.5 | 15.3 | 4.6 | 0.6 | 0.1 | |
| 14 | 72.0 | 7.8 | 15.0 | 3.9 | 1.1 | 0.2 | |
| 15 | 72.2 | 8.0 | 14.5 | 4.1 | 1.1 | 0.1 | |
| 16 | 72.3 | 8.0 | 14.4 | 4.0 | 1.2 | 0.1 | |
| 17 | 72.8 | 7.3 | 16.2 | 3.2 | 0.4 | 0.1 | |
| 18 | 73.0 | 7.4 | 15.2 | 3.8 | 0.6 | 0.1 | |
| 19 | 73.1 | 7.8 | 15.0 | 3.6 | 0.5 | 0.1 | |
| 20 | 73.2 | 7.7 | 15.4 | 3.1 | 0.6 | 0.0 | |
| 21 | 73.3 | 6.8 | 14.7 | 4.4 | 0.7 | 0.1 | |
| 22 | 73.3 | 7.2 | 14.6 | 3.9 | 0.9 | 0.1 | |
| 23 | 73.6 | 6.9 | 15.5 | 3.8 | 0.2 | 0.1 | |
| 24 | 73.6 | 7.4 | 15.1 | 3.0 | 0.7 | 0.2 | |
| 25 | 73.6 | 7.5 | 15.0 | 3.1 | 0.6 | 0.1 | |
| 26 | 73.8 | 6.5 | 16.0 | 3.1 | 0.5 | 0.1 | |
| 27 | 73.9 | 7.6 | 15.0 | 2.8 | 0.7 | 0.1 | |
| 28 | 74.2 | 6.7 | 15.2 | 3.3 | 0.4 | 0.1 | |
| 29 | 74.3 | 7.1 | 14.5 | 3.3 | 0.7 | 0.1 | |
| 30 | 74.3 | 6.4 | 15.2 | 3.4 | 0.5 | 0.1 | |
| 31 | 75.2 | 6.3 | 14.7 | 2.8 | 1.0 | 0.0 | |
| 32 | 75.3 | 6.2 | 14.1 | 3.9 | 0.5 | 0.0 | |
| 33 | 75.4 | 5.4 | 15.5 | 3.4 | 0.3 | 0.1 | |
| 34 | 75.7 | 5.6 | 14.3 | 3.3 | 1.0 | 0.1 | |
| 35 | 76.2 | 5.6 | 14.0 | 3.7 | 0.5 | 0.0 | |
| 36 | 76.7 | 5.6 | 13.5 | 3.2 | 0.8 | 0.1 | |
| 37 | 77.4 | 5.4 | 12.8 | 3.2 | 1.0 | 0.2 | |
| 38 | 77.8 | 5.4 | 12.4 | 2.6 | 1.7 | 0.1 | |
| 39 | 78.2 | 4.9 | 13.2 | 2.8 | 0.8 | 0.1 | |
| 40 | 79.3 | 3.7 | 12.4 | 3.5 | 1.1 | 0.1 | |
| 41 | 79.3 | 4.8 | 12.1 | 2.9 | 0.9 | 0.0 | |
| 42 | 80.6 | 3.5 | 11.3 | 3.0 | 1.5 | 0.3 | |
| 43 | 83.0 | 2.5 | 10.4 | 2.4 | 1.6 | 0.1 | |
| 44 | 83.1 | 2.2 | 9.8 | 3.0 | 1.6 | 0.3 | |
| 45 | 83.7 | 2.2 | 9.8 | 2.7 | 1.5 | 0.2 | |
| Average | 73.6 | 6.7 | 14.3 | 4.3 | 1.0 | 0.1 | |
| Liquid | 1 | 16.0 | 36.0 | 44.3 | 3.6 | 0.1 | 0.1 |
| 2 | 16.9 | 35.7 | 41.5 | 5.3 | 0.6 | 0.1 | |
| 3 | 17.4 | 37.6 | 42.2 | 2.5 | 0.3 | 0.0 | |
| 4 | 18.1 | 39.9 | 37.4 | 4.2 | 0.3 | 0.0 | |
| 5 | 18.6 | 35.0 | 43.7 | 1.9 | 0.8 | 0.0 | |
| 6 | 18.7 | 36.9 | 39.7 | 4.6 | 0.1 | 0.0 | |
| Average | 17.6 | 36.8 | 41.5 | 3.7 | 0.4 | 0.0 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicol, S.; Chen, J.; Qi, W.; Mao, X.; Jak, E.; Hayes, P.C. Measurement of Process Conditions Present in Pilot Scale Iron Ore Sintering. Minerals 2019, 9, 374. https://doi.org/10.3390/min9060374
Nicol S, Chen J, Qi W, Mao X, Jak E, Hayes PC. Measurement of Process Conditions Present in Pilot Scale Iron Ore Sintering. Minerals. 2019; 9(6):374. https://doi.org/10.3390/min9060374
Chicago/Turabian StyleNicol, Stuart, Jiang Chen, Wei Qi, Xiaoming Mao, Evgueni Jak, and Peter C. Hayes. 2019. "Measurement of Process Conditions Present in Pilot Scale Iron Ore Sintering" Minerals 9, no. 6: 374. https://doi.org/10.3390/min9060374